Methylaluminoxane Catalysts
Chih-Feng Huang,1 Shih-Kai Wang,1 Shiao-Wei Kuo,1 Wu-Jang Huang,2 Feng-Chih Chang1 1Institute of Applied Chemistry, National Chiao Tung University, Hsin Chu, Taiwan, Republic of China
2Department of Environmental Science and Engineering, National Pingtung University Science and Technology,
Pingtung, Taiwan, Republic of China
Received 2 August 2003; accepted 12 November 2003
ABSTRACT: The homopolymerization and copolymeriza-tion of tert-butyl methacrylate (tBMA) and norbornene (NB) with nickel(II) acetylacetonate in combination with methyl-aluminoxane were systematically investigated. This catalytic system showed high activity toward the homopolymeriza-tion of both NB and tBMA. For these copolymerizahomopolymeriza-tions, activity was gradually lost with an addition of tBMA to NB or of NB to tBMA. This result was qualitatively explained with the trigger coordination mechanism. Furthermore, the determination of the reactivity ratios indicated a signifi-cantly higher reactivity for NB than for tBMA (rNB⫽ 4.14
and rtBMA⫽ 0.097), and this was interpreted by the coordi-nation mechanism. The synthesized acrylate/NB copoly-mers exhibited glass-transition temperatures of 100 –250°C. The absence of crystallinity and the homogeneous reparti-tion of the monomer units along the main chain yielded products with high transparency and high stability © 2004 Wiley Periodicals, Inc. J Appl Polym Sci 92: 1824 –1833, 2004
Key words:polynorbornene; metallocene; methylaluminox-ane (MAO)
INTRODUCTION
Norbornene (NB) and its derivatives can be polymer-ized through ring-opening metathesis polymeriza-tion,1,2 cationic polymerization,3 and olefin addition polymerization. In the last case, the bicyclic structure of the monomer is retained. Depending on the cata-lytic system used, different polynorbornenes (PNBs) may be obtained, ranging from amorphous ones to more stereoregular and crystalline ones.4 – 6Molecular dynamic simulations have shown that PNBs display a characteristic rigid random-coil conformation and re-stricted rotation around the main chain.7,8According to their peculiar structure, these polymers exhibit very high decomposition temperature, high thermal stabil-ity, excellent dielectric properties, and unusual trans-port properties. Therefore, they are attractive materi-als not only for microelectronics and optical applica-tions7 but also for other potential applications in packing and gas separation.9
However, the PNB homopolymer presents some disadvantages: it is a very brittle material at room temperature, and its solubility in common organic solvents is rather low. In addition, the processability
of the homopolymer is very poor. Therefore, to im-prove the processability, researchers have investigated the copolymerization of NB with other traditional vi-nyl monomers such as ethylene. Through the addition of these monomer units to the polymer chain, the solubility of the polymers in common organic solvents can be increased.
The polymerization of NB can be achieved with some selected catalyst systems based on metal-locenes6,10,11or Pd(II) and Ni(II) complexes.4,5,7–9,12–14
Metallocene-based systems lead to a stereospecific ho-mopolymer, but the monomer conversion rate is rather low. However, palladium catalysts usually give amorphous polymers.
The use of organo-nickel complexes for the poly-merization of NB or substituted functional NB mono-mers has been described in the patent literature.15–17 Recently, it has also been reported that nickel-based methylaluminoxane (MAO) catalysts can polymerize NB with acrylate, diene, NB, and styrene mono-mers.18 –26The polymerization of NB with this type of catalyst system has also been mentioned in Japanese patents,27,28 but no details have been given.
In this article, we report a preliminary study of the homopolymerization and copolymerization of NB with tert-butyl methacrylate (tBMA) in the presence of nickel(II) acetylacetonate [Ni(acac)2] associated with
an MAO catalyst system. The thermal properties of the produced copolymer are also presented in detail.
Correspondence to: F.-C. Chang (changfc@cc.nctu.edu.tw).
Contract grant sponsor: National Science Council of Tai-wan; contract grant number: NSC-91-2216-E-009-018. Journal of Applied Polymer Science, Vol. 92, 1824–1833 (2004) © 2004 Wiley Periodicals, Inc.
EXPERIMENTAL Materials
Toluene, Ni(acac)2, NB, and tBMA with a purity of
99% were supplied by Aldrich Chemical Co. (Milwau-kee, WI). NB and tBMA were distilled under reduced pressure before use. Ni(acac)2was used without
fur-ther purification. Toluene was distilled twice over so-dium/benzophenone before use. MAO (10 wt % in toluene) was used as received.
Polymerization
The polymerization was carried out under dry nitro-gen in toluene at room temperature in a glass flask equipped with a rubber septum. Ni(acac)2 was first dissolved in the reaction solvent, and MAO was then added. After 15 min for the activation of the catalytic system, the monomers were finally introduced to ini-tiate the polymerization. After a give reaction time, the polymerization mixture was poured into a large amount of methanol containing a few percent of hy-drochloric acid to precipitate the polymer. The liquid phase was filtered, and the recovered polymer was dried in vacuo. The percentage of the polymer yield was calculated as the weight function of the converted monomer over the total monomer as follows:
Yield (%) ⫽
Weight of resulting polymer
Weight of total monomers ⫻ 100% (1) The catalytic activity was defined as the weight of the resulting polymer divided by the amount of intro-duced Ni(II) and the polymerization time (h):
Catalytic activity⫽
Weight of resulting polymer WeightNi(II)⫻ Timepolymerization
(2)
Characterization
The thermal properties were measured with a TA Instrument 2920 differential scanning calorimeter (USA) with a mechanical cooling accessory. Each sam-ple was first heated to 250°C and was kept at that temperature for 5 min. It was then quickly quenched to 30°C. The glass-transition temperature (Tg) was
ob-tained as the inflection point of the jump heat capacity with a scanning rate of 20°C/min and within a tem-perature range of 30 –200°C. IR spectra of copolymer films were determined with the conventional NaCl disk method. The film used in this study was suffi-ciently thin to obey the Beer–Lambert law. Fourier transform infrared (FTIR) measurements were
re-corded on a Nicolet Avatar 320 FTIR spectrophotom-eter (USA), and 32 scans were collected with a spectral resolution of 1 cm⫺1. 13C and 1H spectra were re-corded on a Varian spectrometer (USA) and a Bruker AC300 spectrometer (Germany) in C6D6, and
high-resolution solid-state13C-NMR experiments were car-ried out at room temperature with a Bruker DSX-400 spectrometer operating at resonance frequencies of 399.53 and 100.47 MHz for1H and 13C, respec-tively. The 13C cross-polarization/magic-angle spinning (CP–MAS) spectra were measured with a 3.9-s 90° pulse, with a 3-s pulse delay time, an acquisition time of 30 ms, and 2048 scans. All NMR spectra were taken at 300 K with broad-band proton decoupling and a normal cross-polarization pulse sequence. A magic-angle-sample spinning rate of 5.4 kHz was used to avoid absorption overlapping.
RESULTS AND DISCUSSION Polymer syntheses
The polymerization activity of Ziegler–Natta and met-allocene catalysts depends on several parameters, such as the aluminum/transition-metal ratio, the po-lymerization temperature, the transition-metal/mono-mer ratio, and the monotransition-metal/mono-mer composition. In this study, we investigated how these parameters could affect the activity of the Ni(acac)2/MAO catalytic
sys-tem in the homopolymerization and copolymerization of NB with tBMA.
Influence of the catalyst system
Preliminary experiments were made to check the po-lymerization activity of each component. The Ni(a-cac)2catalyst alone in the absence of MAO was
inac-tive for the NB and tBMA polymerization, and MAO alone was able to initiate the NB polymerization with very low activity. Furthermore, in a previous study by
Figure 1 Yield of NB polymerization ([NB] ⫽ 7.4 mol/L, polymerization time⫽ 4 h, temperature ⫽ 0°C, and [Ni] ⫽ 0.0016 mol/L): (■) Ni(acac)2/MAO and (䊐) MAO alone.
Endo et al.,18 MAO alone was unable to polymerize with tBMA because of the cationic mechanism. Figure 1 shows that the polymer yield obtained in the pres-ence of MAO alone was substantially lower than that obtained in the Ni(acac)2/MAO system. This result
reveals that the combination of Ni(acac)2and MAO is
essential in this catalytic system and that the active centers are formed through interactions between these two components.
Influence of the polymerization time
Figure 2 displays the influence of the polymerization time on the yield for both homopolymerization and copolymerization. For homopolymerization, the poly-mer yield increased as the time increased, and two distinct periods could be observed in the polymeriza-tion process. In the first period of 0 –2 h, there was a rapid increase of the yield with the time. In the second period (ca. 2 h), the polymer yield increased only slightly or approached a constant value. For copoly-merization, the overall activity was relatively lower at 0.75% after 0.5 h and remained unchanged thereafter. Influence of the polymerization temperature A series of polymerization runs were performed at temperatures ranging from 0 to 40°C while other
pa-Figure 3 Dependence of the yield on the polymerization temperature (composition feed ratio of the tBMA/NB copol-ymer⫽ 0.41/0.58, [Ni] ⫽ 0.0016 mol/L, Ai/Ni ⫽ 100, and time⫽ 1 h).
Figure 4 Dependence of the yield on the Al/Ni ratio (com-position feed ratio of the tBMA/NB copolymer⫽ 0.41/0.58, [Ni]⫽ 0.0016 mol/L, temperature ⫽ 0°C, and time ⫽ 1 h).
Figure 2 Dependence of the yield on the polymerization time (composition feed ratio of the tBMA/NB copolymer ⫽ 0.41/0.58, [Ni] ⫽ 0.0016 mol/L, Ai/Ni ⫽ 100, and tem-perature⫽ 0°C).
rameters were kept constant. Figure 3 shows no sig-nificant effects of the temperature on the polymer yield or catalyst activity for both homopolymerization and copolymerization.
Influence of the Al/Ni ratio
The dependence of the polymerization yield on the Al/Ni molar ratio is shown in Figure 4. The observed behavior for both homopolymerization and copolymer-ization is common to many Ziegler–Natta and metallo-cene systems, the catalytic activity increasing initially and then remaining nearly constant. These results can be interpreted as an increasing number of active species. At a certain higher Al/Ni molar ratio, it approached its maximum concentration of active species. In the case of NB homopolymerization, the maximum yield was ob-tained at a relatively lower Al/Ni ratio of about 150. For the tBMA homopolymerization, a slightly higher Al/Ni ratio (⬎200) was necessary to obtain the maximum yield. Despite the higher yield obtained in the homopolymer-ization reactions (55% for NB and 60% for tBMA), it was surprising to find that the maximum yield of the copo-lymerization of NB and tBMA, at the same high Al/Ni ratio (⬎200), was only at 0.74%.
Influence of the moles of nickel
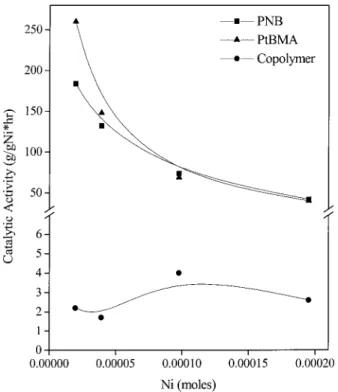
Figure 5 shows the dependence of the catalytic activity on the moles of nickel. For both NB and tBMA
ho-mopolymerizations, an exponential decrease in the activity with the moles of nickel was observed. The activity of the copolymerization of NB and tBMA was extremely low but seemed to be independent of the nickel concentration.
Influence of the monomer composition
A series of copolymerization reactions were per-formed with different feed compositions while other parameters were kept constant, such as the Al/Ni ratio, moles of nickel, temperature, and time. Figure 6 shows the dependence of the polymerization yield on the initial feed composition. For comparison, the yields of the homopolymerizations are also presented. Increasing the initial tBMA feed led to a drastic de-crease in the product yield of the copolymer with respect to the NB homopolymerization. However, de-spite the high activity observed for the tBMA homopo-lymerization, a low content of NB (12%) in the initial feed caused it to lose nearly all its activity in compar-ison with the tBMA homopolymerization. This pecu-liar behavior of the Ni(acac)2/MAO system in the
NB/tBMA copolymerization is discussed later. Polymer characterizations
13C-NMR spectroscopy was used to characterize the
structures and compositions of the homopolymers and copolymers. The spectra obtained for the
ho-Figure 5 Catalyst activity versus the moles of nickel (com-position feed ratio of the tBMA/NB copolymer⫽ 0.41/0.58, amount of Al ⫽ 0.01 mol, temperature ⫽ 0°C, and time ⫽ 1 h).
Figure 6 Influence of the composition of the feed on the yield ([Ni] ⫽ 0.0016 mol/L, Al/Ni ⫽ 100, temperature ⫽ 0°C, and time ⫽ 4 h).
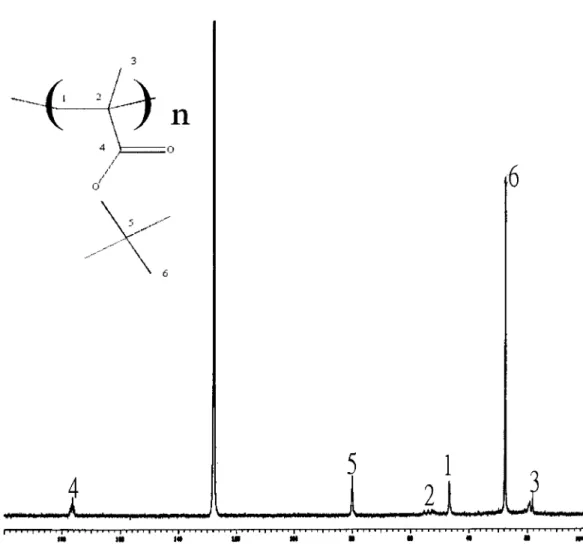
mopolymer of tBMA are shown in Figure 7, and the peak assignments have been reported previously.29 –33 Solid-state 13C-NMR was used to compare the two homopolymers and the copolymer of NB and tBMA because PNB is insoluble in most organic solvents. Figure 8 shows the major peaks of PNB between 20 and 60 ppm, which are essentially the same as those reported in the literature.29,30,34These results indicate that the polymerization of NB with the Ni(acac)2/
MAO system occurred without ring opening and via 2,3-addition. As shown in Figure 8, the obtained co-polymer included major peaks from these homopoly-mers. However, the carbonyl peak of tBMA (176.6 ppm) in the copolymer shifted to a higher field than pure poly(tert-butyl methacrylate) (PtBMA; 171.1 ppm). It is well known that the chemical shift strongly depends on the chemical environment of neighboring molecules and conformation packing.35 In general, a chemical shift to a higher field implies that the car-bonyl group of tBMA is in a diluent segment. In light of the chemical structure of PNB, the nonpolar group of PNB may act as an inert diluent segment for the carbonyl group of tBMA. If PNB and PtBMA were physically mixed, we would not expect any chemical
Figure 7 Liquid-state13C-NMR spectrum of PtBMA in C6D6.
Figure 8 CP–MAS solid-state 13
C-NMR spectra of PNB, PtBMA, and the NB/tBMA copolymer with a feed ratio of 0.2/0.8.
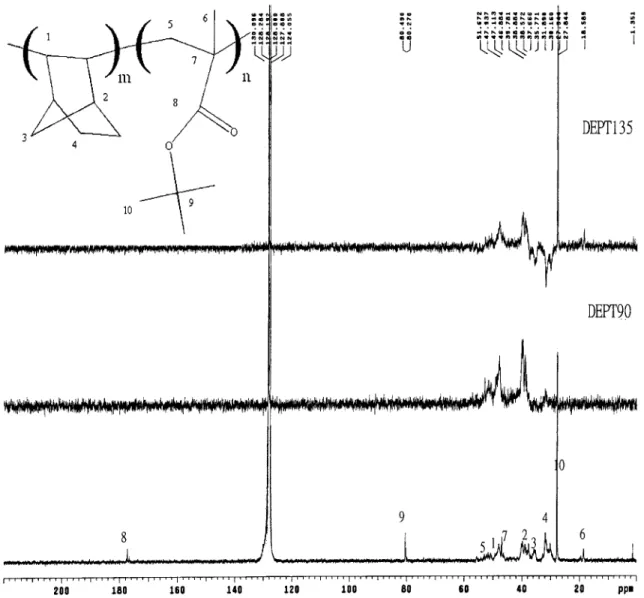
shift of the tBMA carbonyl because of no specific interaction between these two polymers. In Figure 9, the liquid 13C-NMR and distortionless-enhancement-by-polarization-transfer (DEPT) techniques have been used to assign peaks appearing in the spectrum of the copolymer. All these peaks have been labeled accord-ing to the structure of the two monomer units in the polymer chain. The characteristic peak from the CAC double bond is absent from the spectrum, whereas the four usual groups of resonances (1– 4 labeled peaks) appear.29,30 This means that the copolymerization of NB and tBMA with the Ni(acac)2/MAO catalyst
sys-tem took place without ring opening and via an addi-tion polymerizaaddi-tion.
The proportions of these two monomers in the co-polymer prepared at a low conversion were deter-mined through an analysis of these peak areas. The molar fractions of the copolymer were determined through these well-resolved peaks. For example, the molar fraction of tBMA in the NB/tBMA copolymer
was determined from the C10 peak of tBMA and the C3 peak of NB.
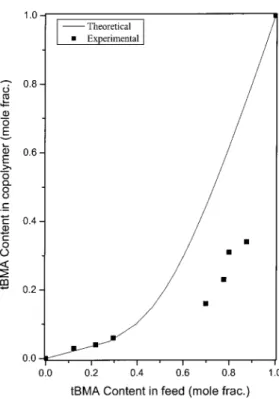
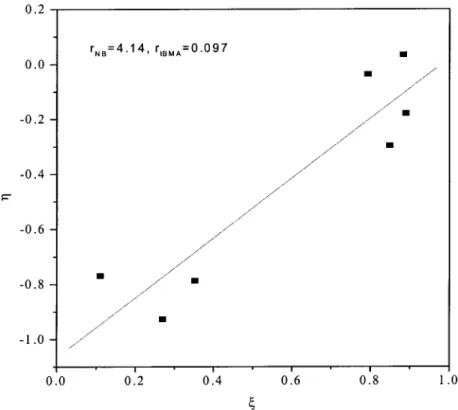
The variation of the copolymer composition deter-mined by 13C-NMR versus the copolymer feed is shown in Figure 10, which indicates that the incorpo-ration of tBMA into the copolymer chain was substan-tially slower than that of NB. In addition, the IR spec-trum of Figure 11 shows the same trend: the carbonyl group of tBMA decreased as NB increased in the feed. The reactivity ratios determined by the Kelen–Tu-dos36,37 method were rtBMA ⫽ 0.097 and rNB ⫽ 4.14
(Fig. 12); they implied that NB had significantly higher reactivity than tBMA. These observed reactivity ratios were rather unusual, clearly not agreeing with a free-radical or cationic polymerization mechanism but in-stead supporting a coordination-type mechanism. The mechanism is discussed in detail later.
The Tgvalues of the tBMA and NB homopolymers
and the corresponding copolymers were measured with differential scanning calorimetry (DSC). Figure
Figure 9 Liquid-state13
C-NMR DEPT spectrum of the NB/tBMA copolymer with a feed ratio of 0.2/0.8.
13 shows that the Tgvalues of pure PtBMA and PNB
were 104 and 230°C, respectively.38 A single Tg was
observed for the copolymer (NB feed ratio⫽ 30%) at 134°C.
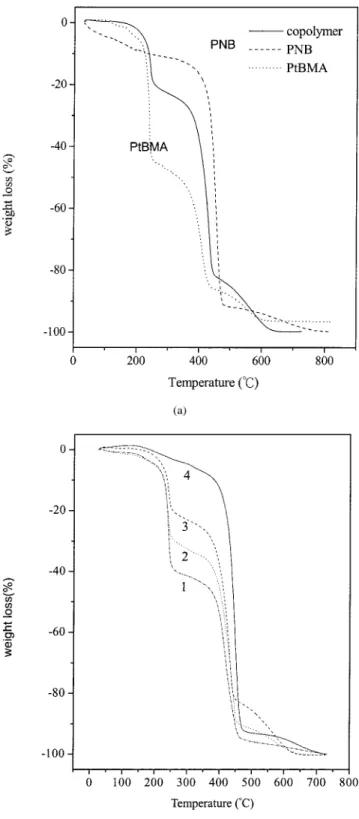
The thermogravimetric analysis (TGA) thermo-grams for the PtBMA and PNB homopolymers and the tBMA/NB copolymers are shown in Figure 14(a,b). PtBMA showed three weight losses, whereas only a one-step weight loss was found for pure PNB. The first-step and second-step weight losses of pure Pt-BMA, at 250 and 400°C, were due to anhydride bond formation between adjacent groups for PtBMA and main-chain decomposition, respectively. The third-step weight loss at 500°C was due to the decomposi-tion of the anhydride bond.33,37The observed one-step weight loss of pure PNB at 450°C was due to the decomposition of the main chain. For the tBMA/NB copolymer, three-step decomposition occurred at 250, 425, and 500°C; this was the same as for their respec-tive homopolymers, as shown in Figure 14(a). In Fig-ure 14, we can see the same tendency of the copolymer compositions determined by NMR and IR spectros-copy. The extent of weight loss at 250°C of the copol-ymer decreased with an increase in the NB constant, as expected. Meanwhile, the extent of weight loss at 425°C increased with the increase in the NB content because of the decrease in the tBMA fraction and the increase in the NB content in the copolymer chain.
Polymerization with nickel-based catalysts can pre-cede by a coordination mechanism.15 In fact, the13
C-NMR spectra of PNB revealed that the polymerization of NB with the Ni(acac)2/MAO system occurred
with-out ring opening and via 2,3-addition; this was similar to a previous study.16The 2,3-addition mode is char-acteristic of a coordination mechanism. On the con-trary, radical or cationic polymerization leads to 2,7-addition.3In addition, the reactivity ratios determined by the Kelen–Tudos method were significantly higher for NB (rNB ⫽ 4.14) than for tBMA (rtBMA⫽ 0.097). It
is well known that the observed reactivity ratio in free-radical polymerization is in reverse order (rtBMA
Ⰷ rNB). This result suggests that a coordination
mech-anism is involved in this Ni(acac)2/MAO system. On
the basis of this mechanism, we can assume that the interaction between the nickel complex and MAO pro-duces a cationic center. In the absence of the mono-mer, MAO and nickel centers closely interact and lead to lower activity. In the presence of monomers, a– bond complex is involved in the cationic nickel center and results in higher reactivity. These complexed monomers are inserted between the alkyl group and the nickel center. The repetition of the complexation– insertion step leads to polymer chain growth and is terminated through  elimination of the hydrogen atom. It is well known that chain termination and transfer reactions can proceed through elimination of the hydrogen atom from the last inserted␣ olefin.39 In contrast, the  elimination of hydrogen would be much more difficult for the last inserted NB because of the strain effect of the cycloring, which is thermody-namically unfavorable. This result can explain the re-activity ratio of NB being much higher than that of tBMA in copolymerization.
Figure 11 FTIR spectra of the polymers synthesized with the following NB/tBMA feed ratios: (a) 100/0, (b) 87/13, (c) 64/36, (d) 58/42, (e) 53/47, (f) 22/78, and (g) 0/100.
Figure 10 Influence of the monomer feed on the tBMA/NB copolymer composition. The theoretical line represents the prediction by the Kelen–Tudos method.
Nevertheless, some results are difficult to explain by the coordination mechanism. A very low content of NB in the initial feed (12 mol %) led to a drastic loss of activity in comparison with tBMA
homopolymeriza-tion, whereas a gradual loss of activity with respect to NB homopolymerization was observed in the pres-ence of a significant amount of tBMA (Figs. 6 and 10). This peculiar behavior can be interpreted in a
qualita-Figure 13 DSC thermograms for the homopolymers and copolymer. The NB/tBMA feed ratio for the copolymer was 0.3/0.7.
Figure 12 Plot for the Kelen–Tudos method.
tive way according to the trigger mechanism proposed by Yestene.40 This mechanism can properly explain the complex copolymerization behavior because the complexation of the first monomer is a step distinctly
different from the propagation step. Therefore, differ-ent monomers may have differdiffer-ent abilities to activate the catalytic center to produce different numbers of active species. These active centers are never free in this mechanism; they are always accompanied by a complexed monomer. The complexed monomer will insert into the growing chain only if another monomer is ready to be complexed. On this basis, we assume that the propagation step may proceed via a trigger mechanism and that the complexed monomer is in-serted only if a new monomer is available to be com-plexed. Considering the strain effect of the cycloring, we may assume that NB has a stronger tendency to complex with the cationic nickel center than tBMA. As a result, the NB monomer is readily available to trig-ger the insertion of previously complexed NB or tBMA into the growing polymer chain and complexes itself with the cationic center. A tBMA monomer is also able to trigger a complexed tBMA, but its ability to force the insertion of already complexed NB into the polymer chain and complex itself with the nickel cen-ter is rather weak. The latcen-ter trigger action is possible, but its probability is very low and increases slowly with an increase in the tBMA concentration.
Therefore, NB displays a much higher rate of incor-porating into the polymer chain than tBMA in NB/ tBMA copolymerization. The use of a higher concen-tration of tBMA may slightly increase the complex-ation and incorporcomplex-ation of tBMA, but the tBMA monomer is unable to trigger and replace the com-plexed NB monomer. As a result, the extent of tBMA incorporation into the polymer is significantly less (in agreement with the experimental reactivity ratio; see Fig. 10) and results in lower reactivity. However, the tBMA polymerization is strongly inhibited by the presence of a small amount of NB. As previously assumed, nickel centers will complex preferentially with NB, but the NB content is rather low. Polymer-ization must proceed by tBMA complexation, as men-tioned previously. On the contrary, the ability of NB complexed with the nickel center is very slow. In agreement with our assumption based on the trigger mechanism involved in this polymerization system, Peruch et al.23and Zhao et al.26reported a similar case for the homopolymerization and copolymerization of NB and styrene in the presence of the nickel-based/ MAO system. However, a detailed kinetic investiga-tion of both the homopolymerizainvestiga-tion and copolymer-ization of NB and tBMA initiated by the Ni(acac)2/
MAO system is necessary to provide further evidence supporting our assumption.
CONCLUSIONS
The homopolymerization and copolymerization of tBMA and NB with Ni(acac)2 in combination with
MAO were successfully performed. For these
copoly-Figure 14 TGA thermograms for (a) the homopolymers and copolymer of NB and tBMA and (b) the copolymers of NB and tBMA with different NB/tBMA feed compositions: (1) 12/88, (2) 22/78, (3) 30/70, and (4) 87/13.
merizations, a gradual loss in the reactivity was found with the addition of tBMA to NB or of NB to tBMA because of the trigger coordination mechanism. The determination of the reactivity ratios indicated a much higher reactivity for NB than for tBMA (rNB⫽ 4.14 and
rtBMA⫽ 0.097); this was interpreted with the
coordi-nation mechanism. References
1. Dragutan, V.; Balaban, A.; Dimonie, M. Olefin Metathesis and Ring-Opening Polymerization of Cyclo-Olefin; Wiley-Inter-science: Chichester, England, 1985.
2. Grubbs, R. H. Comprehensive Organometallic Chemistry; Per-gamon: Oxford, UK, 1982.
3. Gaylord, N. G.; Deohpande, A.; Mandal, B.; Martan, M. J Mac-romol Sci 1977, 11, 1053.
4. Mehler, C.; Risse, W. Makromol Chem Rapid Commum 1991, 12, 255.
5. Mehler, C.; Riss, W. Macromolecules 1992, 25, 4226. 6. Kaminsky, W.; Bark, A.; Steiger, R. J Mol Catal 1992, 74, 109. 7. Haselwander, T.; Heitz, W.; Krugel, S.; Wendoff, J. Macromol
Chem Phys 1996, 197, 3435.
8. Haselwander, T.; Heitz, W.; Krugel, S.; Wendoff, J. Macromol-ecules 1997, 30, 5345.
9. Dorkenoo, K.; Pfrom, P.; Rezac, M. J Polym Sci Part B: Polym Phys 1998, 36, 797.
10. Henschke, O.; Koller, F.; Arnold, M. Macromol Rapid Commun 1997, 18, 617.
11. Bergstrom, C.; Seppala, J. J Appl Polym Sci 1997, 63, 2022. 12. Schulz, R. G. Polym Lett 1966, 4, 541.
13. Tanielian, C.; Kiennemann, O. Can J Chem 1979, 57, 2022. 14. Sen, A.; Lai, T. W. Organometallics 1982, 1, 415.
15. Goodall, B.; Benedikt, G.; Mcintosh, C.; Barnes, D. U.S. Pat. 5,468,819 (1995).
16. Goodall, B.; Benedikt, G.; Mcintosh, C.; Barnes, D. U.S. Pat. 5,569,730 (1995).
17. Goodall, B.; Benedikt, G.; Mcintosh, C.; Barnes, D. U.S. Pat. 5,571,881 (1995).
18. Endo, K.; Masaki, K.; Ronbunshu, K. Chem Abstr 1994, 121, 281314.
19. Endo, K.; Uchida, Y.; Matsuda, Y. Macromol Chem Phys 1996, 197, 3315.
20. Endo, K.; Uchida, Y.; Masaki, K. Polym J 1997, 29, 583. 21. Endo, K.; Inukai, A.; Otsu, T. Macromol Rapid Commum 1994,
15, 893.
22. Endo, K. Macromol Chem Phys 1999, 200, 1722.
23. Peruch, F.; Cramail, H.; Deffieux, A. Macromol Chem Phys 1998, 199, 2221.
24. Geraldo, L.; Carlotta, B.; Ripa, A.; Giarrusso, A.; Porri, L. Mac-romol Rapid Commun 1997, 18, 801.
25. Coutinho, F.; Monteiro, L.; Costa, M. Polym Bull 1998, 40, 423. 26. Zhao, C. T.; Ribeiro, M. D. R.; Portela, M. F. Eur Polym J 2001,
37, 45.
27. Jpn. Pat. 0665,319 [9465,319]; Chem Abstr 121, 58228p (1994). 28. Jpn. Pat. 0718,021 [9518,021]; Chem Abstr 122, 291806f (1995). 29. Michael, A. R.; Beulich, I. Macromolecules 1999, 32, 7335. 30. Rische, T.; Waddon, A. J.; Dickinson, L. C.; Macknight, W. J.
Macromolecules 1998, 31, 1871.
31. Teresa, S. B.; Izabela, C. J Mol Catal A 2000, 160, 133.
32. Wang, J.; Varshney, S. K.; Jerome, R.; Teyssie, P. J Polym Sci Part A: Polym Chem 1992, 30, 2251.
33. Guegon, P.; Cernohous, J. J.; Khandpur, A. K. Macromolecules 1996, 29, 4605.
34. Chu, P. P.; Huang, W. J.; Chang, F. C. Polymer 2002, 41, 401. 35. Tait, P.; Lenz, R. Preparation and Properties of Stereoregular
Polymers; Reidel: Dordrecht, 1980.
36. Joseph, P. J Polym Sci Polym Chem Ed 1975, 13, 2277. 37. Ascenso, A.; Dias, A.; Gomes, P. Macromolecules 1989, 22, 998. 38. Zaho, G. T.; Ribeiro, M. D. R.; Pinho, M. N.; Subrahmanyam,
V. S.; Gil, C. L.; Lima, A. P. Polymer 2001, 42, 2455.
39. Crabtree, R. H. The Organometallic Chemistry of Transition Metals; Wiley: New York, 1994.
40. Ystene, M. Makromol Chem Macromol Symp 1992, 66, 71.
![Figure 1 Yield of NB polymerization ([NB] ⫽ 7.4 mol/L, polymerization time ⫽ 4 h, temperature ⫽ 0°C, and [Ni] ⫽ 0.0016 mol/L): (■) Ni(acac)2/MAO and (䊐) MAO alone.](https://thumb-ap.123doks.com/thumbv2/9libinfo/7639353.137477/2.945.528.808.120.345/figure-yield-polymerization-polymerization-time-temperature-mao-mao.webp)