149
Plasticity in the Hippocampus
Ming-Yuan Min1, Hsiu-Wen Yang2
, and Yi-Wen Lin3
Summary
The cortical noradrenergic (NAergic) system, which originates from the locus coeruleus (LC) located in the pons, plays an important role in cortical plasticity and many other brain functions. In rats in which the NAergic system has been eliminated by 6-hydroxydopamine during the neonatal period, induction of long-term potentiation (LTP) at CA1 synapses in the hippocampus is impaired, whereas induction of long-term depression is unaffected. Bath application of norepineph-rine, a β-adrenergic receptor agonist, or activators of effector molecules down-stream of the β-adrenergic receptor restores LTP. Similarly, activation of β-adrenergic receptors enhances associative LTP induced by paired stimuli to two independent synaptic inputs on the same postsynaptic neuron. The time window within which LTP can be induced by paired stimuli is increased by β-adrenergic receptor activation, but the magnitude of LTP is not affected. The signaling mole-cules involved in enhancement of the homosynaptic and associative LTP following β-adrenergic receptor activation are the same and include protein kinase A and mitogen-activated protein kinases. These experimental results suggest that a simul-taneous increase in the activity of LC neurons during induction protocols may have a permissive role in the induction of homosynaptic and associative LTP in the hippocampus.
Key words LTP, LTD, β-Adrenergic, 6-Hydroxydopamine, Locus coeruleus
1
Department of Life Science & Institute of Zoology, College of Life Science, National Taiwan University, No. 1, Sec. 4, Roosevelt Road, Taipei 106, Taiwan
2Department of Biomedical Science, Chung-Shan Medical University, No. 110, Sec. 1, Chien-Kuo N. Road, Taichung 402, Taiwan
3
Institute of Biomedical Science, Academia Sinica, No. 128, Sec. 2, Academia Road, Taipei 115, Taiwan
exceeds a critical value, referred to as the modifi cation threshold (θm), and are depressed if the total response is greater than zero but less than θm [5, 6].
This idea of regulation of synaptic strength is called the BCM theory, named after the three scientists, Bienenstock, Cooper, and Murp, who proposed it [5]. An example of BCM modifi cation for synaptic effi cacy (or BCM curve) at the CA1 synapse is shown in Fig. 1. According to this theory, a total postsynaptic response greater than θm activates large numbers of NMDA receptors, which leads to LTP by the activation of protein kinases, which phosphorylate AMPA receptors. In contrast, a total postsynaptic response greater than zero but less than θm activates fewer NMDA receptors, which leads to LTD because of the activation of phospha-tases and the subsequent dephosphorylation of AMPA receptors [4, 7].
Norepinephrine (NE), one of the most important neuronal modulators in the brain, is involved in the regulation of many brain functions, including the wake/
Fig. 1. Effect of norepinephrine (NE) on the BCM curve. The BCM curve shows the relation
between the frequency of tetanic stimulation [or the amount of N-methyl-d-aspartate (NMDA) receptor activation by tetanic stimulation] and the resultant long-term change in synaptic strength in control slices (open circles). The arrow indicates the modifi cation threshold (qm). Note the right shift of θm in the control slices to θ′m in the 6-hydroxydopamine (6-OHDA)-treated slices (fi lled circles), suggesting that NE depletion impairs long-term potentiation (LTP) but has no effect on long-term depression (LTD) induction. (From Yang et al. [12], with permission)
sleep cycle [8], memory storage [9], synaptic/cortical plasticity [10–12], autonomic functions [13], and pain modulation [14]. In the neocortex and hippocampus, NE fi bers mainly originate from the locus coeruleus (LC), located in the pons. In vitro studies have suggested that NE, acting on β-adrenergic receptors, has a signifi cant effect on synaptic plasticity at CA1 synapses [10, 12, 15, 16] and mossy fi ber syn-apses [17]. In this chapter, we fi rst discuss the permissive role of NE in modulating synaptic plasticity based on previous studies, followed by some interesting issues that need to be examined further.
Role of the Noradrenergic System in Homosynaptic Plasticity
At hippocampal CA1 synapses, bath application of 10 µM NE during the delivery of conditioned stimulation to induce a long-term change in synaptic strength results in a left shift of θm in the BCM curve. That is, application of NE does not have any effect on the magnitude of LTP induced by high-frequency stimulation at 50 or 100 Hz but blocks induction of LTD by low-frequency stimulation (1 Hz) and enhances the effect of 10-Hz stimulation—which alone does not result in any sig-nifi cant change in synaptic strength in control conditions—to induce LTP [15]. Similar observations have been made at mossy fi ber synapses on CA3 pyramidal neurons [17, 18]. An in vivo study also showed that induction of LTP by mild tetanus stimulation is impaired in animals in which endogenous catecholamine was depleted by injecting 6-hydroxyl dopamine (6-OHDA), but LTP can still be induced by strong tetanus [19]. These observations therefore suggest that the cortical nor-adrenergic (NAergic) system might have a permissive role in modulating LTP induction in the hippocampus.
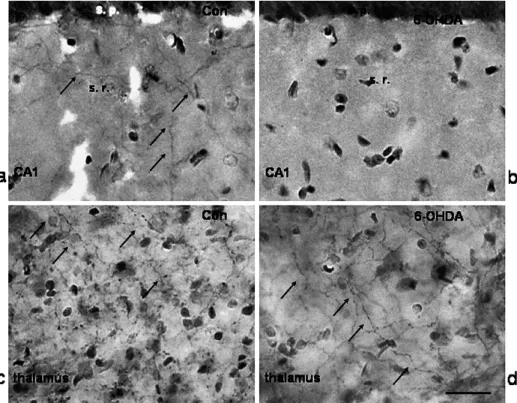
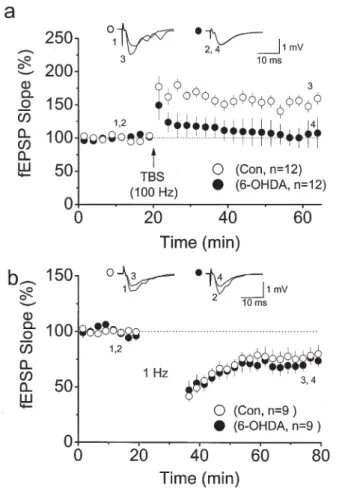
Nevertheless, a critical question remains: How do conditioned stimuli for LTP induction activate the NAergic pathway to enhance LTP induction? A high density of NAergic fi bers is found in the stratum radiatum of the CA1 region (Fig. 2) and the stratum lucidum of the CA3 region [20, 21]. The overlapping distribution of NAergic and glutamatergic fi bers of the Shaffer collateral branches provides an opportunity for simultaneous activation of glutamatergic and NAergic fi bers during tetanus. Consistent with this argument, studies in which electrophysiological and neurochemical data were simultaneously recorded in the hippocampus in vivo showed that the local NE concentration can rise to several times the basal value during tetanus for LTP induction [22, 23]. Another potential approach to addressing this question is to examine synaptic plasticity in hippocampal slices from animals in which endogenous NAergic fi bers in the hippocampus have been depleted. The treatment of neonatal rats with 6-OHDA might provide a model meeting this requirement, as it results in a persistent loss of catecholaminergic fi bers in the cerebral cortex but not in subcortical areas (Fig. 2) [24]. Using hippocampal slices from 6-OHDA-treated rats, we found that the LTP induced by theta burst stimula-tion decays within 15 min (Fig. 3A,C), whereas the LTD induced by 900 pulses at 1 Hz is not affected (Fig. 3B,C) [12]. Taking these results together, it reveals a right
shift of θm in the BCM curve that is consistent with the effect of perfusion slices with 10 µM NE [15]. Interestingly, a similar observation has been reported at mossy fi ber synapses, where the LTP induced by mild tetanus decays within 15 min when β-adrenergic receptors are blocked by timolol [17]. Bath application of NE restores the expression of LTP to normal and blocks LTD induction (Fig. 3A,B) in slices from 6-OHDA-treated animals but has no effect on the magnitude of LTP in slices from control animals (Fig. 3C) [12]. These observations suggest that NAergic fi bers are recruited to enhance LTP induction in normal hippocampal slices, whereas a higher tetanus intensity is required for LTP induction in slices lacking NAergic innervation. Similar conclusions were drawn in a recent study that examined the expression of LTP in several strains of mice with various levels of endogenous NE [16].
The enhancement of LTP and blockage of LTD by NE in the hippocampus appears to occur through the activation of β-adrenergic receptors [12, 15–17]. In
Fig. 2. Animal model in which endogenous NE is depleted by 6-OHDA. Dopamine- β-hydroxylase (DBH) immunohistochemistry performed in the stratum radiatum (s.r.) of the hip-pocampal CA1 area in control (Con) (a) and in 6-OHDA-treated animals (b). Thalamus area in the same section in the control animal (c) and the 6-OHDA-treated animal (d). Note the DBH-immunoreactive fi bers (indicated by arrows) in all photographs except b. s.p., stratum pyramidale.
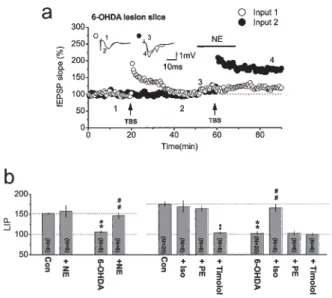
control slices, the effect of NE on the BCM curve is blocked by timolol, a selective β-adrenergic receptor antagonist, but not by phentolamine, a selective α-adrenergic receptor [15]. Similarly, isoproterenol, a selective β-adrenergic receptor agonist, but not phenylephrine, a selective α-adrenergic receptor agonist, restores LTP to normal in slices from 6-OHDA-treated animals (Fig. 4) [12] and from mice with low endogenous NE levels [16]. Interestingly, the blockage of depotentiation (low frequency-induced LTD after LTP induction) at CA1 synapses requires the activa-tion of both α- and β-adrenergic receptors [15], suggesting that the mechanisms underlying LTD induction and depotentiation are different [25].
Fig. 3. LTP and LTD in slices from 6-OHDA-lesioned animals. a LTP at CA1 synapses induced
in slices of control (open circles) and 6-OHDA-treated (fi lled circles) rats using theta burst stimu-lation (TBS), which consists of 10 bursts at 5 Hz with each burst consisting of four pulses at 100 Hz. b LTD induced by 900 pulses at 1 Hz stimulation. Note the rapid decay in the LTP, but not the LTD, induced in slices from 6-OHDA-treated rats. (From Yang et al. [12], with permission)
Role of the NAergic System in Associative Plasticity
A signifi cant weakness in suggesting a physiological role of the conventional LTP and LTD induced by tetanus or prolonged low-frequency stimulation has been the question of whether these stimulating paradigms realistically resemble any physiological function in the brain. Recently, it was demonstrated that LTP/LTD-like changes in synaptic strength can also be induced by the simultaneous spiking of a pair of pre- and postsynaptic neurons within a precise time window, so-called spike timing-dependent plasticity (STDP) [26–28]. Obviously, this form of synaptic plasticity fi ts very well with the Hebbian learning rules that: (1) neurons that fi re in synchrony become wired together (i.e., when the presynaptic axon is active and the postsynaptic neuron simultaneously is strongly activated, the synapse formed by the presynaptic axon is strengthened); and (2) neurons that fi re out of synchrony lose their link (see also Chapter 25 in [29]). It is now generally accepted that STDP pro-vides a more genuine cellular model for experience-driven change in brain function or the Hebbian learning rules than does conventional LTP/LTD [26–28, 30, 31].
LIP
Fig. 4. Activation of β-adrenergic, but not α-adrenergic, receptors restores LTP in slices from
6-OHDA-treated rats. a A typical experiment on a slice from a 6-OHDA-treated rat in which two independent Schaffer collateral branches were stimulated. Theta burst stimulation (TBS) was applied to one of the two inputs (input 1) to induce LTP in normal conditions; 40 min later, TBS was applied to the other input (input 2) to induce LTP with simultaneous application of NE during delivery of TBS. The resultant LTPs are compared. b Summarized results show that application of NE restores LTP in slices from 6-OHDA-treated rats and the summarized results of pharma-cological experiments. Note that in slices from control rats LTP induction is not affected by iso-proterenol (Iso), a β-adrenergic receptor agonist, or phenylephrine (PE), an α-adrenergic receptor agonist; it is blocked by timolol, a β-adrenergic receptor antagonist. Also note in slices from 6-OHDA-treated rats LTP is restored by application of Iso but not PE, and this effect is prevented when the β-adrenergic receptor is blocked by timolol. (From Yang et al. [12], with permission)
STDP is bidirectional; that is, synaptic effi cacy can be either potentiated or depressed by paired pre- and postsynaptic spiking, depending on both the timing interval and the temporal order of the pre- and postsynaptic spiking. Generally speaking, to induce STDP, the timing interval between paired pre- and postsynaptic spiking has to be less than ∼25 ms, whereas a signifi cantly wider time window (up to ∼100 ms) has been suggested for LTD induction [32, 33]. As for the temporal order of pre- and postsynaptic spiking, repeated paired pre/postsynaptic spiking results in LTP if presynaptic stimulation precedes postsynaptic stimulation, whereas it results in LTD if the temporal order of pre/postsynaptic spiking is reversed (Fig. 5).
Fig. 5. Spike-timing-dependent plasticity (STDP) at a synapse of the lateral perforant path on a
granule cell in the dentate gyrus. a The arrangement of the recording electrode (Rec.) and stimu-lating electrodes (Sti. 1, Sti. 2). b Evoked neuronal activity by Sti. 1 is fi eld EPSP ( fEPSP) activity, as it is completed blocked by the AMPA receptor antagonist DNQX. The activity evoked by Sti.
2 is the fi eld somatic spike (fSS), as it is insensitive to DNQX, but is completely blocked by
tetrodotoxin (TTX). c LTP induction by paired fEPSP–fSS stimulation. The upper traces show baseline fEPSP activity (left), fEPSP and fSS during pairing (middle), and fEPSP activity after pairing (right) for one experiment. Note the potentiation of fEPSP activity after paired fEPSP-afSS stimulation with ∆t = 15 ms. The lower plot shows the summarized results for nine experiments, in which the ∆t for the paired fEPSP–afSS stimulation was <30 ms. d LTD induction by paired fSS–fEPSP stimulation. Note the depression of fEPSP activity (compare the left and right insets) after paired afSS–fEPSP stimulation with ∆t = −25 ms (see middle inset). The lower plot shows the summarized results for eight experiments, in which ∆t for the paired fEPSP–afSS stimulation was<|−40|ms. (From Lin et al. [25], with permission)
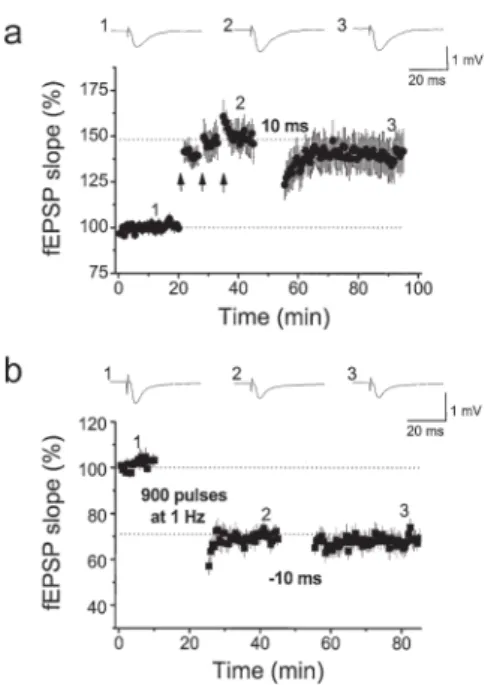
STDP-like LTP and LTD both require activation of NMDA receptors [10, 32, 33]. Again, the voltage-dependent nature of the NMDA receptor makes it an ideal detector for the correlated pre- and postsynaptic spiking activity. Depolarization of the membrane potential by the action potential back-propagated along dendrites of postsynaptic neurons removes magnesium, which blocks the pore site of the NMDA receptor in the resting condition and allows the NMDA receptor to be activated by glutamate released presynaptically during the paired pre- and postsynaptic spiking. We have recently characterized the cellular mechanisms underlying induction of STDP at a synapse of the lateral perforant pathway on a granule cell in the dentate gyrus (Fig. 5). We found that the signaling molecules involved in STDP induction are similar to those involved in conventional LTP and LTD because the saturated synaptic potentiation or depression caused by, respectively, tetanus or low-fre-quency stimulation, occludes the induction of STDP (Fig. 6) [25]. Similar to the homosynaptic plasticity discussed above, STDP is also subject to modifi cation by the NAergic system. We have reported that by acting at β-adrenergic receptors NE enhances STDP by increasing the time window of the pre/postsynaptic activation required for LTP induction without changing the magnitude of LTP (Fig. 7) [10].
Fig. 6. Induction of STDP is occluded by saturated homosynaptic LTP/LTD. a Homosynaptic
LTP induced by three trains of 100 pulses at 100 Hz (intertrain interval 30 s), repeated three times at intervals of 5 min (arrows); following this homosynaptic LTP, induction of LTP by paired stimulation with an interval of 10 ms is occluded. b LTD induced by stimulation with 900 pulses at 1 Hz; following this homosynaptic LTD, induction of LTD by paired stimulation with an inter-val of −10 ms is again occluded. (From Lin et al. [25], with permission)
Molecular Signaling Cascades Activated to Enhance LTP
Activation of β-adrenergic receptors is known to increase the cytoplasmic concen-tration of cyclic adenosine monophosphate (cAMP), which in turn activates cAMP-dependent protein kinase A (PKA). This signaling pathway seems to act in parallel with the signaling pathway involved in LTP induction, which requires activation of calcium/calmodulin-dependent protein kinase II (CaMKII) by calcium infl ux through activated NMDA receptors during tetanus [4]. However, several studies have suggested that an increase in cAMP levels in postsynaptic neurons is required for LTP induction under control conditions [34–36]. Consistent with these observa-tions, we found that bath application of activators of Gs protein or adenylyl cyclase (AC) also restored LTP in slices from 6-OHDA-treated animals (Fig. 8) [12]. The type I and VIII ACs, which are present at high amounts in neurons, are good can-didate proteins for linking these two pathways. Unlike other AC subtypes, which
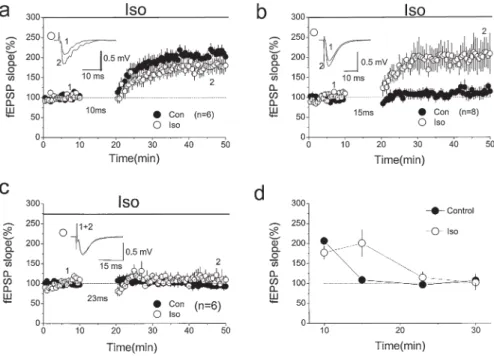
Fig. 7. Enhancement of STDP by activation of β-adrenergic receptors. a Application of 1 µM
isoproterenol (Iso) has no signifi cant effect on the magnitude of LTP induced using the pairing protocol with a 10-ms interval. b Application of Iso enhances LTP induction using the pairing protocol with a 15-ms interval, which does not induce LTP under control conditions. c Application of Iso has no signifi cant effect when the interval is increased to 23 ms. d Summarized results for
a–c and those using a 30-ms interval. In all of the plots, the open circles are results in the presence
of Iso and the fi lled circles results from control experiments (Con). Note that there is no signifi cant difference in the LTP induced in control and Iso experiments, except for that using a 15-ms interval. (From Lin et al. [10], with permission)
require an external signal for their activation, types I and VIII can be directly acti-vated by CaMKII intracellularly and contribute to the cAMP increase in the cyto-plasm [37]. Thus, the cAMP increase required for LTP induction might occur via activation of type I or VIII ACs by CaMKII [38, 39]. In hippocampal slices taken from type VIII AC knockout mice, induction of LTP is impaired, demonstrating the involvement of this protein in LTP induction [40].
It is likely that the permissive effect of the β-adrenergic receptor on homosyn-aptic LTP induction might be through the activation of other types of ACs down-stream of the Gs proteins activated by β-adrenergic receptors, which in turn cause a suffi ciently large increase in cytoplasmic cAMP for LTP induction. Therefore, cooperative activation of other types ACs by β-adrenergic receptors with activation of types I and VIII by NMDA receptor signaling leads to LTP induction. This could also explain why, in hippocampal slices that lack NAergic fi ber innervation, strong tetanus is required for LTP induction, as more type I or VIII ACs must be activated to produce enough cAMP for LTP induction.
At hippocampal CA1 synapses, the expression of LTP is due to enhancement of AMPA receptor function, either by phosphorylation of existing AMPA receptors at synaptic sites or by recruitment of new AMPA receptors to the synaptic activa-tion zone [41]. In addiactiva-tion to being directly involved in modulaactiva-tion of AMPA
cAMP (n= 4) is also observed. The results of these two PKA activators are pooled. (From Yang et al. [12], with permission)
Fig. 9. Signaling cascades involved in the enhancing effect of β-adrenergic receptors (β-R) on synaptic plasticity at CA1 synapses. Top right High-frequency stimulation (HFS) activates many NMDA receptors (NMDA R) and causes a large amount of calcium infl ux and activation of CaMKII. The activated CaMKII either phosphorylates the AMPA receptor (AMPA-R) itself, leading to LTP, or increases cytoplasmic levels of cAMP by stimulating type I or VIII ACs (ACI/VIII), which activate PKA or the more downstream effectors MAP/ERK to cause phosphory-lation of AMPA receptors and LTP induction (black arrows). Middle Activation of β-adrenergic receptors increases cytoplasmic levels of cAMP to enhance LTP induction, either by stimulating type I/VIII ACs or other types of AC (gray arrows). Activation of β-adrenergic receptors also inhibits phosphatase 2B (PP2B) and prevents induction of LTD by low-frequency stimulation (LFS) (bottom). As regards STDP, paired pre-/postsynaptic spiking causes activation of many NMDA receptors and leads to LTP as homosynaptic LTP. In addition, the activated PKA–MAP/ ERK signaling pathway also modulates voltage-dependent potassium (Kv) and sodium (Nav) chan-nels function to shape the back-propagating action potential (bAP), which in turn changes the time window for LTP induction
receptor function, the signaling molecules activated following β-adrenergic recep-tor activation must also target other ion channels or receprecep-tors to enhance the asso-ciative form of LTP or STDP. This is because β-adrenergic receptor activation does not affect the magnitude of LTP but increases the time window of the pre/postsyn-aptic activation required for LTP induction (Fig. 9) [10]. For example, one possible candidate molecule is the Kv4.3 channel, which is responsible for a transient (or
fewer NMDA receptors are activated, there is less calcium infl ux into the postsyn-aptic cytoplasm, and phosphatase 2B (or calcineurin) is activated, rather than CaMKII, causing dephosphorylation of existing AMPA receptors and resulting in LTD [4]. Activation of the β-adrenergic receptor–cAMP–PKA pathway activates inhibitor I, which inhibits calcineurin (phosphatase 2B) and blocks LTD [4, 43, 44]. Interestingly, LTD induction is not affected in slices from 6-OHDA-treated animals [12], suggesting that NAergic fi bers are not recruited during low-frequency stimulation, which may be due to their higher threshold for activa-tion. Consistent with this argument, we found that STDP induction is also not affected [10], as presynaptic stimulation is given at low frequency during its induction.
Other Considerations
If recruitment of NAergic fi bers occurs only during tetanus stimulation, which does not resemble any physiological condition in the neuronal circuit, how is the NAergic system activated to modulate induction of homosynaptic plasticity and STDP under physiological conditions? Neuronal activity of LC neurons undergoes certain rhythms, which are closely related to the wake/sleep cycle of animals. They fi re at highest frequency when animals are active waking, less when animals are quiet waking, and even less when animals are in non-rapid eye movement (REM) sleep and REM sleep [45, 46]. Thus, synaptic plasticity would be enhanced if an increase in synaptic activity in hippocampal circuit occurred simultaneously with an increase in the spiking rate of LC neurons; for example, synaptic plasticity might occur more easily during active waking. Sleep, however, has been suggested to play a signifi -cant role in declarative memory consolidation [47], as sleep deprivation has been shown to impair long-term synaptic plasticity signifi cantly in the hippocampus in rats [48, 49]. These observations suggest that the temporal dissociation of NAergic activity from the neuronal circuit in the cortex and hippocampus during sleep could be essential for memory consolidation.
Like memory, LTP expression has different stages. There is an early-phase LTP (e-LTP), which usually lasts only 3–4 h after its induction and is independent
of protein synthesis. Following e-LTP is late-phase LTP (l-LTP), which can last several hours in slice preparations or even days in vivo and is dependent on protein synthesis [50, 51]. At the cellular level, e-LTP might resemble the acquisi-tion of new memory, and the conversion of e-LTP to l-LTP might resemble memory consolidation. As discussed above, the NAergic system, acting on β-adrenergic receptors, may have a permissive effect on the induction of e-LTP, and it may have a similar effect on memory acquisition. In line with this argument, a behavioral study showed that coeruleocortical NAergic lesions produced by intracerebral injection of 6-OHDA impairs learning behavior in animals [52]. What about the effect of the NAergic system on l-LTP? Does it also enhance l-LTP expression? Given that sleep is important for memory consolidation, which resembles the conversion of e-LTP to l-LTP at the cellular level, and that neuronal activity of LC neurons is low during sleep, it might appear that lower endogenous levels of NE would be the ideal situation for e-LTP to convert to l-LTP. However, experi-mental results do not support this argument. At mossy fi ber synapses, homosynaptic e-LTP and l-LTP are both enhanced by β-adrenergic receptor activation [17]. One important factor that should be borne in mind is that mossy fi ber LTP is NMDA receptor-independent [53, 54]; furthermore, the effect of endogenous levels of NE must be taken into account. Thus, at CA1 synapses, where LTP is NMDA receptor-dependent, the role of the NAergic system on l-LTP requires further investigation.
It has been shown that slow-wave sleep and sleep spindles are possible candidate mechanisms for sleep to enhance memory directly [55]. In hippocampal slices, it is also been demonstrated that somatic spiking at theta frequency [56] or ripple complex activity can directly enhance the conversion of e-LTP to l-LTP [57]. Thus, at the cellular and molecular levels, changes in the synchronization of the neuronal spiking pattern might be a crucial factor in enhancing expression of l-LTP and memory. Thalamic neurons can change their spiking patterns and degree of syn-chronized fi ring depending on the extracellular levels of NE, and the modifi cation of the functions of ionic channels by NE can account for the changed spiking activ-ity [52]. If the NAergic system did have a signifi cant effect on l-LTP, would it involve a similar mechanism and allow somatic activity-dependent modifi cation of l-LTP? Again, this would be an interesting question to answer.
Conclusions
A signifi cant role of the NAergic system in modulating homosynaptic and associa-tive forms of synaptic plasticity has been confi rmed and the underlying molecular mechanism explored in detail. However, some questions remain to be answered, in particular the possible differential effects of the NAergic system on cortical plasticity during different states of brain functions, for example, during active awaking and/or sleep.
tivity: orientation specifi city and binocular interaction in visual cortex. J Neurosci 2:32–48. 6. Bear MF, Kirkwood A (1996) Bidirectional plasticity of cortical synapses. In: Fazeli MS,
Collingridge GL (eds) Cortical plasticity: LTP and LTD. BIOS Scientifi c, Oxford, pp 191–205.
7. Bear MF, Malenka RC (1994) Synaptic plasticity: LTP and LTD. Curr Opin Neurol 4:389–399.
8. Aston-Jones G, Cohen JD (2005) An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci 28:403–450. 9. Selden NR, Robbins TW, Everitt BJ (1990) Enhanced behavioral conditioning to context and
impaired behavioral and neuroendocrine response to conditioned stimuli following ceruleo-cortical noradrenergic lesion: support for an attentional hypothesis of central noradrenergic function. J Neurosci 10:531–539.
10. Lin Y-W, Min M-Y, Chiu T-H, et al (2003) Enhancement of associative long-term potentia-tion by activapotentia-tion of β-adrenergic receptors at CA1 synapses in rat hippocampal slices. J Neurosci 23:4173–4181.
11. Kasamatsu T (1991) Adrenergic regulation of visual cortical plasticity; a role of locus coe-ruleus system. Prog Brain Res 88:599–616.
12. Yang H-W, Lin Y-W, Yen C-D, et al (2002) Change in bi-directional plasticity at CA1 syn-apses in hippocampal slices taken from 6-hydroxy-dopamine treated rats: the role of endog-enous norepinephrine. Eur J Neurosci 16:1117–1128.
13. Richerson GB (2003) The autonomic nervous system. In: Boron WF, Boulpaep EL (eds) Medical physiology. Saunders, Philadelphia, pp 378–398.
14. Pertovaara A (2006) Noradrenergic pain modulation. Prog Neurobiol 80:53–83.
15. Katsuki H, Izumi Y, Zorumski CF (1997) Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol 77:3013–3020.
16. Schimanski LA, Ali DW, Baker GB, et al (2007) Impaired hippocampal LTP in inbred mouse strains can be rescued by β-adrenergic receptor activation. Eur J Neurosci 25:1589–1598. 17. Huang Y-Y, Kandel ER (1996) Modulation of both the early and the late phase of mossy
fi ber LTP by activation of β-adrenergic receptors. Neuron 16:611–617.
18. Hopkins W, Johnston D (1988) Noradrenergic enhancement of long-term potentiation of mossy fi ber synapses in the hippocampus. J Neurophysiol 59:667–678.
19. Bliss TVP, Goddard GV, Riives M (1983) Reduction of long-term potentiation in the dentate gyrus of the rat following selective depletion of monoamines. J Physiol (Lond) 334: 475–491.
20. Loy R, Koziell DA, Lindsey D, et al (1980) Noradrenergic innervation of adult rat hippo-campal formation. J Comp Neurol 189:699–710.
21. Moore RY, Bloom FE (1979) Central catecholamine neuron systems: anatomy and physio-logy of the norepinephrine and epinephrine systems. Annu Rev Neurosci 2:113–168. 22. Bronzino JD, Kehoe P, Mallinson K, et al (2001) Increased extracellular release of
hippo-campal NE is associated with tetanization of the medial perforant pathway in the freely moving adult male rat. Hippocampus 11:423–429.
23. Harley CW, Lalies MD, Nutt DJ (1996) Estimating the synaptic concentration of norepineph-rine in dentate gyrus which produces beta receptor mediated long-lasting potentiation in vivo using microdialysis and intracerebroventricular norepinephrine. Brain Res 710:293–298. 24. Harik SI (1984) Locus coeruleus lesion by local 6-hydroxydopamine infusion causes marked
and specifi c destruction of noradrenergic neurons, long term depletion of norepinephrine and enzymes that synthesize it, and enhanced dopaminergic mechanisms in the ipsilateral cerebral cortex. J Neurosci 4:699–707.
25. Lin Y-W, Yang H-W, Wang H-J, et al (2006) Spike-timing-dependent plasticity (STDP) at resting and conditioned lateral perforant path synapses on granule cells in the dentate gyrus: different roles of NMDA and group I metabotropic glutamate receptors. Eur J Neurosci 23:2362–2374.
26. Bi G, Poo M-M (2001) Synaptic modifi cation by correlated activity: Hebb’s postulate revised. Annu Rev Neurosci 24:139–166.
27. Dan Y, Poo M-M (2004) Spike-timing-dependent plasticity of neuronal circuits. Neuron 44:23–30.
28. Dan Y, Poo MM (2006) Spike timing-dependent plasticity: from synapse to perception. Physiol Rev 863:1033–1048.
29. Bea MF, Connors BW, Paradiso MA (2007) Neuroscience: exploring the brain (3rd ed). Lippincott Williams & Wilkins, Philadelphia.
30. Song S, Abbott LF (2001) Cortical development and remapping through spike timing-dependent plasticity. Neuron 32:339–350.
31. Song S, Miller KD, Abbott LF (2000) Competitive Hebbian learning through spike-timing-dependent plasticity. Nat Neurosci 3:919–926.
32. Debanne D, Gahwiler BH, Thomson SM (1998) Long term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol (Lond) 507:237–247.
33. Feldman DE (2000) Timing-based LTP and LTD at vertical inputs to layer II / III pyramidal cells in rat barrel cortex. Neuron 27:45–56.
34. Chetaovich DM, Sweatt JD (1993) NMDA receptor activation increases cyclic AMP in area CA1 of the hippocampus via calcium/calmodulin stimulation of adenylyl cyclase. J Neuro-chem 61:1933–1942.
35. Makhinson M, Chotiner JK, Watson JB, et al (1999) Adenylyl cyclase activation modulates activity-dependent changes in synaptic strength and Ca2−/calmodulin-dependent kinase II autophosphorylation. J Neurosci 19:2500–2510.
36. Otmakhova NA, Otmakhov N, Mortenson LH, et al (2000) Inhibition of cAMP pathway decreases early long-term potentiation at CA1 hippocampal synapses. J Neurosci 20:4446–4451.
37. Cooper DMF, Mons N, Karpen JW (1995) Adenylyl cyclase and the interaction between calcium and cAMP signalling. Nature 374:421–424.
38. Liauw J, Wu LJ, Zhuo M (2005) Calcium-stimulated adenylyl cyclases required for long-term potentiation in the anterior cingulate cortex. J Neurophysiol 94:878–882.
39. Wang H, Ferguson GD, Pineda VV, et al (2004) Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci 7: 635–642.
40. Wang H, Pineda VV, Chan GC, et al (2003) Type 8 adenylyl cyclase is targeted to excitatory synapses and required for mossy fi ber long-term potentiation. J Neurosci 23:9710–9718. 41. Malenka RC, Nicoll RA (1997) Silent synapses speak up. Neuron 19:473–476.
42. Yuan LL, Adams JP, Swank M, et al (2002) Protein kinase modulation of dendritic K− channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci 22:4860–4868.
43. Mulkey RM, Endo S, Shenolikar S, et al (1994) Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369:486–488.
44. Mulkey RM, Herron CE, Malenka RC (1993) An essential role for protein phosphatases in hippocampal long term depression. Science 261:1051–1055.
50. Frey U, Huang YY, Kandel ER (1993) Effects of cAMP stimulate a late stage of LTP in hippocampal CA1 neuron. Science 260:1661–1664.
51. Huang YY, Kandel ER (1994) Recruitment of long lasting and protein kinase A dependent long term potentiation in CA1 region of hippocampus requires repeated tetanization. Learn Mem 1:74–82.
52. Steriade M, McCarley RW (1990) Brainstem control of wakefulness and sleep. Plenum, New York.
53. Weisskopf MG, Castillo PE, Zalutsky RA, et al (1994) Mediation of hippocampal mossy fi ber long-term potentiation by cyclic AMP. Science 265:1878–1882.
54. Weisskopf MG, Nicoll RA. (1995) Presynaptic changes during mossy fi bre LTP revealed by NMDA receptor-mediated synaptic responses. Nature 376:256–259.
55. Gais S, Molle M, Helms K, et al (2002) Learning-dependent increases in sleep spindle density. J Neurosci 22:6830–6834.
56. Dudek SM, Fields RD (2002) Somatic action potential are suffi cient for late-phase LTP-related cell signaling. Proc Natl Acad Sci U S A 99:3962–3967.
57. Behrens CJ, van den Boom LP, de Hoz L, et al (2005) Induction of sharp wave-ripple com-plexes in vitro and reorganization of hippocampal networks. Nat Neurosci 8:1560–1567.
![Fig. 1. Effect of norepinephrine (NE) on the BCM curve. The BCM curve shows the relation between the frequency of tetanic stimulation [or the amount of N-methyl-d-aspartate (NMDA) receptor activation by tetanic stimulation] and the resultant long-term cha](https://thumb-ap.123doks.com/thumbv2/9libinfo/8847476.241014/2.659.205.452.574.773/norepinephrine-frequency-stimulation-aspartate-receptor-activation-stimulation-resultant.webp)