Klysimplexins I–T, eunicellin-based diterpenoids from the cultured soft coral
Klyxum simplex†
Bo-Wei Chen,
aChih-Hua Chao,
aJui-Hsin Su,
bChung-Wei Tsai,
cWei-Hsien Wang,
a,bZhi-Hong Wen,
aChiung-Yao Huang,
aPing-Jyun Sung,
a,bYang-Chang Wu
cand Jyh-Horng Sheu*
a,dReceived 30th June 2010, Accepted 6th October 2010
DOI: 10.1039/c0ob00351d
New eunicellin-base diterpenoids, klysimplexins I–T (1–12), were isolated from a cultured soft coral
Klyxum simplex. Their structures were elucidated by spectroscopic methods, particularly in 1D and 2D
NMR experiments. The absolute stereochemistry of 4 was determined by Mosher’s method. Compounds 9 and 12 have been shown to exhibit cytotoxicity toward a limited panel of cancer cell lines. Compounds 2–6, 10 and 11 were found to display significant in vitro anti-inflammatory activity in LPS-stimulated RAW264.7 macrophage cells by inhibiting the expression of the iNOS protein. Compounds 10 and 11 also could effectively reduce the level of COX-2 protein.
Introduction
During the course of our investigation on new natural substances from the cultured and wild-type soft corals K. simplex, new metabolites klysimplexins A–H1and klysimplexin sulfoxides A–
C2were isolated from the cultured soft coral, and simplexins A–I
were obtained from the wild-type soft coral.3Previously reported
eunicellin-based diterpenoids were isolated mostly from octoco-rals (Alcyonaceae) belonging to the genera Acalycigorgia,4
Al-cyonium,5 Astrogorgia,6Briareum,7Cladiella,8 Eleutherobia,9
Eu-nicella,10Klyxum,11Litophyton,12Muricella,13Pachyclavularia,14,15
Sclerophytum,16 Sinularia,17 and Solenopodium.18 Some of these
metabolites have been shown to exhibit cytotoxic activity against the growth of various cancer cell lines.1,2,7–9,14–16In continuation
of our recent effort on discovering novel and bioactive substances from marine invertebrates,19–24the chemical constituents of the
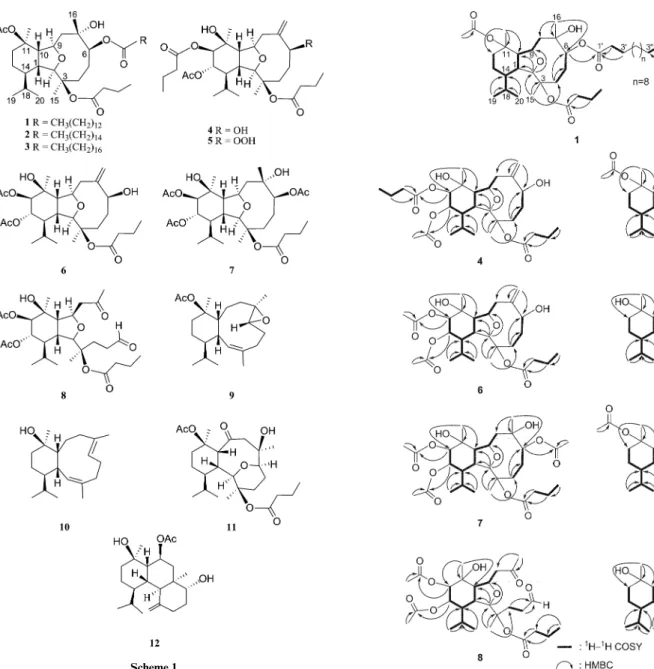
cul-tured soft coral Klyxum simplex were further studied. In this paper, we report the isolation, structure determination, and biological activity of twelve new eunicellin-based metabolites, klysimplexins I–T (1–12, Scheme 1), from K. simplex. The relative structures of 1–12 were established by extensive spectroscopic analysis, including 2D NMR (1H–1H COSY, HSQC, HMBC, and NOESY)
spectroscopy, and the absolute structure of 4 was determined by Mosher’s method. Besides the normal THF-containing eunicellins
1–7 which are similar to those isolated previously from this soft
coral,1–19this investigation also led to the isolation of eunicellins
containing a long-chain ester substitution at C-6 (compounds 1–
3), a 6,7-secoeunicellin 8, two 2,9-deoxygenated derivatives 9 and 10, and a tetradecahydrophenanthrene-type diterpene 12 for the
first time from the genus Klyxum. Cytotoxicity of metabolites 1–12
aDepartment of Marine Biotechnology and Resources, National Sun Yat-sen
University, Kaohsiung 804, Taiwan
bTaiwan Coral Research Center, National Museum of Marine Biology &
Aquarium, Checheng, Pingtung 944, Taiwan
cGraduate Institute of Natural Products, Kaohsiung Medical University,
Kaohsiung 807, Taiwan
dAsia-Pacific Ocean Research Center, National Sun Yat-sen University,
Kaohsiung 804, Taiwan
† Electronic supplementary information (ESI) available:1H and13C NMR
and HRESIMS spectra of 1–12. See DOI: 10.1039/c0ob00351d
against a limited panel of human tumor cell lines including human liver carcinoma (Hep G2 and Hep G3B), human breast carcinoma (MDA-MB-231 and MCF-7) human lung carcinoma (A-549), and human oral cancer cells (Ca9-22) are also discussed, and the ability of 1–12 to inhibit up-regulation of the pro-inflammatory iNOS (inducible nitric oxide synthase) and COX-2 (cyclooxygenase-2) proteins in LPS (lipopolysaccharide)-stimulated RAW264.7 macrophage cells was also evaluated.
Results and discussion
The soft coral (1.5 kg fresh wt) was collected and freeze-dried. The freeze-dried material was minced and extracted exhaustively with EtOH (3¥ 10 L). The organic extract was concentrated to an aque-ous suspension and was further partitioned between CH2Cl2and
water. The combined CH2Cl2-soluble fraction was concentrated
under reduced pressure and the residue was repeatedly purified by chromatography to yield metabolites 1–12.
Klysimplexin I (1) was obtained as a colorless oil. The HRES-IMS of 1 exhibited a [M + Na]+ peak at m/z 701.4974 and
established a molecular formula C40H70O8, implying six degrees
of unsaturation. The IR spectrum of 1 revealed the presence of hydroxy and carbonyl functionalities from absorptions at 3460 and 1738 cm-1. The13C NMR data of 1 was made up 40 carbon
signals in total (Table 1), which were assigned by the DEPT spectrum to eight methyls, nineteen sp3 methylenes, seven sp3
methines (including three oxymethines), three sp2carbonyls and
three sp3 oxygenated quaternary carbons. Three ester carbonyl
carbons (dC 174.7, 172.6 and 170.1) were HMBC correlated by
the methylene protons (dH2.31 m, 2H and 1.61, m, 2H) of a
long-chain ester unit, methylene protons (dH 2.38 m, 2H and 1.67 m,
2H) of an n-butyrate and protons of an acetate (dH1.98, 3H, s),
respectively. The long-chain ester was found to be myristate as the negative ESIMS of 1 exhibited a peak at m/z 227.2, consistent with the molecular formula C14H27O2. Therefore, the remaining
three degrees of unsaturation identified metabolite 1 as a tricyclic compound. In the1H NMR spectrum of 1 (Table 3), two doublets
atdH0.94 and 0.80 (each 3H, d, J= 7.2 Hz) arose from two methyls
of an isopropyl group. Signals resonating atdH2.15 (1H, dd, J=
11.6, 7.2 Hz), 3.13 (1H, br t, J= 7.2), 3.54 (1H, s) and 4.08 (1H,
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011
Scheme 1
ddd, J= 11.7, 8.0 and 4.0), and at dC 42.3, 52.8, 92.0 and 75.5,
indicated the presence of a tetrahydrofuran structural unit.1,3The
molecular framework was established mainly by1H–1H COSY
and HMBC experiments (Fig. 1). The placement of a myristate and an n-butyrate at C-6 and C-3, respectively, was proven from the HMBC correlations from H-6 (d 5.58) and H-2 (d 3.54) to
the carbonyl carbons resonating atd 174.7 (qC) and d 172.6 (qC).
The proton resonances for H3-17 (d 1.48) and H3-16 (d 1.16) also
determined the positions of the acetate and hydroxy groups at C-11 and C-7, respectively. Thus, the molecular framework of 1 was established unambiguously, and was found to be similar to that of a known compound 13 (Scheme 2).3The relative structure
of 1 also was found to be the same as that of 13 by comparison of the chemical shifts and coupling constants for protons of both compounds, and by analysis of NOE correlations. In order to unambiguously confirm the structure, a base-catalyzed hydrolysis of 1 was performed and the reaction was found to afford 13. The structure of 1 was thus fully established.
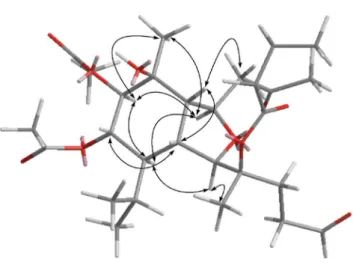
Fig. 1 Key1H-1H COSY and HMBC correlations of 1, 4, and 6–12.
Scheme 2
Klysimplexins J (2) and K (3) were also isolated as colorless oils. The molecular formulae of C42H74O8and C44H78O8, 28 and 56
mass units higher than that of 1, were determined by HRESIMS, respectively. The negative mode ionization of 2 and 3 by LC-ESI MS/MS fragmentation exhibited [M - H]- peaks at m/z 255.6
and 283.5, consistent with the molecular formulae C16H31O2and
C18H35O2, and indicated the presence of palmitate and stearate in
2 and 3, respectively. The1H and13C NMR spectroscopic data of
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011
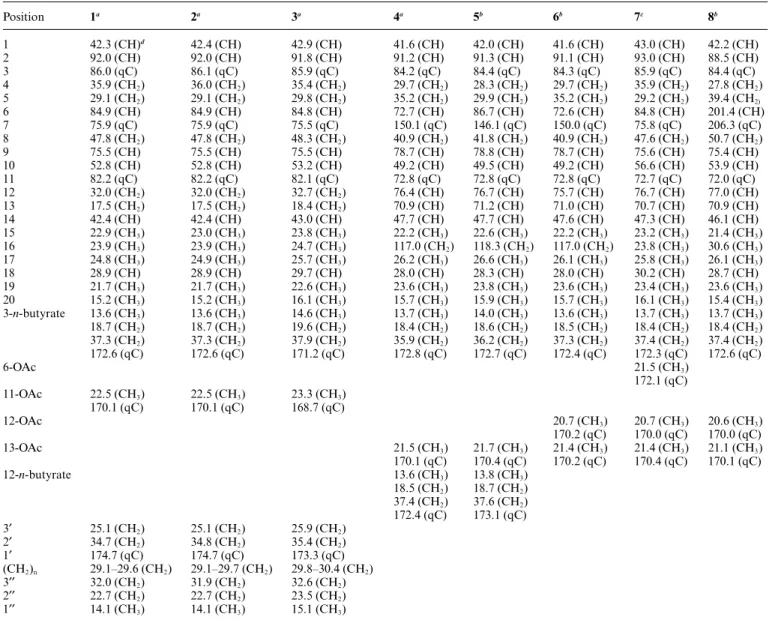
Table 1 13C NMR Data for compounds 1–8 Position 1a 2a 3a 4a 5b 6b 7c 8b 1 42.3 (CH)d 42.4 (CH) 42.9 (CH) 41.6 (CH) 42.0 (CH) 41.6 (CH) 43.0 (CH) 42.2 (CH) 2 92.0 (CH) 92.0 (CH) 91.8 (CH) 91.2 (CH) 91.3 (CH) 91.1 (CH) 93.0 (CH) 88.5 (CH) 3 86.0 (qC) 86.1 (qC) 85.9 (qC) 84.2 (qC) 84.4 (qC) 84.3 (qC) 85.9 (qC) 84.4 (qC) 4 35.9 (CH2) 36.0 (CH2) 35.4 (CH2) 29.7 (CH2) 28.3 (CH2) 29.7 (CH2) 35.9 (CH2) 27.8 (CH2) 5 29.1 (CH2) 29.1 (CH2) 29.8 (CH2) 35.2 (CH2) 29.9 (CH2) 35.2 (CH2) 29.2 (CH2) 39.4 (CH2) 6 84.9 (CH) 84.9 (CH) 84.8 (CH) 72.7 (CH) 86.7 (CH) 72.6 (CH) 84.8 (CH) 201.4 (CH) 7 75.9 (qC) 75.9 (qC) 75.5 (qC) 150.1 (qC) 146.1 (qC) 150.0 (qC) 75.8 (qC) 206.3 (qC) 8 47.8 (CH2) 47.8 (CH2) 48.3 (CH2) 40.9 (CH2) 41.8 (CH2) 40.9 (CH2) 47.6 (CH2) 50.7 (CH2) 9 75.5 (CH) 75.5 (CH) 75.5 (CH) 78.7 (CH) 78.8 (CH) 78.7 (CH) 75.6 (CH) 75.4 (CH) 10 52.8 (CH) 52.8 (CH) 53.2 (CH) 49.2 (CH) 49.5 (CH) 49.2 (CH) 56.6 (CH) 53.9 (CH) 11 82.2 (qC) 82.2 (qC) 82.1 (qC) 72.8 (qC) 72.8 (qC) 72.8 (qC) 72.7 (qC) 72.0 (qC) 12 32.0 (CH2) 32.0 (CH2) 32.7 (CH2) 76.4 (CH) 76.7 (CH) 75.7 (CH) 76.7 (CH) 77.0 (CH) 13 17.5 (CH2) 17.5 (CH2) 18.4 (CH2) 70.9 (CH) 71.2 (CH) 71.0 (CH) 70.7 (CH) 70.9 (CH) 14 42.4 (CH) 42.4 (CH) 43.0 (CH) 47.7 (CH) 47.7 (CH) 47.6 (CH) 47.3 (CH) 46.1 (CH) 15 22.9 (CH3) 23.0 (CH3) 23.8 (CH3) 22.2 (CH3) 22.6 (CH3) 22.2 (CH3) 23.2 (CH3) 21.4 (CH3) 16 23.9 (CH3) 23.9 (CH3) 24.7 (CH3) 117.0 (CH2) 118.3 (CH2) 117.0 (CH2) 23.8 (CH3) 30.6 (CH3) 17 24.8 (CH3) 24.9 (CH3) 25.7 (CH3) 26.2 (CH3) 26.6 (CH3) 26.1 (CH3) 25.8 (CH3) 26.1 (CH3) 18 28.9 (CH) 28.9 (CH) 29.7 (CH) 28.0 (CH) 28.3 (CH) 28.0 (CH) 30.2 (CH) 28.7 (CH) 19 21.7 (CH3) 21.7 (CH3) 22.6 (CH3) 23.6 (CH3) 23.8 (CH3) 23.6 (CH3) 23.4 (CH3) 23.6 (CH3) 20 15.2 (CH3) 15.2 (CH3) 16.1 (CH3) 15.7 (CH3) 15.9 (CH3) 15.7 (CH3) 16.1 (CH3) 15.4 (CH3) 3-n-butyrate 13.6 (CH3) 13.6 (CH3) 14.6 (CH3) 13.7 (CH3) 14.0 (CH3) 13.6 (CH3) 13.7 (CH3) 13.7 (CH3) 18.7 (CH2) 18.7 (CH2) 19.6 (CH2) 18.4 (CH2) 18.6 (CH2) 18.5 (CH2) 18.4 (CH2) 18.4 (CH2) 37.3 (CH2) 37.3 (CH2) 37.9 (CH2) 35.9 (CH2) 36.2 (CH2) 37.3 (CH2) 37.4 (CH2) 37.4 (CH2) 172.6 (qC) 172.6 (qC) 171.2 (qC) 172.8 (qC) 172.7 (qC) 172.4 (qC) 172.3 (qC) 172.6 (qC) 6-OAc 21.5 (CH3) 172.1 (qC) 11-OAc 22.5 (CH3) 22.5 (CH3) 23.3 (CH3) 170.1 (qC) 170.1 (qC) 168.7 (qC) 12-OAc 20.7 (CH3) 20.7 (CH3) 20.6 (CH3) 170.2 (qC) 170.0 (qC) 170.0 (qC) 13-OAc 21.5 (CH3) 21.7 (CH3) 21.4 (CH3) 21.4 (CH3) 21.1 (CH3) 170.1 (qC) 170.4 (qC) 170.2 (qC) 170.4 (qC) 170.1 (qC) 12-n-butyrate 13.6 (CH3) 13.8 (CH3) 18.5 (CH2) 18.7 (CH2) 37.4 (CH2) 37.6 (CH2) 172.4 (qC) 173.1 (qC) 3¢ 25.1 (CH2) 25.1 (CH2) 25.9 (CH2) 2¢ 34.7 (CH2) 34.8 (CH2) 35.4 (CH2) 1¢ 174.7 (qC) 174.7 (qC) 173.3 (qC) (CH2)n 29.1–29.6 (CH2) 29.1–29.7 (CH2) 29.8–30.4 (CH2) 3¢¢ 32.0 (CH2) 31.9 (CH2) 32.6 (CH2) 2¢¢ 22.7 (CH2) 22.7 (CH2) 23.5 (CH2) 1¢¢ 14.1 (CH3) 14.1 (CH3) 15.1 (CH3) a100 MHz in CDCl
3.b125 MHz in CDCl3.c100 MHz in CDCl3.dMultiplicities deduced by DEPT.
2 and 3 were found to be very close to those of 1 (Tables 1 and 3),
indicating the very similar structures for these three metabolites. The relative stereochemistries of 2 and 3 were suggested to be the same as that of 1 due to the biogenetic consideration, NMR spectroscopic data, as well as the same sign of specific optical rotations.
Klysimplexin L (4) was obtained as a colorless oil that gave a pseudomolecular ion peak at m/z 575.3193 [M + Na]+ in the
HRESIMS, consistent with the molecular formula C30H48O9and
implying seven degrees of unsaturation. The NMR spectra data of
4 (Tables 1 and 3) showed the appearance of an 1,1-disubstituted
carbon–carbon double bond (dC 150.1, qC and 117.0, CH2;dH
5.46 s and 5.12 s). Three ester carbonyls (dC 172.8, 172.4 and
170.1) were also assigned from the13C NMR spectrum and were
HMBC correlated with the methylenes (dH2.32 m, 2H and 1.66 m,
2H; 2.13 m, 2H and 1.58 m, 2H) of two n-butyrate units and an acetate methyl (dH 2.00 s, 3H), respectively. Therefore, the
remaining three degrees of unsaturation identified compound 4 as a tricyclic compound. In the1H NMR spectrum of 4 (Table 3),
two doublets atdH0.99 and 0.92 (each 3H, d, J= 7.2 Hz) arose from
two methyls of an isopropyl group. The molecular framework was established by 1H–1H COSY and HMBC experiments (Fig. 1).
The placement of the acetate at C-13 was confirmed from the HMBC correlations of acetate methyl (dH2.00 s, 3H) and H-13
(d 5.49) with the carbonyl carbon resonating at dC 170.1 (qC).
Also, the location of an n-butyryloxy group at C-12 was proven from the HMBC correlations of H-12 (d 5.04) to the carbonyl
carbon resonating atdC172.4 (qC). The downfield chemical shifts
for H3-15 (d 1.61) and C-3 (d 84.2), and the upfield chemical shifts
of H3-17 (d 1.17) and C-11 (d 72.8), determined the positions
of the other n-butyrate and hydroxy group at C-3 and C-11, respectively. From the above results, the structure of compound
4 was shown to be very similar to that of a known compound,
klysimplexin sulfoxide C.2 Therefore, the molecular framework
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011
Table 2 13C NMR data for compounds 9–12 Position 9a 10b 11a 12a 1 36.7 (CH)c 36.8 (CH) 50.1 (CH) 31.4 (CH) 2 129.6 (CH) 130.6 (CH) 78.0 (CH) 51.7 (CH) 3 133.8 (qC) 133.4 (qC) 81.0 (qC) 144.9 (qC) 4 29.0 (CH2) 32.3 (CH2) 28.5 (CH2) 30.4 (CH2) 5 26.1 (CH2) 25.8 (CH2) 21.5 (CH2) 32.2 (CH2) 6 64.9 (CH) 124.6 (CH) 80.3 (CH) 69.8 (CH) 7 60.9 (qC) 138.3 (qC) 85.3 (qC) 38.9 (qC) 8 39.4 (CH2) 39.9 (CH2) 50.0 (CH2) 36.2 (CH2) 9 23.5 (CH2) 24.0 (CH2) 209.0 (qC) 70.3 (CH) 10 42.5 (CH) 47.0 (CH) 56.2 (CH) 46.6 (CH) 11 85.8 (qC) 73.5 (qC) 83.3 (qC) 71.4 (qC) 12 32.3 (CH2) 36.1 (CH2) 31.3 (CH2) 38.6 (CH2) 13 19.9 (CH2) 20.4 (CH2) 20.3 (CH2) 21.1 (CH2) 14 42.9 (CH) 45.6 (CH) 37.2 (CH) 40.9 (CH) 15 25.8 (CH3) 25.0 (CH3) 24.5 (CH3) 111.9 (CH2) 16 19.2 (CH3) 17.0 (CH3) 23.9 (CH3) 23.2 (CH3) 17 23.8 (CH3) 26.1 (CH3) 25.2 (CH3) 28.5 (CH3) 18 27.5 (CH) 26.5 (CH) 28.5 (CH) 26.8 (CH) 19 22.7 (CH3) 22.1 (CH3) 22.6 (CH3) 22.8 (CH3) 20 16.6 (CH3) 18.5 (CH3) 14.9 (CH3) 21..8 (CH3) 3-n-butyrate 15.6 (CH3) 19.6 (CH2) 37.9 (CH2) 171.1 (qC) 9-OAc 22.7 (CH3) 169.0 (qC) 11-OAc 23.5 (CH3) 23.4 (CH3) 168.8 (qC) 168.0 (qC) a100 MHz in CDCl 3.b125 MHz in CDCl3.cMultiplicities deduced by DEPT.
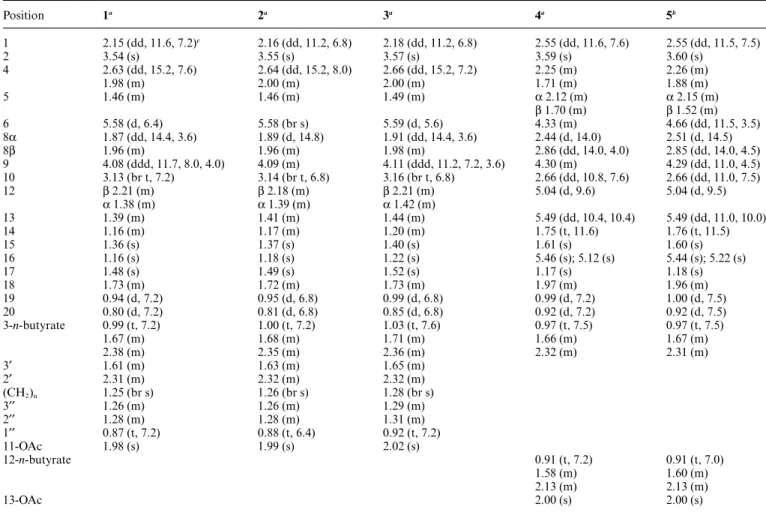
of 4 was established. In the NOESY spectrum of 4 (Fig. 2), observation of the NOE correlations between 10 and both H-8b (d 2.86) and H-1; and H-1 and H-13 suggested that H-1, H-10 and H-13 areb-oriented. Also, correlations between H-2 and both H3-15 and H-14; H-9 and H-12, H-14 and H3-17; and H-6 and
both H-8a (d 2.44) and H3-15 suggested that all of H-2, H-6, H-9,
H-12, H-14, H3-15 and H3-17 area-oriented. Thus, the relative
Fig. 2 Key NOESY correlations of 4.
structure of diterpenoid 4 was established. In order to resolve the absolute structure of 4, we determined the absolute configuration at C-6 using Mosher’s method.25,26The (S)- and
(R)-a-methoxy-a-(trifluoromethyl) phenylacetic (MTPA) esters of 4 (4a and 4b, respectively) were prepared by using the corresponding R-(-)-and S-(+)-a-methoxy-a-(trifluoromethyl) phenylacetyl chlorides, respectively. The values of Dd [d(S-MTPA ester) - d(R-MTPA
ester)] for H-8, H-9 and H2-16 were positive, while the values
of Dd for H-4, H2-5 and H3-15 were negative, revealing the
S-configuration at C-6 (Fig. 3).
Fig. 3 1H NMR chemical shift differences Dd (d
S- dR) in ppm for the
MTPA esters of 4.
Klysimplexin M (5) was isolated as a colorless oil and exhibited a pseudomolecular ion peak at m/z 591.3146 [M + Na]+ by
HRESIMS, appropriate for a molecular formula of C30H48O10,
with one more oxygen atom than that of 4. The NMR spectra data of 5 were found to be very similar to those of 4 (Tables 1 and 3), except for those of CH-6, which were downfield shifted (dC86.7
anddH4.66) relative to these of 4 (dC72.7 anddH4.33). Therefore,
the hydroxy group attached at C-6 in 4 was assumed to be replaced by a hydroperoxy group in 5. The NOE correlations of 5 also showed that the stereochemistry of this metabolite is similar to that of 4. A structure related metabolite, klysimplexin N (6), was also isolated as a colorless oil with a molecular formula of C28H44O9,
implying seven degrees of unsaturation. NMR spectroscopic data of 6 (Tables 1 and 4) showed the presence of an n-butyryloxy group (dC 172.4, qC; 37.3, CH2; 18.5, CH2; 13.6, CH3;dH2.12 m, 2H,
1.63 m, 2H, and 0.93 t, J= 7.0, 3H) and two acetoxy groups (dC
170.2, qC; 21.4, CH3;dC170.2, qC and 20.7, CH3;dH2.10, s and
2.01, s). Comparison of the 1D and 2D NMR data of 6 with those of 4 revealed that the only difference between both compounds arose from the replacement of the n-butyryloxy moiety at C-12 in
4 by an acetoxy group in 6, as confirmed by HMBC correlations of
both acetate methyl (d 2.10) and H-12 (d 5.01) with the carbonyl
carbon resonating atd 170.2 (qC).
The HRESIMS spectrum of 7 exhibited a pseudomolecular ion peak at m/z 607.3095 [M + Na]+, consistent with a molecular
formula C30H48O11 and implying seven degrees of unsaturation.
By comparison of the NMR data of 7 with those of 6 (Tables 1 and 4), it was found that a C-7/C-16 double bond in 6 was replaced by a quaternary carbon bearing a methyl and a hydroxy group in 7. Moreover, the hydroxy group attached at C-6 of 6 was replaced by an acetoxy group in 7. This was further evidenced by the HMBC correlations observed from H3-16 (d 1.19, 3H, s) to C-6 (d 84.8,
CH), C-7 (d 75.8, qC), and C-8 (d 47.6, CH2); and from H-6
(d 5.61) to the carbonyl carbon resonating at dC172.1 (qC). The
more detailed analysis on the1H and13C NMR spectroscopic data
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011
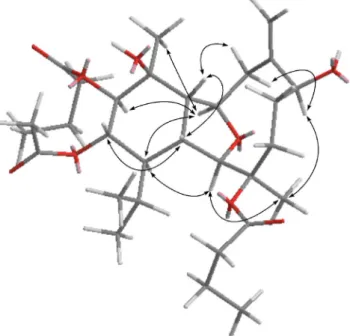
Table 3 1H data for compounds 1–5 Position 1a 2a 3a 4a 5b 1 2.15 (dd, 11.6, 7.2)c 2.16 (dd, 11.2, 6.8) 2.18 (dd, 11.2, 6.8) 2.55 (dd, 11.6, 7.6) 2.55 (dd, 11.5, 7.5) 2 3.54 (s) 3.55 (s) 3.57 (s) 3.59 (s) 3.60 (s) 4 2.63 (dd, 15.2, 7.6) 2.64 (dd, 15.2, 8.0) 2.66 (dd, 15.2, 7.2) 2.25 (m) 2.26 (m) 1.98 (m) 2.00 (m) 2.00 (m) 1.71 (m) 1.88 (m) 5 1.46 (m) 1.46 (m) 1.49 (m) a 2.12 (m) a 2.15 (m) b 1.70 (m) b 1.52 (m) 6 5.58 (d, 6.4) 5.58 (br s) 5.59 (d, 5.6) 4.33 (m) 4.66 (dd, 11.5, 3.5) 8a 1.87 (dd, 14.4, 3.6) 1.89 (d, 14.8) 1.91 (dd, 14.4, 3.6) 2.44 (d, 14.0) 2.51 (d, 14.5) 8b 1.96 (m) 1.96 (m) 1.98 (m) 2.86 (dd, 14.0, 4.0) 2.85 (dd, 14.0, 4.5) 9 4.08 (ddd, 11.7, 8.0, 4.0) 4.09 (m) 4.11 (ddd, 11.2, 7.2, 3.6) 4.30 (m) 4.29 (dd, 11.0, 4.5) 10 3.13 (br t, 7.2) 3.14 (br t, 6.8) 3.16 (br t, 6.8) 2.66 (dd, 10.8, 7.6) 2.66 (dd, 11.0, 7.5) 12 b 2.21 (m) b 2.18 (m) b 2.21 (m) 5.04 (d, 9.6) 5.04 (d, 9.5) a 1.38 (m) a 1.39 (m) a 1.42 (m) 13 1.39 (m) 1.41 (m) 1.44 (m) 5.49 (dd, 10.4, 10.4) 5.49 (dd, 11.0, 10.0) 14 1.16 (m) 1.17 (m) 1.20 (m) 1.75 (t, 11.6) 1.76 (t, 11.5) 15 1.36 (s) 1.37 (s) 1.40 (s) 1.61 (s) 1.60 (s) 16 1.16 (s) 1.18 (s) 1.22 (s) 5.46 (s); 5.12 (s) 5.44 (s); 5.22 (s) 17 1.48 (s) 1.49 (s) 1.52 (s) 1.17 (s) 1.18 (s) 18 1.73 (m) 1.72 (m) 1.73 (m) 1.97 (m) 1.96 (m) 19 0.94 (d, 7.2) 0.95 (d, 6.8) 0.99 (d, 6.8) 0.99 (d, 7.2) 1.00 (d, 7.5) 20 0.80 (d, 7.2) 0.81 (d, 6.8) 0.85 (d, 6.8) 0.92 (d, 7.2) 0.92 (d, 7.5) 3-n-butyrate 0.99 (t, 7.2) 1.00 (t, 7.2) 1.03 (t, 7.6) 0.97 (t, 7.5) 0.97 (t, 7.5) 1.67 (m) 1.68 (m) 1.71 (m) 1.66 (m) 1.67 (m) 2.38 (m) 2.35 (m) 2.36 (m) 2.32 (m) 2.31 (m) 3¢ 1.61 (m) 1.63 (m) 1.65 (m) 2¢ 2.31 (m) 2.32 (m) 2.32 (m) (CH2)n 1.25 (br s) 1.26 (br s) 1.28 (br s) 3¢¢ 1.26 (m) 1.26 (m) 1.29 (m) 2¢¢ 1.28 (m) 1.28 (m) 1.31 (m) 1¢¢ 0.87 (t, 7.2) 0.88 (t, 6.4) 0.92 (t, 7.2) 11-OAc 1.98 (s) 1.99 (s) 2.02 (s) 12-n-butyrate 0.91 (t, 7.2) 0.91 (t, 7.0) 1.58 (m) 1.60 (m) 2.13 (m) 2.13 (m) 13-OAc 2.00 (s) 2.00 (s) aSpectra recorded at 400 MHz in CDCl
3at 25◦C.bSpectra recorded at 500 MHz in CDCl3at 25◦C.cJ values in Hz in parentheses.
and the detected 2D correlations in1H–1H COSY and HMBC
spectra led to the establishment of the molecular framework of
7 (Fig. 1). The relative configurations of all chiral centers except
that of C-7 in 7 were confirmed to be mostly the same as those of 4 by analysis of NOE correlations. H3-16 was found to exhibit
an NOE correlation with H-5b but not with H-6, revealing the b-orientation of the acetoxy group at C-6 and a-orientation of the hydroxy group at C-7. Thus, the structure of diterpenoid 7 was established.
Klysimplexin P (8) was obtained as a colorless oil. The HRES-IMS (m/z 563.2833 [M + Na]+) of 8 established a molecular
for-mula of C28H44O10, appropriate for seven degrees of unsaturation.
Inspection of the NMR spectroscopic data of 8 by the assistance of DEPT spectrum revealed the presence of eight methyls, five methylenes, eight methines (including four oxymethines), two sp3
oxygenated carbons, and five carbonyl carbons (including one ketone, one aldehyde, and three ester carbonyls). Three ester carbonyls (dC 172.6, 170.1 and 170.0) were also assigned from
the 13C NMR spectrum and were HMBC correlated with the
methylenes (dH2.26 m, 2H and 1.63 m, 2H) of an n-butyrate unit
and two acetate methyls (dH2.01 s, 3H; 2.09 s, 3H), respectively.
Therefore, the remaining two degrees of unsaturation identified metabolite 8 as a bicyclic compound. The1H NMR data of 8
(Table 4) showed a methyl (d 2.23, 3H, s) attached to a carbonyl
carbon, two tertiary methyls attached to oxygenated carbons (d
1.42 and 1.14, each 3H, s), and two secondary methyls (d 1.01
and 0.88, each 3H, d, J = 7.5 Hz) of an isopropyl moiety. Two oxymethines observed atdC77.0 (CH) and 70.9 (CH) anddH5.04
(1H, d, J= 9.5 Hz) and 5.45 (1H, dd, J = 11.0, 10.0 Hz) indicated the presence of two acetoxy substituents in the six-membered ring, as those of compounds 6 and 7. By comparison of the1H NMR
and13C NMR spectroscopic data of 8 with those of 1–7, signals
resonating atdH2.47 (1H, m), 2.48 (1H, m), 3.73 s and 4.46 (1H,
br t, J= 8.5), and at dC 42.2, 53.9, 88.5 and 75.4 also indicated
the presence of a tetrahydrofuran structural unit in 8. The 1D NMR and HSQC data showed signals atdC206.3 (qC) and 201.4
(CH); dH 9.70 (1H, br s), and further supported the presence
of a ketone and an aldehyde. The above findings together with careful analysis of1H–1H COSY and HMBC correlations (Fig. 1),
led to the establishment of the 6,7-secoeunicellin skeleton of 8, as confirmed by key HMBC correlations from H3-16 to C-7 (d
206.3) and C-8 (d 50.7), and one proton of H2-4 (d 2.44) and H2-5
(d 2.50) to aldehyde carbon (d 201.4). Therefore, the molecular
framework of 8 was established. In the NOESY spectrum of 8 (Fig. 4), the NOE correlations of H-10 with both H2-8 and
H-1; and H-1 with H-13 suggested that H-1, H-10 and H-13 are
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011
Table 4 1H data for compounds 6–10 Position 6a 7b 8a 9c 10a 1 2.55 (dd, 11.5, 7.5)d 2.41 (m) 2.47 (m) 2.38 (m) 2.82 (m) 2 3.59 (s) 3.55 (s) 3.73 (s) 5.17 (d, 6.4) 5.29 (d, 9.0) 4 2.24 (m) 2.66 (m) 2.44 (m) 2.34 (m) 2.10 (m) 1.71 (m) 1.98 (m) 2.19 (m) 1.95 (m) 1.83 (m) 5 a 2.18 (m) a 1.57 (m) 2.50 (m) b 2.19 (m) 2.25 (m) b 1.70 (m) b 1.47 (m) a 1.40 (m) 2.03 (m) 6 4.33 (dd, 11.0, 4.0) 5.61 (d, 5.4) 9.70 (br s) 3.28 (dd, 11.2, 4.0) 5.24 (dd, 11.0, 4.5) 8a 2.44 (d, 14.5) 1.81 (m) 2.73 (m) 1.99 (m) 2.19 (m) 8b 2.86 (dd, 14.0, 5.0) 1.93 (m) 1.03 (m) 1.85 (m) 9 4.28 (dd, 11.5, 3.5) 4.29 (m) 4.46 (br t, 8.5) 1.33 (m) 1.57 (m) 1.29 (m) 10 2.67 (dd, 11.0, 7.0) 2.63 (br t, 8.4) 2.48 (m) 2.96 (m) 1.94 (m) 12 5.01 (d, 10.0) 5.02 (d, 9.6) 5.04 (d, 9.5) 1.90 (m) 1.48 (m) 1.56 (m) 13 5.49 (dd, 11.0, 10.0) 5.48 (dd, 10.9, 9.9) 5.45 (dd, 11.0, 10.0) 1.37 (m) 1.53 (m) 1.42 (m) 14 1.73 (m) 1.74 (m) 1.83 (t, 11.0) 1.09 (m) 0.94 (m) 15 1.60 (s) 1.39 (s) 1.42 (s) 1.70 (s) 1.72 (s) 16 5.46 (s); 5.12 (s) 1.19 (s) 2.23 (s) 1.17 (s) 1.53 (s) 17 1.18 (s) 1.12 (s) 1.14 (s) 1.44 (s) 1.25 (s) 18 1.98 (m) 1.72 (m) 1.73 (m) 1.92 (m) 1.88 (m) 19 0.99 (d, 7.5) 1.01 (d, 7.0) 1.01 (d, 7.5) 0.92 (d, 6.8) 0.98 (d, 7.0) 20 0.92 (d, (7.5) 0.96 (d, 7.0) 0.88 (d, 7.5) 0.69 (d, 6.8) 0.77 (d, 7.0) 3-n-butyrate 0.93 (t, 7.0) 0.99 (t, 7.1) 0.99 (t, 7.5) 1.63 (m) 1.69 (m) 1.63 (m) 2.12 (m) 2.37 (m); 2.28 (m) 2.26 (m) 6-OAc 2.09 (s) 11-OAc 2.01 (s) 12-OAc 2.10 (s) 2.08 (s) 2.09 (s) 13-OAc 2.01 (s) 1.99 (s) 2.01 (s) aSpectra recorded at 500 MHz in CDCl
3at 25◦C.bSpectra recorded at 300 MHz in CDCl3at 25◦C.cSpectra recorded at 400 MHz in CDCl3at 25◦C.
dJ values in Hz in parentheses.
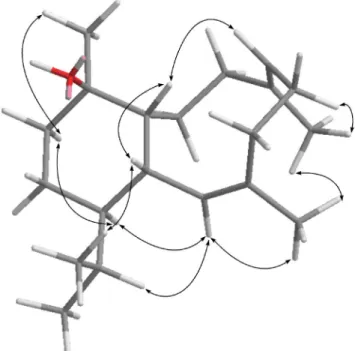
Fig. 4 Key NOESY correlations of 8.
b-oriented. Also, correlations between H-2 and both H3-15 and
H-14; H-9 and H-12, H-14 and H3-17; and H-12 and both H-14
and H3-17, suggested that all of H-2, H-9, H-12, H-14, H3-15 and
H3-17 area-oriented. Thus, the structure of diterpenoid 8 was
unambiguously established.
Klysimplexin Q (9) was found to possess a molecular formula of C22H36O3, as revealed from its HRESIMS (m/z 371.2560
[M + Na]+). Thus, the compound possesses five degrees of
unsaturation. The 3H singlet appearing atd 2.01 in the1H NMR
spectrum and the carbonyl signal at d 168.8 in the13C NMR
spectrum were ascribable to an acetate. The twenty-two carbon signals appearing in the13C NMR spectrum of 9 (Table 2) were
identified by DEPT spectrum to six methyls, six methylenes, six methines (including one vinylic CH and one epoxide CH), two sp3oxygenated quaternary carbons, one sp2 quaternary carbon
and one ester carbonyl carbon. Moreover, the13C signals at d
133.8 (qC), 129.6 (CH), 64.9 (CH), and 60.9 (qC) assigned one trisubstituted double bond and one epoxide in the molecule. Three 3H singlets appearing in the1H NMR spectrum atd 1.70, 1.44, and
1.17 were assigned to an olefinic methyl, one methyl attached to a quaternary oxycarbon, and one methyl of a trisubstituted epoxide in the molecule, respectively. Also, two doublets atdH0.92 and 0.69
(each 3H, d, J= 6.8 Hz) arose from two methyls of an isopropyl group. The above functionalities revealed that compound 9 is a bicyclic compound. The more detailed analysis on the1H and 13C NMR spectroscopic data and the detected 2D correlations in 1H–1H COSY and HMBC spectra led to the establishment of the
molecular framework of 9 (Fig. 1). In the NOESY spectrum of compound 9 (Fig. 5), the NOE correlations between H3-15 and
H-2 revealed the Z geometry of the double bond at C-2 and C-3. In addition, H-6 showed NOE interactions with H-10, H-5b (d 2.19), and H-4b (d 2.34), but not with H3-16; and H-1 showed
NOE responses with both H-10 and H-4b (d 2.34), but not with H-14, indicating theb-orientation for all of H-10, H-6, and H-1. The above observations and correlations between H-14 and both H-12a (d 1.56) and H-2; H-12a (d 1.56) and H3-17; H-2 and both
H3-16 and H3-15; and H3-16 and H3-15 suggested that all of H-14,
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011
Fig. 5 Key NOESY correlations of 9.
H3-16, and H3-17 area-oriented. The structure of diterpenoid 9
was thus fully established.
Klysimplexin R (10) was isolated as a colorless oil and exhibited a pseudomolecular ion peak at m/z 290.2607 [M]+by HREIMS,
appropriate for a molecular formula of C20H34O and implying
four degrees of unsaturation. The IR spectrum of 10 revealed the presence of hydroxy group (3398 cm-1). The 13C NMR
spectroscopic data of 10 were found to be very similar to those of 9, except that the 6,7-epoxide (d 64.9, CH and 60.9, qC) in 9
was converted to an olefinic group (d 138.3, qC and 124.6, CH)
in 10, as also confirmed by HMBC correlations (H3-16 to C-6,
C-7, and C-8), and the acetoxy group at C-11 in 9 was replaced by a hydroxy group in 10. It was further observed that the NOE correlations of 10 (Fig. 6) are very similar to those of 9 and the
E geometry 6,7-double bond in 10 was established by the NOE
correlation between H3-16 and one proton of H2-5 (d 2.25) and the
upfield chemical shift of C-16 (d 17.0) in 10. Thus, the structure
of 10 was determined unambiguously.
Klysimplexin S (11) was obtained as a colorless oil. The HRESIMS of 11 established the molecular formula C26H42O7,
implying 6 degrees of unsaturation. The IR absorption bands at 3347, 1731, and 1716 cm-1revealed the presence of hydroxy and
carbonyl functionalities. The13C NMR spectrum of 11 showed
the presence of a ketone (dC209). Two ester carbonyl carbons (dC
171.1 and 167.9) were HMBC correlated with the methylenes (dH
2.45 m, 2H and 1.75 m, 2H) of an n-butyrate and an acetate methyl (dH2.02 s, 3H), respectively. Therefore, 11 is a tricyclic diterpenoid.
The molecular framework was also confirmed by1H–1H COSY
and HMBC experiments (Fig. 1). It was shown that the NMR data of 11 (Tables 2 and 5) were almost identical to those of australin A (14),8except that the hydroxy group at C-3 in 14 (Scheme 2) was
replaced by an n-butyryloxy in 11, as confirmed by the downfield shiftedd value of H3-15 (d 1.55) of 11, relative to that of 14 (d
1.23), and the HMBC correlation from H-2 (d 3.90) to the carbonyl
carbon resonating atd 171.1 (qC).
Fig. 6 Key NOESY correlations of 10.
Table 5 1H data for compounds 11–12
Position 11a 12a 1 2.56 (dd, 12.0, 4.4)b 2.34 (dd, 12.8, 6.4) 2 3.90 (s) 2.22 (d, 13.2) 4 2.98 (dd, 13.6, 4.0) 2.15 (m) 1.41 (dd, 13.6, 7.6) 5 1.70 (m) a 1.86 (m) b 1.51 (m) 6 3.85 (dd, 11.2, 6.0) 4.35 (dd, 12.0, 5.2) 8 a 2.02 (d, 12.0) a 2.30 (m) b 2.79 (d, 12.0) b 1.42 (m) 9 5.31 (m) 10 4.06 (d, 4.4) 1.94 (d, 5.6) 12 2.26 (dd, 9.6, 3.6) 1.59 (m) 1.54 (m) 13 1.65 (m) 1.57 (m) 1.23 (m) 1.37 (m) 14 1.98 (m) 1.15 (m) 15 1.55 (s) 4.84 (s) 4.66 (s) 16 1.16 (s) 0.86 (s) 17 1.49 (s) 1.32 (s) 18 1.92 (m) 1.80 (m) 19 1.01 (d, 6.8) 0.90 (d, 6.8) 20 0.76 (d, 6.8) 0.87 (d, 6.8) 3-n-butyrate 1.05 (t, 7.2) 1.75 (m) 2.45 (m) 9-OAc 2.08 (s) 11-OAc 2.02 (s) 7-OH 4.88 (s) aSpectra recorded at 400 MHz in CDCl 3 at 25◦C.bJ values in Hz in parentheses.
The HRESIMS (m/z 387.2509, [M + Na]+) of klysimplexin T
(12) established the molecular formula C22H36O4Na, consistent
with five degrees of unsaturation. The IR absorptions of 12 indicated the presence of hydroxy (3641 cm-1) and carbonyl
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011
(1735 cm-1) functionalities. The NMR spectra of 12 (Tables 2 and
5) showed signals of an 1,1-disubstituted carbon–carbon double bond (dC 144.9, qC and 111.9, CH2;dH 4.84 s and 4.66 s). The
presence of an acetoxy group was indicated by the1H NMR signal
atd 2.08 (s, 3H) and13C NMR signals atd 22.7 (CH
3) and 169.0
(qC).1H NMR data of 12 (Table 5) showed the presence of a methyl
(dH1.32) attached to an oxygenated carbon, and a methyl (dH0.86)
bound to a quaternary carbon, respectively. Also, two doublets at
dH0.90 and 0.87 (each 3H, d, J= 6.8 Hz) arose from two methyls
of an isopropyl group. The above functionalities revealed that 12 is a tricyclic terpenoidal compound. The molecular framework of 12 was further established by 2D NMR studies, in particular1H–1H
COSY and HMBC correlations (Fig. 1). In the NOESY spectrum of 12 (Fig. 7), NOE correlations of H-1 and all of H-6, H-10, H-18 and H3-19 were observed, suggesting that H-1, H-6, H-10
and the C-14 isopropyl group areb-oriented, and H-14 should be placeda-oriented. Also, correlations of H-2 and H-14, H3-16 and
H3-17; and H-9 and H3-17 suggested that of all of H-2, H-9, H-14,
H3-16 and H3-17 area-oriented. Thus, the relatively structure of
diterpenoid 12 could be established.
Fig. 7 Key NOESY correlations of 12.
Compound 12 could be assumed to be biosynthesized via the acid-catalyzed ring opening of 6,7-epoxide to form a C-7 carbonium ion followed by carbon–carbon bond formation between C-2 and C-7 of a corresponding eunicellin, presumably the deacetyl derivative of 9, and hydroxylation at C-9 and subsequent acetylation (Scheme 3). Alternatively, 1–3 and 7 might arise from the acid-catalyzed reaction of a 6,7-diastereomer of 9, which was not found in this study, to form the cation at C-7 and the subsequent addition of water froma face in the next step.
Cytotoxicity of metabolites 1–12 toward a limited panel of cancer cell lines was evaluated. The results showed that compound
Scheme 3 Proposed biosynthetic pathway of 12.
9 exhibited cytotoxicity toward Hep G2 and Hep 3B (human
hepatocellular carcinoma), MDA-MB-231 and MCF-7 (human breast carcinoma), A549 (human lung carcinoma), and Ca9-22 (human gingival carcinoma) cell lines with IC50’s of 53.2, 35.1, 44.0,
36.5, 40.5, and 40.5mM, respectively. Also, metabolite 12 showed cytotoxicity (IC50’s 34.3, 26.4, 44.0, 27.2, 42.0 and 37.4mM) against
the growth of Hep G2, Hep 3B, MDA-MB-231, MCF-7, A549, and Ca9-22 cells, respectively. Other metabolites were found to be inactive against the growth of the above six cancer cells.
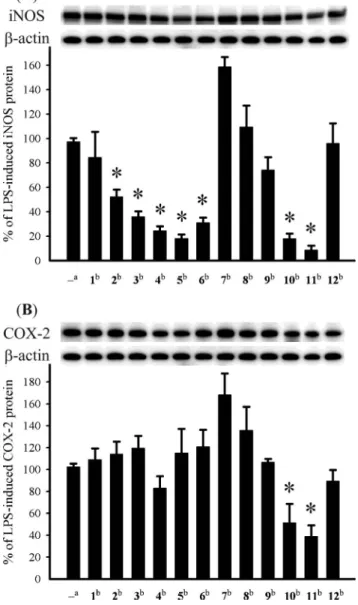
The in vitro anti-inflammatory effects of compounds 1–12 were also tested. In this assay, the inhibition of LPS-induced up-regulation of pro-inflammatory proteins, iNOS and COX-2 in RAW264.7 macrophage cells was measured by immunoblot analysis. At a concentration of 10mM, compounds 2–6, 10 and 11 were found to significantly reduce the expression of iNOS protein, relative to the control cells stimulated with LPS only. Furthermore, at the same concentration, metabolites 10 and 11 also could effectively reduce COX-2 expression in the same macrophage cells with LPS treatment. On the other hand, 7 could enhance the expression of both iNOS and COX-2 which might arise from the presence of acetoxy and hydroxy groups at C-6 and C-7, respectively. Thus, compounds 2–6, 10 and 11 might be useful anti-inflammatory agents, while 11 is a promising anti-anti-inflammatory lead compound as it showed potent inhibitory activity against the expression of both iNOS and COX-2 proteins (Fig. 8)
Conclusion
Our investigation demonstrated that the cultured soft coral,
K. simplex, could be a good source of bioactive substances.
Several of the isolated compounds, in particular 10 and 11, are potential anti-inflammatory agents. Also, it is worthwhile to note here that 8 is a 6,7-secoeunicellin, while 12 is a tricarbocyclic compound which might be derived from the carbon–carbon bond formation between C-2 and C-7 of a corresponding 2,9-deoxygenated eunicellin.
Experimental
General experimental procedures
Optical rotations were measured on a JASCO P-1020 polarimeter. IR spectra were recorded on a JASCO FT/IR-4100 infrared spectrophotometer. ESIMS spectra were obtained with a Bruker
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011
Fig. 8 Effect of compounds 1–12 on iNOS and COX-2 protein expression of RAW264.7 macrophage cells by immunoblot analysis. (A) Immunoblots of iNOS andb-actin; (B) Immunoblots of COX-2 and b-actin. The values are mean± SEM. (n = 6). Relative intensity of the LPS alone stimulated group was taken as 100%. Under the same experimental condition CAPE (caffeic acid phenylethyl ester, 10mM) reduced the levels of the iNOS and COX-2 to 2.5± 3.7% and 67.2 ± 13.4%, respectively. *Significantly different from LPS alone stimulated group (*P< 0.05).astimulated with
LPS,bstimulated with LPS in the presence of 1–12 (10mM).
APEX II mass spectrometer. LC-ESI MS/MS spectrometry anal-ysis was carried out using an Applied Biosystem API 4000 tandem quadrupole mass spectrometer. NMR spectra were recorded on a Varian Unity INOVA 500 FT-NMR at 500 MHz for1H
and 125 MHz for 13C or on a Varian 400 MR FT-NMR at
400 MHz for1H and 100 MHz for13C, or on a Bruker
AVANCE-DPX 300 FT-NMR at 300 MHz for1H and 75 MHz for13C,
respectively. Silica gel (Merck, 230–400 mesh) was used for column chromatography. Precoated silica gel plates (Merck, Kieselgel 60 F-254, 0.2 mm) were used for analytical TLC. High-performance liquid chromatography was performed on a Hitachi L-7100 HPLC apparatus with a ODS column (250¥ 21.2 mm, 5 mm).
Extraction and isolation
Specimens of the cultured soft coral K. simplex were collected by hand in a 30 ton cultivating tank located in the National Museum of Marine Biology and Aquarium, Taiwan, in July 2005. A voucher sample (CSC-2) was deposited at the Department of Marine Biotechnology and Resources, National Sun Yat-sen University. The octocoral (1.5 kg fresh wt) was collected and freeze-dried. The freeze-dried material was minced and extracted exhaustively with EtOH (3¥ 10 L). The EtOH extract of the frozen organism was partitioned between CH2Cl2and H2O. The CH2Cl2
-soluble portion (15.2 g) was subjected to column chromatography on silica gel and eluted with EtOAc in n-hexane (0–100% of EtOAc, gradient) and then further with MeOH in EtOAc with increasing polarity to yield 40 fractions. Fraction 10, eluted with n-hexane–EtOAc (15 : 1), was rechromatographed over a Sephadex LH-20 column, using acetone as the mobile phase to afford five subfractions (A1–A4). Subfraction A3 was separated by reverse-phase HPLC (CH3CN–H2O, 6 : 1 to 3 : 1) to afford compounds
9 (6.0 mg) and 10 (2.2 mg). Fraction 21, eluted with n-hexane–
EtOAc (9 : 1), was rechromatographed over a Sephadex LH-20 column, using acetone as the mobile phase to afford five subfractions (B1–B5). Subfraction B3 was separated by reverse-phase HPLC (CH3CN, 100%) to afford compounds 1 (15.5 mg),
2 (4.2 mg), and 3 (1.1 mg), respectively. Fraction 23, eluted with
n-hexane–EtOAc (5 : 1), was rechromatographed over a Sephadex LH-20 column, using acetone as the mobile phase to afford five subfractions (C1–C5). Subfractions C3 and C4 were separated by reverse-phase HPLC (CH3CN–H2O, 4 : 1 to 1 : 1) to afford
compounds 4 (1.2 mg), 5 (1.1 mg), 6 (1.0 mg), and 12 (1.1 mg), respectively. Fraction 26, eluted with n-hexane–EtOAc (2 : 1), was rechromatographed over a Sephadex LH-20 column, using acetone as the mobile phase to afford five subfractions (D1–D4). Subfraction D3 was separated by reverse-phase HPLC (CH3CN–
H2O, 3 : 1 to 1 : 2) to afford compounds 7 (15.3 mg), 8 (1.2 mg),
and 11 (2.3 mg).
Klysimplexin I (1). Colorless oil; [a]25
D -38 (c 1.55, CHCl3);
IR (neat) vmax3460, 1738 cm-1;13C and1H NMR data (400 MHz;
CHCl3), see Tables 1 and 3; ESIMS m/z 701 [M + Na]+; HRESIMS
m/z 701.4974 [M + Na]+(calcd. 701.4968 for C
40H70O8Na).
Klysimplexin J (2). Colorless oil; [a]25
D -40 (c 0.42, CHCl3);
IR (neat) vmax3463, 1723 cm-1;13C and1H NMR data (400 MHz;
CHCl3), see Tables 1 and 3; ESIMS m/z 730 [M + Na]+; HRESIMS
m/z 729.5277 [M + Na]+(calcd. 729.5281 for C
42H74O8Na).
Klysimplexin K (3). Colorless oil; [a]25
D -38 (c 0.11, CHCl3);
IR (neat) vmax3437, 1734 cm-1;13C and1H NMR data (400 MHz;
CDCl3), see Tables 1 and 3; ESIMS m/z 757.55 [M + Na]+;
HRES-IMS m/z 757.5590 [M + Na]+(calcd. 757.5594 for C
44H78O8Na).
Klysimplexin L (4). Colorless oil; [a]25
D -64 (c 0.12, CHCl3);
IR (neat) vmax3452, 1734 cm-1;13C and1H NMR data (400 MHz;
CDCl3), see Tables 1 and 3; ESIMS m/z 575 [M + Na]+; HRESIMS
m/z 575.3193 [M + Na]+(calcd. 575.3196 for C
30H48O9Na).
Klysimplexin M (5). Colorless oil; [a]25
D -74 (c 0.11, CHCl3);
IR (neat) vmax3452, 1738 cm-1;13C and1H NMR data (500 MHz;
CDCl3), see Tables 1 and 3; ESIMS m/z 591 [M + Na]+; HRESIMS
m/z 591.3146 [M + Na]+(calcd. 591.3145 for C
30H48O10Na).
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011
Klysimplexin N (6). Colorless oil; [a]25
D -53 (c 0.10, CHCl3);
IR (neat) vmax3467, 1738 cm-1;13C and1H NMR data (500 MHz;
CDCl3), see Tables 1 and 4; ESIMS m/z 547 [M + Na]+; HRESIMS
m/z 547.2885 [M + Na]+(calcd. 547.2883 for C
28H44O9Na).
Klysimplexin O (7). Colorless oil; [a]25
D -27 (c 1.53, CHCl3);
IR (neat) vmax3478, 1734 cm-1;13C and1H NMR data (300 MHz;
CDCl3), see Tables 1 and 4; ESIMS m/z 607 [M + Na]+; HRESIMS
m/z 607.3095 [M + Na]+(calcd. 607.3094 for C
30H48O11Na).
Klysimplexin P (8). Colorless oil; [a]25
D -23 (c 0.12, CHCl3);
IR (neat) vmax3460, 1738 and 1711 cm-1;13C and1H NMR data
(500 MHz; CDCl3), see Tables 1 and 4; ESIMS m/z 563 [M +
Na]+; HRESIMS m/z 563.2833 [M + Na]+ (calcd. 563.2832 for
C28H44O10Na).
Klysimplexin Q (9). Colorless oil; [a]25
D+56 (c 0.60, CHCl3); IR
(neat) vmax1734 cm-1;13C and1H NMR data (400 MHz; CDCl3),
see Tables 2 and 4; ESIMS m/z 371 [M + Na]+; HRESIMS m/z
371.2560 [M + Na]+(calcd. 371.2562 for C
22H36O3Na).
Klysimplexin R (10). Colorless oil; [a]25
D +30 (c 0.22, CHCl3);
IR (neat) vmax3398 cm-1;13C and1H NMR data (500 MHz; CDCl3),
see Tables 2 and 4; EIMS m/z 290 [(5.9) M]+, 272 [(9.9) M- H 2O]+,
257 [(5.9) M- Me - H2O]+; HREIMS m/z 290.2607 [M]+(calcd.
290.2610 for C20H34O).
Klysimplexin S (11). Colorless oil; [a]25
D -43 (c 0.23, CHCl3);
IR (neat) vmax3347 1731 and 1716 cm-1;13C and1H NMR data
(400 MHz; CDCl3), see Tables 2 and 5; ESIMS m/z 489 [M +
Na]+; HRESIMS m/z 489.2831 [M + Na]+ (calcd. 489.2828 for
C26H42O7Na).
Klysimplexin T (12). Colorless oil; [a]25
D -56 (c 0.11, CHCl3);
IR (neat) vmax 3641 and 1735 cm-1; 13C and 1H NMR data
(400 MHz; CDCl3), see Tables 2 and 5; ESIMS m/z 387 [M +
Na]+; HRESIMS m/z 387.2509 [M + Na]+ (calcd. 387.2511 for
C22H36O4Na).
Base-catalyzed hydrolysis of 1
A solution of 1 (10.1 mg) was dissolved in 5% methanolic NaOH solution (2.7 mL), and the mixture was stirred at 0◦C for 12 h. The mixture was then neutralized with diluted HCl (0.1 N) and the resulted solution was evaporated. The afforded residue was extracted with CHCl3 (2.0 mL ¥ 3). The CHCl3-soluble layers
were combined, dried over anhydrous NaSO4 and evaporated.
The residue was subjected to column chromatograph over silica gel using EtOAc–n-hexane (1 : 1) to yield 13 (4 mg, 57.4%).
Preparation of (S)-and (R)-MTPA esters of 4
To a solution of 4 (0.5 mg) in pyridine (0.4 mL) was added R-(-)-a-methoxy-a-(trifluoromethyl)-phenylacetyl (MPTA) chloride (25mL), and the mixture was allowed to stand for 24 h at room temperature. The reaction was quenched by addition of 1.0 mL of water, and the mixture was subsequently extracted with EtOAc (3¥ 1.0 mL). The EtOAc-soluble layers were combined, dried over anhydrous MgSO4and evaporated. The residue was subjected to
column chromatography over silica gel using n-hexane–EtOAc (6 : 1) to yield the (S)-MTPA ester, 4a (0.6 mg, 86%). The same procedure was used to prepare the (R)-MTPA ester, 4b (0.6 mg, 86%) from the reaction of (S)-MTPA chloride with 4 in pyridine.
Selective1H NMR (CDCl 3, 400 MHz) of 4a: 5.425 (1H, m, H-6), 5.415 (1H, s, H-16a), 5.196 (1H, s, H-16b), 4.312 (1H, dd, J= 10.8 and 4.8, H-9), 2.467 (1H, d, J= 13.2 Hz, H-8a), 2.252 (1H, m, H-4a), 2.084 (1H, m, H-5a), 1.847 (1H, m, H-5b), 1.610 (3H, s, H3-15). Selective1H NMR (CDCl3, 400 MHz) of 4b:d 5.391 (1H, dd, J= 10.4 and 3.6 Hz, H-6), 5.215 (1H, s, H-16a), 5.097 (1H, s, H-16b), 4.306 (1H, dd, J= 10.8 and 4.8, H-9), 2.459 (1H, d, J = 13.6 Hz, H-8a), 2.253 (1H, m, H-4a), 2.145 (1H, m, H-5a), 1.867 (1H, m, H-5b), 1.623 (3H, s, H3-15).
Cytotoxicity testing
Cell lines were purchased from the American Type Culture Collec-tion (ATCC). Cytotoxicity assays were performed using the MTT [3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide] colorimetric method.27,28
In vitro anti-inflammatory assay
Macrophage (RAW264.7) cell line was purchased from ATCC.
In vitro anti-inflammatory activity of compounds 1–12 was
mea-sured by examining the inhibition of lipopolysaccharide (LPS)-induced upregulation of iNOS (inducible nitric oxide synthetase) and COX-2 (cyclooxygenase-2) proteins in macrophage cells using western blotting analysis.29,30
Acknowledgements
Financial support awarded to J.-H. Sheu was provided by the National Museum of Marine Biology & Aquarium (NMM98001), the National Science Council of Taiwan (NSC-98-2113-M-110-002-MY3), and the Ministry of Education of Taiwan (98C031702).
Notes and references
1 B.-W. Chen, Y.-C. Wu, M. Y. Chiang, J.-H. Su, W.-H. Wang, T.-Y. Fan and J.-H. Sheu, Tetrahedron, 2009, 65, 7016–7022.
2 B.-W. Chen, C.-H. Chao, J.-H. Su, Z.-H. Wen, P.-J. Sung and J.-H. Sheu, Org. Biomol. Chem., 2010, 8, 2363–2366.
3 S.-L. Wu, J.-H. Su, Z.-H. Wen, C.-H. Hsu, B.-W. Chen, C.-F. Dai, Y.-H. Kuo and J.-H. Sheu, J. Nat. Prod., 2009, 72, 994–1000.
4 K. Kyeremeh, T. C. Baddeley, B. K. Stein and M. Jaspars, Tetrahedron, 2006, 62, 8770–8778.
5 J. Su, Y. Zheng and L. Zeng, J. Nat. Prod., 1993, 56, 1601–1604. 6 N. Fusetani, H. Nagata, H. Hirota and T. Tsuyuki, Tetrahedron Lett.,
1989, 30, 7079–7082.
7 C. A. Ospina and A. D. Rodr´ıguez, J. Nat. Prod., 2006, 69, 1721–1727. 8 A. F. Ahmed, M.-H. Wu, G.-H. Wang, Y.-C. Wu and J.-H. Sheu, J. Nat.
Prod., 2005, 68, 1051–1055.
9 S. Ketzinel, A. Rudi, M. Schleyer, Y. Benayahu and Y. Kashman, J. Nat. Prod., 1996, 59, 873–875.
10 M. J. Ortega, E. Zub´ıa and J. Salv´a, J. Nat. Prod., 1997, 60, 485–487. 11 L. Chill, N. Berrer, Y. Benayahu and Y. Kashman, J. Nat. Prod., 2005,
68, 19–25.
12 T. Miyamoto, K. Yamada, N. Ikeda, T. Komori and R. Higuchi, J. Nat. Prod., 1994, 57, 1212–1219.
13 Y. Seo, J.-R. Rho, K. W. Cho and J. Shin, J. Nat. Prod., 1997, 60, 171–174.
14 G.-H. Wang, J.-H. Sheu, M. Y. Chiang and T.-J. Lee, Tetrahedron Lett., 2001, 42, 2333–2336.
15 G.-H. Wang, J.-H. Sheu, C.-Y. Duh and M. Y. Chiang, J. Nat. Prod., 2002, 65, 1475–1478.
16 P. Sharma and M. Alam, J. Chem. Soc., Perkin Trans. 1, 1988, 2537– 2540.
17 T. Kusumi, H. Uchida, M. O. Ishitsuka, H. Yamamoto and H. Kakisawa, Chem. Lett., 1988, 1077–1078.
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011
18 S. J. Bloor, F. J. Schmitz, M. B. Hossain and D. V. D. Helm, J. Org. Chem., 1992, 57, 1205–1216.
19 H.-C. Huang, Z.-H. Wen, C.-H. Chao, A. F. Ahmed, M. Y. Chiang, Y.-H. Kuo, C.-H. Hsu and J.-H. Sheu, Tetrahedron Lett., 2006, 47, 8751–8755.
20 Y.-J. Tseng, Z.-H. Wen, C.-F. Dai, M. Y. Chiang and J.-H. Sheu, Org. Lett., 2009, 11, 5030–5032.
21 A. F. Ahmed, J.-H. Su, Y.-H. Kuo and J.-H. Sheu, J. Nat. Prod., 2004,
67, 2079–2082.
22 C.-H. Chao, Z.-H. Wen, I.-M. Chen, J.-H. Su, H.-C. Huang, M. Y. Chiang and J.-H Sheu, Tetrahedron, 2008, 64, 3554–3560.
23 Y. Lu, C.-Y. Huang, Y.-F. Lin, Z.-H. Wen, J.-H. Su, Y.-H. Kuo, M. Y. Chiang and J.-H. Sheu, J. Nat. Prod., 2008, 71, 1754– 1759.
24 C.-H. Chao, Z.-H. Wen, J.-H. Su, I.-M. Chen, H.-C. Huang, C.-F. Dai and J.-H. Sheu, Steroids, 2008, 73, 1353–1358.
25 I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092–4096.
26 I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Org. Chem., 1991, 56, 1296–1298.
27 M. C. Alley, D. A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589–601.
28 D. A. Scudiero, R. H. Shoemaker, K. D. Paull, A. Monks, S. Tierney, T. H. Nofziger, M. J. Currens, D. Seniff and M. R. Boyd, Cancer Res., 1988, 48, 4827–4833.
29 Y.-H. Jean, W.-F. Chen, C.-S. Sung, C.-Y. Duh, S.-Y. Huang, C.-S. Lin, M.-H. Tai, S.-F. Tzeng and Z.-H. Wen, Br. J. Pharmacol., 2009, 158, 713–725.
30 Y.-H. Jean, W.-F. Chen, C.-Y. Duh, S.-Y. Huang, C.-H. Hsu, C.-S. Lin, C.-S. Sung, I.-M. Chen and Z.-H. Wen, Eur. J. Pharmacol., 2008, 578, 323–331.
Downloaded by CHINA MEDICAL UNIVERSITY on 17 March 2011