The Fragment Transformation Method to Detect the Protein
Structural Motifs
Chih-Hao Lu,1Yeong-Shin Lin,2Yu-Ching Chen,1Chin-Sheng Yu,2Shi-Yu Chang,1and Jenn-Kang Hwang1,2,3* 1Institute of Bioinformatics, National Chiao Tung University, Hsinchu, Taiwan, Republic of China
2Department of Biological Science & Technology, National Chiao Tung University, Hsinchu, Taiwan, Republic of China 3Core Facility for Structural Bioinformatics, National Chiao Tung University, Hsinchu, Taiwan, Republic of China
ABSTRACT To identify functional structural motifs from protein structures of unknown function becomes increasingly important in recent years due to the progress of the structural genomics initia-tives. Although certain structural patterns such as the Asp-His-Ser catalytic triad are easy to detect because of their conserved residues and stringently constrained geometry, it is usually more challeng-ing to detect a general structural motifs like, for example, the␣-metal binding motif, which has a much more variable conformation and sequence. At present, the identification of these motifs usually relies on manual procedures based on different structure and sequence analysis tools. In this study, we develop a structural alignment algorithm com-bining both structural and sequence information to identify the local structure motifs. We applied our method to the following examples: the ␣-metal binding motif and the treble clef motif. The ␣-metal binding motif plays an important role in nonspecific DNA interactions and cleavage in host defense and apoptosis. The treble clef motif is a zinc-binding motif adaptable to diverse functions such as the binding of nucleic acid and hydrolysis of phosphodiester bonds. Our results are encouraging, indicating that we can effectively identify these structural motifs in an automatic fashion. Our method may provide a useful means for automatic functional annotation through detecting structural motifs associated with particular functions. Proteins 2006;63:636 – 643. ©2006 Wiley-Liss, Inc.
Key words: local structure alignment; DNA-binding proteins; structural motif; structural genomics
INTRODUCTION
Due to the progress of structural genomics research,1a
significant number of protein structures of unknown func-tion are solved in recent years—more than 700 structures annotated as “Structural Genomics Unknown Protein” are currently deposited in Protein Data Bank (PDB).2Hence,
the need of function prediction tools from protein tures becomes increasingly important in the present struc-tural genomic era. Sequence alignment and global fold comparison are valuable tools in inferring protein function through comparing sequence or fold similarity; however, these methods may not be applicable in certain situations:
because nonhomologous proteins with different folds may evolve convergently similar spatial dispositions of func-tional residues for similar functions, sequence or fold alignment may fail to identify the particular functional sites. It may also happen that nonhomologous proteins evolve similar overall conformations as a scaffold for different functions. The apparent fold similarity may lead to erroneous prediction of function. Hence, the ability to identify local structural motifs is important in predicting protein function and in providing a complementary tool to the fold alignment method.
The structural motif may be roughly divided into two types. The first type is characterized by a conserved spatial arrangement of a number of conserved residues that are functionally important. The distances among these resi-dues are usually quite conserved, showing little variations. Hence, the sequence and distance constraints provide stringent criteria to identify this type of motif. A typical example is given by the catalytic triad (Asp-His-Ser) of serine protease. Study3 showed that the majority of the
catalytic triads are within a root-mean-square (RMS) distance of 2.0 Å from the consensus template. Using this distance, that is, 2.0 Å, as a cutoff can effectively separate the catalytic triad from other noncatalytic Ser-His-Asp associations. Another example is given by the active site of enolase superfamily, which can be accurately character-ized by the spatial arrangement of five residues.4 A
number of methods5–16 were developed to identify this
type of structural motif, taking advantage of the distance constraints of the conserved residues.
The second type of structural motifs is composed of a number of secondary structure elements (SSEs) with each SSE arranged in a particular orientation. These SSEs are usually separated by variable lengths of intervening se-quences in different protein families. The motif sese-quences are usually much less conserved than those of the first type, and the RMS distances of the motif residues from the
Grant sponsor: National Science Council, National Research Pro-gram of Genomic Medicine. Grant sponsor: the University System at Taiwan-Veteran General Hospital Grant.
*Correspondence to: Jenn-Kang Hwang, Institute of Bioinformatics, National Chiao Tung University, Hsinchu 30050, Taiwan, ROC. E-mail: jkhwang@cc.nctu.edu.tw
Received 2 August 2005; Revised 14 November 2005; Accepted 28 November 2005
Published online 7 February 2006 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/prot.20904
PROTEINS: Structure, Function, and Bioinformatics 63:636 – 643 (2006)
consensus template are more widely distributed. Ex-amples are the ␣-metal binding motif,17–23 the treble
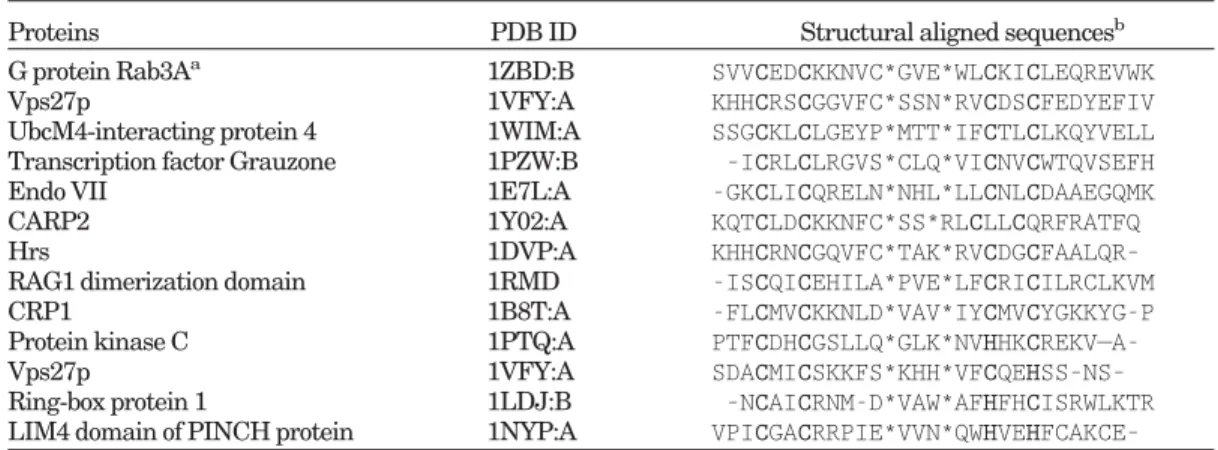
clef finger motif,24and the helix-turn-helix motif.25Figure
1 shows a typical ␣-metal binding motif. The motif sequences are highly variable—there is only one strictly conserved His residue, which serves as a general base to activate a water nucleophile.23 There do exist a special
case of the ␣-metal motif that can be identified by its particular sequence motif, the so-called the H-N-H mo-tif,26,27 characterized by the consensus sequence of
EXHHX14NX8HX3H;28 however, this instance does not
reflect the general cases. At present, the identification of the␣-metal motifs mainly relies on manual inspection assisted with a number of structure and sequence analysis tools.22Here, we present a structural alignment algorithm
combining both structural and sequence information to identify the local structure motifs. As a practical applica-tion, we applied our method to detecting the ␣-metal motif and the treble clef finger motif.
METHODS
In this section, we will first define some basic notations of our method. The symbol S denotes the query structural motif of length n, which may comprise several discontinu-ous peptide fragments. The C␣coordinates of S are given
by (x1,x2,…xn). The symbol T is used to denote the target
protein of length m with the C␣ coordinates given by (y1,y2,…ym). The basic unit of the structural alignment is
a triplet composed of three consecutive C␣ atoms. The structures S and T can be expressed in terms of these triplets, that is, S⫽ {1,2,…n⫺2} and T⫽ {1,2,…m⫺2},
where
i⫽ 共xi,xi⫹1,xi⫹2兲 and i⫽ 共yi,yi⫹1,yi⫹2兲.
We can construct an (m⫺ 2) ⫻ (n ⫺ 2) matrix between the and triplets, M⫽
冨
M11 M12 . . . M1,n⫺2 M21 M22 . . . M2,n⫺2 . . . . Mm⫺2,1 Mm⫺2,2 . . . Mm⫺2,n⫺2冨
, (1)where the element Mi,j is a rigid-body transformation
matrix form i to j, that is, Mi,ji ⫽ j (see Fig. 2 for
schematic illustration).
Triplet Clustering
We use the symbol Dklij to denote the Cartesian distance
between the transformed triplet Mi,jk and the target
triplet l. The distance Dklij is used as a measure of
orientation similarity between the triplet pairs (i,j) and
(k,l). We cluster the triplet fragments using the
single-linkage algorithm.29The procedures go as follows: for two
triplets (i,j) and (k,l) under the criteria: Dklij ⬍ D0, and i⫽ k and j ⫽ l, the triplets will be clustered in the same
group. Let G1and G2be two cluster groups, the former
containing (i,j) and (k,l), the latter containing (i⬘,j⬘).
If Dk⬘l⬘ ij
⬍ D0, then G1and G2will be merged to form a new
cluster G3⫽ G1艛 G2. The procedures will be carried out
iteratively until no more new cluster groups are formed. For the final cluster G, we can get the aligned substruc-ture pair: S⫽ 艛 k僆G k and T⫽ 艛 k僆G k.
Note that the substructure is not necessarily continuous in space. We define for each cluster Gthe alignment ratio⑀ as f/N, where f is the number of triplets of S, that is, the aligned atoms, and N is the number of the triplets of the query structure.
The Scoring Function
To assess the alignment quality of a given cluster G, we define the following scoring function
C⫽ w1RANKa共CR兲 ⫹ w2RANKd共CB兲 ⫹ w3RANKd共CS兲
(2) where RANKa(x) is a ranking function of x in the ascending
order, RANKd(x) is a ranking function in the descending
order, and wiis the weight of the associated rank score.
The weights wiof Equation (2) are positive and satisfies
Fig. 1. The ribbon representation of colicin ColE7 (PDB code: 7CEI). The␣-metal motif is rendered in colors according to the secondary structural elements (helices in red, -strands in yellow, and loops in green), while other parts of the protein in gray. The Zn metal ion is represented by a cyan sphere. The␣-metal motif of the colicin family (i. e., ColE2, ColE7, ColE8, and ColE9) contains the so-called conserved H-N-H motif, which is shown in a ball-and-stick model. However, only the first His residue of the sequence motif (i.e., on the first-strand) is strictly conserved in the general␣-metal motif of other proteins. All molecular images in this work are created by PyMOL (http://www.pymol.org).
the condition that⌺i 3w
i⫽ 1. The ␣-metal motifs
identi-fied by Li et al.22are used as the positive examples for the
manual adjustment of the parameters: the weight wi, and
the clustering parameters D0and⑀. The final values of
these parameters are: (w1,w2,w3)⫽ (0.4,0.2,0.4), D0⫽ 3 Å
and⑀⬎ 0.75. The same parameters are used throughout all calculations in this work. The alignment scores CR, C
B,
and CSare defined as follows,
CR⫽ 1
εRMSD共S,T兲 (3)
where RMSD(S,T) is the root-mean-square deviation of
C␣atoms between S and T;
CB⫽
1
ε共BLOSUM共S,T兲 ⫹ d0
B兲 (4)
where BLOSUM(S,T) is the sequence alignment score based on BLOSUM6230 between S and T
and d0 B is a
constant to make CBnegative;
CS⫽ 1
ε共SS共S,T兲 ⫹ d0
s兲 (5)
where SS(S,T) is the secondary structure alignment score between S and T and d0
S is a constant to make C S
negative. The secondary structure assignment is based on the DSSP method,31which assigns secondary structures
according to their hydrogen bonding patterns. The DSSP method defines eight secondary structures: ␣-helix (H), 310-helix (G),-helix (I), extended -strand (E), isolated
-strand (B), turn (T), bend (S), and coil (U). The substitu-tion matrix is given in Table I.
Fig. 2. iandkare two arbitrary triplet units of the query structural motif S, andiandlare two arbitrary
triplet units of the target protein T. The tripletiis transformed tojthrough the transformation matrix Mi,j.
TABLE I. Substitution Matrix of Secondary Structural Elements H G I E B T S U H 5 3 3 ⫺5 ⫺5 0 0 0 G 3 5 3 ⫺5 ⫺5 0 0 0 I 3 3 5 ⫺5 ⫺5 0 0 0 E ⫺5 ⫺5 ⫺5 5 3 0 0 0 B ⫺5 ⫺5 ⫺5 3 5 0 0 0 T 0 0 0 0 0 3 0 0 S 0 0 0 0 0 0 3 0 U 0 0 0 0 0 0 0 1
TABLE II. The Top Ranking Hits of the␣-Metal Motif Proteins PDB ID Structural aligned sequencesb Colicin E7a 7CEI:B SFELHHE.(13).NISVV-TPKRHIDI Colicin E9 1EMV:B VYELHHD.(13).NISVV-TPKRHIDI Vnn 1OUO:A RIEWEHV.(36).NLTPA-IGEVNGDR I-HmuI 1U3E:M GLVVDHK.(10).NLRWV-TQKINVEN I-PpoI 1EVW:A TCTASHL.(10).HLCWE-SADDNKGR Serratia 1Q10:A KVDRGHQ.( 9).NITPQ-KSDLNGAW EndoVII 1E71:A ANHLDHD.(10).VRGLL-CNLCDAAE CAD/DFF40 1V0D:A TWNLDHI.(34).NLKLV-HIACHKKT HIV integrase 1K6Y:A QLDCTHL.( 2).KVILV*IESMNKEL
Only the representative proteins of each protein family are listed.
aThe ␣-metal motif of this protein is used as the structural
template.
bIn the alignment, The symbol “.(n).” indicates a sequence of length, n,
“-” a gap and “*” a sequence of unspecified length. The letter in bold face indicates the strictly conserved residue the His residue, while the letter underlined the less conserved His, Asn or Glu residue.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
␣-metal motifs (using ColE9 (1EMV:B) as the reference) in Vvn (1OUO:A), I-HmuI (1U3E:M), I-PpoI (1EVW), Serratia nuclease (1QL0), the T4 Endo VII (1E7L:A), CAD/DFF40 (1V0D), and HIV IN (1EX4:A). The conserved His residue, the ligand and the binding residue (His, Asn or Asp), which is the underlined residue in Table II, are shown in the ball-and-stick model.
Fig. 4. The identified treble clef finger motifs [using G protein Rab3A (1ZBD:B) as the reference in Vps27p (1VFY:A(1)], UbcM4-interacting protein 4 (1WIM:A), transcription factor Grauzone (1PZW:B), the T4 Endo VII (1E7L:A), CARP2 (1Y02:A), Hrs (1DVP:A), RAG1 dimerization domain (1RMD), CRP1 (1B8T:A), Protein kinase C (1PTQ:A), Vps27p (1VFY:A(2), ring-box protein 1 (1LDJ:B), and LIM4 (1NYP:A).
RESULTS AND DISCUSSION The␣-Metal Motif
The␣-metal motif has recently received much attention due to its important role in nonspecific DNA interactions and cleavage in host defense and apoptosis.17–22,27,32,23 The
␣-metal binding motif is characterized by one ␣-helix and an antiparallel -sheet, which are separated from each other by a variable length of intervening sequences from 9 to 36. This motif usually binds to divalent metal ions like Zn or Mg. The lengths and sequences of the ␣-metal motif vary greatly in different proteins. For example, there is only one strictly conserved His residue located at the first-strand in the motif sequence. We used the␣-metal motif of the nuclease domain of colicin E7 (PDB ID: 7CEI)33as the query motif, which is composed of
three fragments: E542-H545 (-strand), I561–V564(-strand) and P566 –D571 (␣-helix). The query fragments are compared against the nonredundant PDB (nrpdb) chain set composed of nonidentical proteins from NCBI (http://www.ncbi.nlm.nih.gov/Structure/VAST/nrpdb.html). Table II lists the top ranking hits of proteins and the corresponding structure-aligned sequences. We list only the representative proteins of the protein families, which are colicin E9 (ColE9),34the periplasmic nuclease Vvn,22
the HNH homing endonuclease I-HmuI,35the His–Cys box
endonuclease I-PpoI,18the endonucleases such as Serratia
nuclease,17 CAD/DFF40,32the T4 Endo VII,20 and HIV
integrase (IN).36The lengths of the intervening sequence
between the two-strands vary from 2 to 39. The struc-tures of the␣-metal binding motifs of these proteins are shown in Figure 3. We have identified all proteins that have experimentally confirmed␣-metal binding motifs. The only exception is HIV IN, which has not been reported to have a structural motif similar to the␣-metal binding motif. The identified structural motif is located in the catalytic core domain of HIV IN.36 Two of the three
catalytic residues36of HIV IN, that is, D64 and E152, are
located in this motif, while the third one (D116) is in the vicinity of the motif. Although we did not find a divalent metal ion in the center of the motif in the crystal structures of HIV IN, we did find a phosphate group (PO4⫺) in 1K6Y,
which is close to the supposed metal-binding site. How-ever, conspicuous differences do exist: in HIV IN, the -strands of the structural motif are separated by only two residues, while the usual intervening length is much longer in the range between 9 –36 (Table II and Fig. 3); furthermore, the second-strand does not directly connect to the␣-helix like others do. Obviously, the question of whether HIV IN did contain a authenticate ␣-metal motif needs further study.
The Treble Clef Finger Motifs
The treble clef finger motif24is a zinc-binding structural
motif composed of a zinc knuckle, a loop, a-hairpin and an␣-helix. It is so named because of its projection of the domain C␣trace resemble the treble clef sign.24The treble
clef finger structures show highly variable sequences and appear in many folds. For example, in terms of the SCOP protein folds, the treble clef finger motif occurs in a number of folds such as the RING/U-box, glucocorticoid receptor-like folds, FYVE/PHD zinc fingers, His-Me finger endonucleases, C2H2 and C2HC zinc fingers, and cysteine-rich domains. It has been noted37 that fold similarity
search programs such as DALI,38and CE,39and VAST40
are not consistent in detecting the treble clef finger motif due to its relatively smaller size. Previously, a combination of fold alignment methods, together with the PSI-BLAST and the manual procedures, was used to identify the treble clef finger motifs.24We used the zinc finger motif of the G
protein Rab3A (PDB ID: 1ZDB:B)33as the query motif,
which is composed of three fragments: S108 –C119, G123– E125, and W135–K148. The top ranking hits are shown in Table III, and the corresponding structures of the zinc fingers are shown in Figure 4. Both sequence and
struc-TABLE III. Top Ranking Hits of the Treble Clef Finger Motif
Proteins PDB ID Structural aligned sequencesb
G protein Rab3Aa 1ZBD:B SVVCEDCKKNVC*GVE*WLCKICLEQREVWK
Vps27p 1VFY:A KHHCRSCGGVFC*SSN*RVCDSCFEDYEFIV
UbcM4-interacting protein 4 1WIM:A SSGCKLCLGEYP*MTT*IFCTLCLKQYVELL Transcription factor Grauzone 1PZW:B –ICRLCLRGVS*CLQ*VICNVCWTQVSEFH
Endo VII 1E7L:A -GKCLICQRELN*NHL*LLCNLCDAAEGQMK
CARP2 1Y02:A KQTCLDCKKNFC*SS*RLCLLCQRFRATFQ
Hrs 1DVP:A KHHCRNCGQVFC*TAK*RVCDGCFAALQR–
RAG1 dimerization domain 1RMD -ISCQICEHILA*PVE*LFCRICILRCLKVM
CRP1 1B8T:A -FLCMVCKKNLD*VAV*IYCMVCYGKKYG-P
Protein kinase C 1PTQ:A
PTFCDHCGSLLQ*GLK*NVHHKCREKV—A-Vps27p 1VFY:A SDACMICSKKFS*KHH*VFCQEHSS–NS–
Ring-box protein 1 1LDJ:B –NCAICRNM-D*VAW*AFHFHCISRWLKTR LIM4 domain of PINCH protein 1NYP:A VPICGACRRPIE*VVN*QWHVEHFCAKCE–
Only the representative proteins are listed. In addition, we list the proteins (the bottom 4), although not among the highest ranks, that contain motifs other than the C2C2 sequence motif of the query proteins, that is, C2HC, C2CH, and C2H2. The residue in bold indicates the conserved residues of the motif.
a
The treble clef finger motif of this protein is used as the structural template.
bIn the alignment, The symbol “.(n).” indicates a sequence of length n, “-” a gap and “*” a sequence of unspecified
length.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
Fig. 5. The ribbon drawing of the two overlapping zinc fingers in the FYVE domain of Hrs (1DVP:A). The finger motifs are colored in violet. Shown in the bottom are the sequences of the first zinc finger (G161–C196, which are underlined in the upper sequence) and second zinc finger (K176 –R219, which are underlined in the lower sequence). The zinc ligands are shown in bold face.
Fig. 6. The ribbon drawing of the consecutive zinc fingers in the tandem LIM domains of LMO4 (1RUT:X). The finger motifs are colored in violet. Shown in the bottom are the motif sequences of the first zinc finger (the sequence underlined in the upper row—W20-L57) and the second zinc finger (the sequence underlined in the upper row—L48 –A96). The zinc ligands are shown in bold face. Note that LMO4 has an unusual CCCD Zn-binding module.
tures of the treble clef zinc fingers show significant varia-tions; for example, the motif sequences show little similar-ity except the sequence motif like C2C2, C2HC, C2CH, C2H2, or C2CD (see next section) involved in zinc binding.
The Double Treble Clef Finger Motif
The treble clef finger motif in many cases occurs as an overlapping treble-clef structure,24 where the two zinc
binding fingers, each finger showing a typical treble clef motif, overlap each other over a significant fraction of the whole motif sequence. In the overlapping zinc binding module, the first and the third Cys pairs bind to one zinc ion, while the second and the forth Cys pairs bind to the other zinc ion. The overlapping treble-clef structure is typical of the FYVE domains.41Our method successfully
identified this structural motif in the FYVE domain of the proteins such as endosomal autoantigen 1 (1JOC:B), the Vps27p protein (1VFY:A), Hrs (1DVP:A), and the protein 19 from Mus musculus (1WFK). As an example, Figure 5 shows the detected overlapping treble-clef structure in the FYVE domain of Hrs.42Note that, although the two zinc
fingers overlap over more than two-thirds of the motif sequence, our method is still able to identify these two overlapping fingers.
The treble-clef fingers also occurs as the so-called double treble-clef structure24where two fingers are arranged in a
consecutive, instead of overlapping, fashion. In contrast to the previous overlapping treble-clef structure, the first two pair of the ligands bind to one zinc ion, while the last two pairs bind to a second zinc ion. The double treble-clef structure is typical of LIM domains.43Our method detects
an interesting case of such motif in the tandem LIM domains of the LMO4 protein shown in Figure 6. Note that LIMO4 has an unusual CCCD Zn-binding sequence motif.
Comparison with Other Approaches
Currently, there are three popular methods for struc-ture alignment available on the Internet: DALI, CE, and VAST. However, because DALI and CE are mainly de-signed for the structural alignment of global folds, they failed to return results for both the␣-metal and the zinc finger motifs, which are composed of relatively short and discontinuous fragments. On the other hand, the VAST algorithm, using a hierarchical alignment based on the SSEs, is able to identify local structural motifs. In the case of␣-metal motifs, the VAST tool, using the same query fragment defined in the previous section against the medium-redundancy subset of PDB from NCBI, correctly identifies 4 out of the top 10 ranks (40%) based on the VAST score, while our approach yields 7 of 10 (70%). In the case of the zinc finger motifs, VAST correctly identify 5 out the top 10 ranks (50%), while our approach is able to yield 9 of 10 (90%). It should be noted that our program can be easily extended to identify the type 1 structural motifs such as the catalytic triads, which can be identified through stringent distance constraints. Using the data set comprising 88 serine hydrolase entries in enzyme class E.C. 3.4.21 from Chou and Cai,44our approach can achieve
a overall success rate of 6187/6189 (99.92%) for the active
site identification, which is comparable to that of the covariant discriminant approach designed for the identifi-cation of the catalytic triad.
In summary, we have developed an approach to identify the local structural motifs, which may be useful in the automatic functional annotation for structural genomics research. The Linux binary codes of our method are available upon request.
ACKNOWLEDGMENTS
We are grateful to both the hardware and software supports of the Structural Bioinformatics Core Facility at National Chiao Tung University. We are also grateful to Dr. Hanna Yuan for helpful discussion on the␣-metal binding motif, and to the anonymous reviewer for the valuable suggestions.
REFERENCES
1. Brenner SE. A tour of structural genomics. Nat Rev Genet 2001;2:801– 809.
2. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res 2000;28:235–242.
3. Wallace AC, Laskowski RA, Thornton JM. Derivation of 3D coordinate templates for searching structural databases: applica-tion to Ser-His-Asp catalytic triads in the serine proteinases and lipases. Protein Sci 1996;5:1001–1013.
4. Meng EC, Polacco BJ, Babbitt PC. Superfamily active site tem-plates. Proteins 2004;55:962–976.
5. Artymiuk PJ, Poirrette AR, Grindley HM, Rice DW, Willett P. A graph-theoretic approach to the identification of three-dimen-sional patterns of amino acid side-chains in protein structures. J Mol Biol 1994;243:327–344.
6. Wallace AC, Borkakoti N, Thornton JM. TESS: a geometric hashing algorithm for deriving 3D coordinate templates for search-ing structural databases. Application to enzyme active sites. Protein Sci 1997;6:2308 –2323.
7. Pennec X, Ayache N. A geometric algorithm to find small but highly similar 3D substructures in proteins. Bioinformatics 1998; 14:516 –522.
8. Russell RB. Detection of protein three-dimensional side-chain patterns: new examples of convergent evolution. J Mol Biol 1998;279:1211–1227.
9. Kleywegt GJ. Recognition of spatial motifs in protein structures. J Mol Biol 1999;285:1887–1897.
10. Barker JA, Thornton JM. An algorithm for constraint-based structural template matching: application to 3D templates with statistical analysis. Bioinformatics 2003;19:1644 –1649.
11. Jambon M, Imberty A, Deleage G, Geourjon C. A new bioinfor-matic approach to detect common 3D sites in protein structures. Proteins 2003;52:137–145.
12. Tendulkar AV, Wangikar PP, Sohoni MA, Samant VV, Mone CY. Parameterization and classification of the protein universe via geometric techniques. J Mol Biol 2003;334:157–172.
13. Brakoulias A, Jackson RM. Towards a structural classification of phosphate binding sites in protein–nucleotide complexes: an automated all-against-all structural comparison using geometric matching. Proteins 2004;56:250 –260.
14. Ivanisenko VA, Pintus SS, Grigorovich DA, Kolchanov NA. PDB-SiteScan: a program for searching for active, binding and posttrans-lational modification sites in the 3D structures of proteins. Nucleic Acids Res 2004;32:W549 –W554.
15. Weskamp N, Kuhn D, Hullermeier E, Klebe G. Efficient similarity search in protein structure databases by k-clique hashing. Bioin-formatics 2004;20:1522–1526.
16. Torrance JW, Bartlett GJ, Porter CT, Thornton JM. Using a library of structural templates to recognise catalytic sites and explore their evolution in homologous families. J Mol Biol 2005;347: 565–581.
17. Miller MD, Tanner J, Alpaugh M, Benedik MJ, Krause KL. 2.1 A structure of Serratia endonuclease suggests a mechanism for binding to double-stranded DNA. Nat Struct Biol 1994;1:461– 468.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
18. Flick KE, Jurica MS, Monnat RJ Jr, Stoddard BL. DNA binding and cleavage by the nuclear intron-encoded homing endonuclease I-PpoI. Nature 1998;394:96 –101.
19. Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS. The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Struct Fold Des 1999;7:91–102.
20. Raaijmakers H, Vix O, Toro I, Golz S, Kemper B, Suck D. X-ray structure of T4 endonuclease VII: a DNA junction resolvase with a novel fold and unusual domain-swapped dimer architecture. EMBO J 1999;18:1447–1458.
21. Cheng YS, Hsia KC, Doudeva LG, Chak KF, Yuan HS. The crystal structure of the nuclease domain of colicin E7 suggests a mecha-nism for binding to double-stranded DNA by the H-N-H endonucle-ases. J Mol Biol 2002;324:227–236.
22. Li CL, Hor LI, Chang ZF, Tsai LC, Yang WZ, Yuan HS. DNA binding and cleavage by the periplasmic nuclease Vvn: a novel structure with a known active site. EMBO J 2003;22:4014 – 4025. 23. Hsia KC, Li CL, Yuan HS. Structural and functional insight into sugar-nonspecific nucleases in host defense. Curr Opin Struct Biol 2005;15:126 –134.
24. Grishin NV. Treble clef finger—a functionally diverse zinc-binding structural motif. Nucleic Acids Res 2001;29:1703–1714. 25. Shanahan HP, Garcia MA, Jones S, Thornton JM. Identifying
DNA-binding proteins using structural motifs and the electro-static potential. Nucleic Acids Res 2004;32:4732– 4741.
26. Kuhlmann UC, Moore GR, James R, Kleanthous C, Hemmings AM. Structural parsimony in endonuclease active sites: should the number of homing endonuclease families be redefined? FEBS Lett 1999;463:1–2.
27. Hsia KC, Chak KF, Liang PH, Cheng YS, Ku WY, Yuan HS. DNA binding and degradation by the HNH protein ColE7. Structure (Camb) 2004;12:205–214.
28. Sui MJ, Tsai LC, Hsia KC, Doudeva LG, Ku WY, Han GW, Yuan HS. Metal ions and phosphate binding in the H-N-H motif: crystal structures of the nuclease domain of ColE7/Im7 in complex with a phosphate ion and different divalent metal ions. Protein Sci 2002;11:2947–2957.
29. Gower JC, Ross GJS. Minimum spanning trees and single-linkage cluster analysis. Appl Stat 1969;18:54 – 64.
30. Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA 1992;89:10915–10919. 31. Kabsch W, Sander C. Dictionary of protein secondary structure:
pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983;22:2577–2637.
32. Woo EJ, Kim YG, Kim MS, Han WD, Shin S, Robinson H, Park SY, Oh BH. Structural mechanism for inactivation and activation of CAD/DFF40 in the apoptotic pathway. Mol Cell 2004;14:531–539. 33. Ku WY, Liu YW, Hsu YC, Liao CC, Liang PH, Yuan HS, Chak KF. The zinc ion in the HNH motif of the endonuclease domain of colicin E7 is not required for DNA binding but is essential for DNA hydrolysis. Nucleic Acids Res 2002;30:1670 –1678.
34. Kuhlmann UC, Pommer AJ, Moore GR, James R, Kleanthous C. Specificity in protein–protein interactions: the structural basis for dual recognition in endonuclease colicin–immunity protein com-plexes. J Mol Biol 2000;301:1163–1178.
35. Shen BW, Landthaler M, Shub DA, Stoddard BL. DNA binding and cleavage by the HNH homing endonuclease I-HmuI. J Mol Biol 2004;342:43–56.
36. Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, Stroud RM. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc Natl Acad Sci USA 2000;97:8233– 8238.
37. Grishin NV. C-terminal domains of Escherichia coli topoisomer-ase I belong to the zinc-ribbon superfamily. J Mol Biol 2000;299: 1165–1177.
38. Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci 1995;20:478 – 480.
39. Shindyalov IN, Bourne PE. Protein structure alignment by incre-mental combinatorial extension (CE) of the optimal path. Protein Eng 1998;11:739 –747.
40. Madej T, Gibrat JF, Bryant SH. Threading a database of protein cores. Proteins 1995;23:356 –369.
41. Misra S, Hurley JH. Crystal structure of a phosphatidylinositol 3-phosphate-specific membrane-targeting motif, the FYVE do-main of Vps27p. Cell 1999;97:657– 666.
42. Mao Y, Nickitenko A, Duan X, Lloyd TE, Wu MN, Bellen H, Quiocho FA. Crystal structure of the VHS and FYVE tandem domains of Hrs, a protein involved in membrane trafficking and signal transduction. Cell 2000;100:447– 456.
43. Deane JE, Ryan DP, Sunde M, Maher MJ, Guss JM, Visvader JE, Matthews JM. Tandem LIM domains provide synergistic binding in the LMO4:Ldb1 complex. EMBO J 2004;23:3589 –3598. 44. Chou KC, Cai YD. A novel approach to predict active sites of
enzyme molecules. Proteins 2004;55:77– 82. THE FRAGMENT TRANSFORMATION METHOD

![Fig. 4. The identified treble clef finger motifs [using G protein Rab3A (1ZBD:B) as the reference in Vps27p (1VFY:A(1)], UbcM4-interacting protein 4 (1WIM:A), transcription factor Grauzone (1PZW:B), the T4 Endo VII (1E7L:A), CARP2 (1Y02:A), Hrs (1DVP:A), RAG](https://thumb-ap.123doks.com/thumbv2/9libinfo/7631066.135090/4.891.180.714.70.540/identified-finger-protein-reference-interacting-protein-transcription-grauzone.webp)