Yaw-Kuen Li and Chun-Jung Chen
Phimonphan Chuankhayan, Wen-guey Wu,
Shrestha, Cheng-Fu Chao, Yen-Chieh Huang,
Chiang, Chueh-Yuan Kuo, Keshab Lal
Yin-Cheng Hsieh, Yue-Jin Wu, Tzu-Ying
INSERTION DOMAINS
WITHOUT CHITIN BINDING AND
Binding for Catalysis: A CHITINASE
Chitooligomers Reveal Novel Substrate
NCTU2 Chitinase Complexes with
Bacillus cereus
Crystal Structures of
doi: 10.1074/jbc.M110.149310 originally published online August 4, 2010 2010, 285:31603-31615.
J. Biol. Chem.
10.1074/jbc.M110.149310 Access the most updated version of this article at doi:
. JBC Affinity Sites Find articles, minireviews, Reflections and Classics on similar topics on the
Alerts:
When a correction for this article is posted •
When this article is cited •
to choose from all of JBC's e-mail alerts Click here
Supplemental material:
http://www.jbc.org/content/suppl/2010/08/04/M110.149310.DC1.html http://www.jbc.org/content/285/41/31603.full.html#ref-list-1This article cites 59 references, 15 of which can be accessed free at
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
Downloaded from
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
Crystal Structures of Bacillus cereus NCTU2 Chitinase
Complexes with Chitooligomers Reveal Novel Substrate
Binding for Catalysis
A CHITINASE WITHOUT CHITIN BINDING AND INSERTION DOMAINS
*□SReceived for publication, June 3, 2010, and in revised form, July 27, 2010Published, JBC Papers in Press, August 4, 2010, DOI 10.1074/jbc.M110.149310
Yin-Cheng Hsieh‡§1, Yue-Jin Wu¶1, Tzu-Ying Chiang‡储, Chueh-Yuan Kuo‡§, Keshab Lal Shrestha¶, Cheng-Fu Chao¶, Yen-Chieh Huang‡, Phimonphan Chuankhayan‡, Wen-guey Wu§, Yaw-Kuen Li¶2, and Chun-Jung Chen‡储**3
From the‡Life Science Group, Scientific Research Division, National Synchrotron Radiation Research Center, Hsinchu 30076, Taiwan, the¶Department of Applied Chemistry, National Chiao Tung University, Hsinchu 30010, Taiwan, the§Institute of Bioinformatics and Structural Biology,储Department of Physics, National Tsing Hua University, Hsinchu 30043, Taiwan, and the **Institute of Biotechnology, National Cheng Kung University, 1 University Road, Tainan City 701, Taiwan
Chitinases hydrolyze chitin, an insoluble linear polymer of
N-acetyl-D-glucosamine (NAG)n, into nutrient sources. Bacillus
cereus NCTU2 chitinase (ChiNCTU2) predominantly produces
chitobioses and belongs to glycoside hydrolase family 18. The crystal structure of wild-type ChiNCTU2 comprises only a cat-alytic domain, unlike other chitinases that are equipped with additional chitin binding and insertion domains to bind sub-strates into the active site. Lacking chitin binding and chitin insertion domains, ChiNCTU2 utilizes two dynamic loops (Gly-67—Thr-69 and Ile-106 –Val-112) to interact with (NAG)n,
gen-erating novel substrate binding and distortion for catalysis. Gln-109 is crucial for direct binding with substrates, leading to conformational changes of two loops with a maximum shift of ⬃4.6 A˚ along the binding cleft. The structures of E145Q, E145Q/ Y227F, and E145G/Y227F mutants complexed with (NAG)n
reveal (NAG)2, (NAG)2, and (NAG)4in the active site,
respec-tively, implying various stages of reaction: before hydrolysis, E145G/Y227F with (NAG)4; in an intermediate state, E145Q/
Y227F with a boat-form NAG at theⴚ1 subsite, ⴚ1-(NAG); after hydrolysis, E145Q with a chair formⴚ1-(NAG). Several residues were confirmed to play catalytic roles: Glu-145 in cleavage of the glycosidic bond betweenⴚ1-(NAG) and ⴙ1-(NAG); Tyr-227 in the conformational change ofⴚ1-(NAG); Asp-143 and Gln-225 in stabilizing the conformation ofⴚ1-(NAG). Additionally, Glu-190 acts in the process of product release, and Tyr-193 coordi-nates with water for catalysis. Residues Asp-143, E145Q, Glu-190, and Tyr-193 exhibit multiple conformations for functions.
The inhibitors zinc ions and cyclo-(L-His-L-Pro) are located at
various positions and confirm the catalytic-site topology. Together with kinetics analyses of related mutants, the struc-tures of ChiNCTU2 and its mutant complexes with (NAG)n
pro-vide new insights into its substrate binding and the mechanistic action.
Chitin, derived primarily from exoskeletons of insects and crustaceans, is a -1,4-linked insoluble linear polymer of
N-acetyl-D-glucosamine (NAG)n.
4 It is not only the second most abundant organic compound next to cellulose on the planet but also the major source of amino sugars in nature (1). Chitin can be hydrolyzed by chitinases (EC 3.2.1.14) (2); the hydrolyzed products serve as sources of nutrients. Chitinases are found in diverse organisms, including bacteria, fungi, and higher plants, and play varied roles in their origins (2– 4). Plant chitinases are involved in a mechanism to defend against infec-tion by phytopathogenic fungi (4); fungal chitinases are required for hyphal growth (5); bacterial chitinases are consid-ered primarily to digest and utilize chitin as a carbon and nitro-gen nutrient (1).
Chitinases of glycoside hydrolase family 18 (GH-18) exhibit three distinct structural features (6, 7) upon the commonly existing catalytic domain (CaD) that generally comprise a (␣8/ 8) TIM barrel-fold with a conserved glutamate residue that presumably acts as an acid during catalysis. One structural fea-ture, such as that of chitinase B1 from Aspergillus fumigatus (8), contains an extra chitin insertion domain (CID) (9) that com-prises⬃70–90 amino acids and inserts into the TIM-barrel of CaD. The CID provides residues interacting with a substrate and increases the depth of the substrate binding cleft. Another feature, such as that of chitinase A from Serratia marcescens (10), contains a CID and an additional N- or C-terminal chitin binding domain (CBD) with several aligned aromatic residues to increase the specific activity of hydrolysis (11). With the *This work was supported in part by National Science Council (NSC) Grants
95-2923-B-213-001-MY3 and 95-2313-B-009-001-MY3, National Synchro-tron Radiation Center Grants 973RSB02 and 983RSB02 (to C.-J. C.), and the MOE-AU Program and NSC Grant 96-2113-M-009-012-MY3 (to Y.-K. L.). □S The on-line version of this article (available at http://www.jbc.org) contains
supplemental Figs. 1–3.
The atomic coordinates and structure factors (codes3N11,3N12,3N13,3N15, 3N17,3N18, and3N1A) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
1Both authors contributed equally to this work.
2To whom correspondence may be addressed. Tel.: 886-3-573-1985; Fax:
886-3-572-3764; E-mail: ykl@cc.nctu.edu.tw.
3To whom correspondence may be addressed: Life Science Group, Scientific
Research Division, National Synchrotron Radiation Research Center, Hsin-chu 30076, Taiwan. Tel.: 886-3-578-0281 (ext. 7330); Fax: 886-3-578-3813; E-mail: cjchen@nsrrc.org.tw.
4The abbreviations used are: NAG
n, N-acetyl-D-glucosamine; ChiNCTU2, B. cereus NCTU2 chitinase; GH, glycoside hydrolase; CID, chitin insertion
domain; CaD, catalytic domain; CBD, chitin binding domain; MAD, multiple wavelength anomalous diffraction.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
additional domains of CID or CBD, the enzymes commonly function as exochitinases. The other feature comprises only a CaD without the CBD and CID and generally forms a shallow substrate binding cleft to hydrolyze substrates randomly. Enzymes of this kind are typically endochitinases, such as hevamine from Hevea brasiliensis (12, 13).
Bacillus cereusis a large, Gram-positive, endospore-forming bacterium that exists commonly in soils and plants (14 –17). A chitin-degrading Bacillus strain, designated NCTU2, was screened and identified from soils. An extracellular chitinase, designed ChiNCTU2, from this strain was purified from the culture filtrate. The full gene of ChiNCTU2 from the genomic DNA encodes a signal peptide (27 amino acids) and a mature protein (333 amino acids) (18). The multiple-sequence align-ment of GH-18 enzymes shows the conservation of the (D/N)G(L/I/V/M/F)(D/N)(L/I/V/M/F)(D/N)XE motif (resi-dues 138 –145 in ChiNCTU2) and the tyrosine residue (Tyr-227 in ChiNCTU2) (19), indicating that Glu-145 and Tyr-(Tyr-227 are key residues for mediating catalysis of ChiNCTU2. Lacking the CBD and CID in the sequence, ChiNCTU2 is supposed to comprise the third structural feature of GH-18 as mentioned above (6, 20). The enzyme can hydrolyze colloidal chitin, chi-tohexaose, and chitotetraose into the predominant product, the chitobiose (18).
As little information exists about the three-dimensional structure of a chitinase without the CBD and CID, we have determined the structure of ChiNCTU2 at sufficient resolution to elucidate its substrate binding for the catalytic mechanism. We introduced several point mutations that allowed us to obtain crystal structures of ChiNCTU2 mutants E145Q, E145Q/Y227F, and E145G/Y227F complex with several short-form NAG substrates. The structures of the wild-type enzyme and mutant complexes together with activity assays of related mutants might provide insight into novel substrate binding for catalysis of ChiNCTU2.
EXPERIMENTAL PROCEDURES
Materials—All chemicals were purchased from Sigma unless specified otherwise.
Protein Preparation and Site-directed Mutagenesis—The protein purification and preliminary crystallographic analy-sis of wild-type ChiNCTU2 were described previously (18, 21). Site-directed mutagenesis for mutations was performed according to the QuikChange method (Stratagene). The syn-thetic oligonucleotides used for mutagenesis were listed in Table 1. The basic procedure involved PCR amplification with ChiNCTU2/pET-22b(⫹) as the template and two synthetic oli-gonucleotides containing the desired mutation as the primers. The DNA sequencing of full genes confirmed the desired muta-tions. Purification steps for mutants are similar to that for the wild-type enzyme.
Enzymatic Activity Assay—Chitinase activity was analyzed by estimating the reducing ends of sugars. The assay was per-formed on mixing the enzyme with colloidal chitin (1%) in phosphate buffer (20 mM, pH 7.0, 0.3 ml). After individual incu-bations for 10, 20, and 30 min at 37 °C, hydrolysis was termi-nated and analyzed on the addition of dinitrosalicylic acid (0.6 ml). The mixture was boiled for 15 min, chilled, and centrifuged
to remove insoluble chitin. The resulting adduct of reducing sugars was measured spectrophotometrically at 540 nm. The extinction coefficient of the resulting adduct (at 540 nm) was determined to be 788M⫺1䡠cm⫺1when N-acetyl-D-glucosamine was the reducing sugar. One unit of chitinase activity is defined as the amount of enzyme required to release 1mol of detect-able reducing sugars in 1 min at 37 °C. Tdetect-able 2 shows the data of wild-type ChiNCTU2 and mutants.
Kinetic Study—The enzymatic activity was assayed by the increase of the optical density at 400 nm at 25 °C in reaction mixtures containing the pure enzyme at a constant concentra-tion in MES buffer (50 mM, pH 6.5) and increasing concentra-tions of 4-nitrophenyl--chitobioside from 1.0 Mto 2.5 mM. The initial rate of hydrolysis was determined for each assay, and
Kmand kcatwere calculated from an average of three measure-ments according to the Michaelis-Menten equation. Table 2 shows the data of wild-type and mutant enzymes.
Crystallization of Wild-type ChiNCTU2, Mutant-(NAG)n
Complexes—All crystallizations were achieved with the hang-ing-drop vapor diffusion method in a 24-well ADX plate (Hampton Research) at 291 K (21). 1-l drops of protein (⬃10 mg ml⫺1) in phosphate buffer (20 mM, pH 7.0) were mixed with TABLE 1
Primers used for site-directed mutagenesis
The mutated bases resulting in changes of amino acids are italic and underlined.
Mutants Primers Q109G Forward 5⬘-CTTTCAATAGGTGGAGGGAATGGAGTCGTTTTA-3⬘ Reverse 5⬘-TAAAACGACTCCATTCCCTCCACCTATTGAAAG-3⬘ D143A Forward 5⬘-GATGGAATAGATATTGCCCTTGAATCAGGTATT-3⬘ Reverse 5⬘-AATACCTGATTCAAGGGCAATATCTATTCCATC-3⬘ E145Q Forward 5⬘-GATGGAATAGATATCGACCTTCAGTCAGGTA-3⬘ Reverse 5⬘-TACCTGACTGAAGGTCGATATCTATTCCATC-3⬘ E145G Forward 5⬘-ATAGATATTGACCTTGGATCAGGTATTTAC-3⬘ Reverse 5⬘-GTAATACCTGATCCAAGGTCAATATCTAT-3⬘ E190Q Forward 5⬘-ATTAAGCATGGCTCCTCAAACAGCTTATGT-3⬘ Reverse 5⬘-ACATAAGCTGTTTGAGGAGCCATGCTTAAT-3⬘ Q225G Forward 5⬘-CATTCATGTTGGACACTACAACGCTGGTAG-3⬘ Reverse 5⬘-CTACCAGCGTTGTAGTGTCCAACATGAATG-3⬘ Y227F Forward 5⬘-TGTTCAACACTTCAACGCTGGTAGC-3⬘ Reverse 5⬘-GCTACCAGCGTTGAAGTGTTGAACA-3⬘ TABLE 2
Kinetic parameters of wild type and mutants of ChiNCTU2
Kinetic assays were performed with 4-nitrophenyl--chitobioside as described under “Experimental Procedures.” Results and values were confirmed and averaged from three independent experiments. Relative activity of wild type chitinase and mutants were measured at pH 6.0. ND, not detectable.
Protein type Km kcat Relative kcat/Km Relative activity M s⫺1 % Wild type 74 20.94 1.000 100 Q109G ND D143A ND E145G ND E145Q 70 0.04 0.002 0.2 E190Q 112 10.01 0.316 31.6 Q225G 69 0.17 0.008 0.8 Y227F 113 2.37 0.073 7.3
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
1-l drops of the reservoir solution and equilibrated against reservoir solution (0.5 ml). The zinc binding crystals used to determine phases were grown in the zinc acetate dihydrate (50 mM) and PEG8000 (22%, w/v) in sodium cacodylate buffer (100 mM, pH 6.0). The crystals of wild-type ChiNCTU2 without zinc binding were obtained from potassium phosphate monobasic (50 mM, pH 6.5) and PEG8000 (20%, w/v). Mutants D143A, E145Q, and E145Q/Y227F complex with (NAG)2(molar ratio 1:3) were crystallized using PEG8000 (22%, w/v) in potassium phosphate monobasic (50 mM, pH 6.5) buffer (100 mM, pH 6.0). The E145G/Y227F complex with (NAG)4and cyclo-(L-His-L -Pro) (molar ratio 1 to 3) were obtained under conditions of PEG8000 (20%, w/v) and calcium acetate (200 mM) in MES buffer (100 mM, pH 6.0). Single crystals (0.4⫻ 0.2 ⫻ 0.2 mm3) were transferred to the crystallization solution containing glyc-erol (20%) and flash-cooled in liquid nitrogen for x-ray diffrac-tion experiment.
Data Collection and Processing—X-ray diffraction data from crystals of wild-type ChiNCTU2 in the conditions containing zinc atoms were first collected at 110 K using a CCD detector (Q-4R, ADSC) at wavelength 1.0 Å on Taiwan contract beam-line BL12B2 at SPring-8 in Japan. A 360° rotation with a 1.0° oscillation was measured with an exposure duration of 15 s and a distance of 180 mm from the crystal to the detector at 110 K using a cryo-system (X-Stream, Rigaku/MSC). Because of the presence of zinc ions in the crystallization solution, we under-took a zinc multiple wavelength anomalous diffraction (MAD) method to solve the structure. The x-ray energy scan and fluo-rescence measurement on the crystal showed a typical zinc absorption spectrum.
All x-ray diffraction data were collected at 110 K at beamline BL13B1 equipped with a CCD detector (Q315, ADSC) at the National Synchrotron Radiation Research Center in Taiwan and BL12B2 at SPring-8. All data sets were indexed, integrated, and scaled using HKL2000 (22). Details of data statistics appear in Table 3.
Determination and Refinement of Structures—The structure of wild-type ChiNCTU2 was solved with the zinc MAD method. The positions and occupancies of four zinc atoms were determined with SOLVE Version 2.09 at 2.3 Å resolution. Phases were improved by density modification with RESOLVE (23, 24). An initial structure with⬃76% complete model was automatically traced into electron density maps with RESOLVE and ARP/WARP (25), and the remaining model was manually built with O (26). Throughout the refinement, a random selec-tion (10%) of the data were set aside as a “free data set,” and the model was refined against the remaining data with Fⱖ 0 as a working data set (27). After rigid-body refinement and simu-lated annealing with CNS (28), several rounds of model build-ing with Coot (29) and refinement with CNS were performed to improve the quality and completeness of the structure. The refinement proceeded through another cycle of individual
B-factor refinement and the PICKWATER from CNS and Ref-mac5 (30) to define peaks in difference maps to locate 302 water molecules. The refinement converged to a final Rwork19.8% (Rfree21.3%) at resolution 1.20 Å. The structures of zinc-free chitinase and various mutant-saccharide complexes were determined by molecular replacement with MOLREP (31)
using the wild-type structure as the search model. The struc-tures were refined as described above. Table 3 shows details of refinement statistics.
Model Validation—The correctness of the stereochemistry of the models was verified using PROCHECK (32). The calcu-lations of root mean square deviations from ideality (33) for bonds, angles, and dihedral and improper angles performed in CNS Version 1.2 showed satisfactory stereochemistry. In a Ramachandran plot (34) all residue main-chain dihedral angles are in the most favored and additionally allowed regions with only glycine exceptions. Model statistics for wild-type zinc-bound, zinc-free ChiNCTU2, and mutant complexes are given in Table 3.
RESULTS
Overall Structure of Wild-type Chitinase—The structure of wild-type ChiNCTU2 was determined by the zinc MAD method and refined to 1.2 Å resolution (Fig. 1A). The overall structure comprises 10-strains and 12 ␣-helices, of which /␣ motifs form a TIM-barrel structure with eight parallel-strains (123456910) forming the core of the enzyme, in which strain 10 is hydrogen-bonded to strand 1. In general, -strains alternate along the sequence with the ␣-helices as the “return stroke” in the order1␣12␣23␣34␣45␣5␣667 8␣7␣89␣9␣1010␣11␣12 (Fig. 1B). There are two ␣-helices on the turn between5 and 6 and two ␣-helices between 8 and9. In addition to the -sheets of the core, an antiparallel two-stranded sheet labeled7 and 8 is formed within the con-necting loop between6 and ␣7 of the barrel. The dimensions of ChiNCTU2 are⬃52 ⫻ 54 ⫻ 35 Å3with the surface area of 12,483 Å2accessible to solvents. Of 16 prolines in total, one (Pro-181) has cis-peptide geometries in the monomer struc-ture. The electrostatic surface shows that ChiNCTU2 is a glob-ule enzyme with a continuous negatively charged cleft on the surface with dimensions⬃40 ⫻ 15 ⫻ 7 Å3, which is suitable for the entrance and binding of (NAG)n(Fig. 1C). The negatively
charged residues around this cleft are Glu-68, Glu-77, Asp-143, Glu-145, and Glu-190.
As predicted from the sequence alignment, a structural comparison of ChiNCTU2 with other GH-18 chitinases reveals that ChiNCTU2 comprises only a compact CaD, without CBD and CID, of which the structure differs from those of hevamine and other chitinases (Fig. 1D). Among distinct structural features surrounding the TIM-barrel in ChiNCTU2 are two anti-parallel-sheets (7 and 8) and long loops between4-␣4, ␣7-␣8, and ␣10-10 that elongate the binding cleft (Fig. 1C). A further structural feature is the ␣5 helix in the surface cleft of ChiNCTU2. From a compar-ison with endochitinases, such an ␣5 helix exists only in structures of exochitinases.
Probing Essential Catalytic Residues; Sequence Alignment, Mutagenesis, and Kinetic Analysis—To locate the essential res-idues of ChiNCTU2, we aligned amino acid sequences of vari-ous chitinases to analyze their diversity (Fig. 2) (8, 10, 13, 35– 44). The sequences share several highly conserved frag-ments among all chitinases in which three regions are impor-tant. The first fragment, located near3, with the sequence 107GGXXX111 (numbers based on ChiNCTU2), is related to
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
TABLE 3 Statistics of diffraction data and structure refinement ChiNCTU2: Zn-MAD Native with zinc PDB 3N12 Native without zinc PDB 3N11 D143A ⴙ (NAG) 2 PDB 3N13 2E145Q ⴙ (NAG) 2 PDB 3N15 E145Q/Y227F ⴙ (NAG) 2 PDB 3N17 E145G/Y227F ⴙ (NAG) 4 PDB 3N18 E145G/Y227F ⴙ cyclo-( L -His-LPro) PDB 3N1A Peak Inflection High-remote Data collection Wavelength (Å) 1.28228 1.28284 1.25233 1.000 1.000 1.000 1.000 1.000 1.000 1.000 Temperature (K) 110 110 110 110 110 110 110 110 110 110 Space group P 21 P 21 P 21 P 21 P 21 21 21 P 21 21 21 P 21 21 21 P 21 21 21 P 21 21 21 P 21 21 21 Cell dimensions (Å) a 50.76 50.76 50.77 50.82 48.52 48.72 48.54 48.64 48.68 48.74 b 48.61 48.61 48.62 48.75 76.05 76.75 76.19 76.36 75.07 75.22 c 66.63 66.6 66.64 66.65 76.61 78.5 76.31 76.91 79.41 79.45  99.10 99.09 99.10 98.89 Resolution range (Å) a 30–2.3 (2.38–2.3) 30–2.3 (2.38–2.3) 30–2.24 (2.32–2.24) 30–1.1 (1.16–1.1) 30–1.35 (1.40–1.35) 30–1.7 (1.76–1.7) 30–1.94 (2.01–1.94) 30–1.2 (1. 24–1.2) 30–1.6 (1.66–1.6) 30–2.0 (2.07–2.0) Completeness (%) a 99.9 (99.7) 99.9 (99.3) 99.5 (95.2) 94.5 (91.3) 98.8 (96.5) 99.9 (100) 99.1 (99.9) 97.2 (88.1) 98.2 (96.9) 99.9 (99.9) Redundancy a 7.4 (7.3) 7.4 (7.2) 7.4 (7.1) 2.6 (2.6) 5.9 (5.5) 3.7 (3.6) 4.8 (5.2) 6.6 (5.2) 6.2 (6.3) 6.6 (7.0) 具I/ (I )典 a 20.4 (8.3) 28.5 (11.42) 27.1 (10.69) 16.5 (2.95) 34.7 (6.2) 24.1 (3.6) 20.2 (11.3) 43.3 (8.5) 39.5 (5.6) 27.35 (6.09) Rsym (%) a,b 9.5 (26.0) 6.9 (18.2) 7.1 (19.1) 5.5 (42.7) 4.9 (28.0) 5.3 (37.5) 6.5 (15.0) 4.7 (19.9) 4.5 (33.3) 7.5 (42.4) Refinement Resolution range (Å) 30–1.2 30–1.35 30–1.7 30–1.94 30–1.2 30–1.6 30–2.0 Rwork c/R free d(%) 19.8/21.3 20.2/21.5 18.6/21.0 18.0/22.2 17.5/18.7 19.7/22.7 19.9/23.7 No. of atoms Protein 2558 2501 2506 2509 2534 2511 2511 Ligand/ion 82 9 2 9 2 9 5 7 1 7 Water molecules 302 342 146 191 291 321 96 B -factors (Å 2) Protein 11.12 11.90 15.76 18.76 7.39 16.1 27.6 Ligand/ion 31.54 19.08 19.29 8.58 48.4 60.0 Water molecules 20.21 25.25 22.87 26.10 15.84 30.6 30.8 Root mean square deviations Bond lengths (Å) 0.0034 0.0047 0.0057 0.0067 0.0042 0.0044 0.0033 Bond angles (°) 0.8934 0.9376 1.0034 1.1713 0.9803 0.9669 0.7548 aValues in parentheses are for highest resolution shells. bR sym ⫽⌺ h⌺ i关 兩Ii (h )⫺具 I(h) 典兩 /⌺ h⌺ iIi (h )兴 , where Ii is the ith measurement, and 具I (h )典 is the weighted mean of all measurements of I( h ). cR work ⫽⌺ h兩 Fo ⫺ Fc 兩/⌺ hF o , where Fo and Fc are the observed and calculated structure factor amplitudes of reflection h . dR free is as Rwork but calculated with 10% of randomly chosen reflections omitted from refinement.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
substrate binding (10); the second fragment, located in4, with the sequence141DXDXE145, serves a catalytic center of GH-18 chitinases (45); the third fragment, located between6 and 7, with the sequence225QXYN228, is involved in the substrate hydrolysis (46).
The conserved aspartic acid (Asp-143), glutamic acid (Glu-145) and tyrosine (Tyr-227) of ChiNCTU2, located at the active site (Fig. 3A), were selected as primary mutants based on the structure of wild-type ChiNCTU2 and the sequence alignment of GH-18 enzymes. The kinetic parameters of wild-type and mutant enzymes were determined to show that the related mutants, D143A, E145G, E145Q, and Y227F, exhibit the defect
in catalysis (Table 2). The sigmoid kinetics was not observed for wild-type ChiNCTU2 using 4-nitrophenyl--chitobioside as a substrate with the present data. The mutant E145G decreased the activity to an undetectable level in kcat, confirming its essen-tial catalytic role (47– 49). Glu-145-related mutants, including the single mutant E145Q and double mutants E145Q/Y227F and E145G/Y227F, were generated for co-crystallization with (NAG)n.
The Active Site and Conformations of (NAG)n in Complex
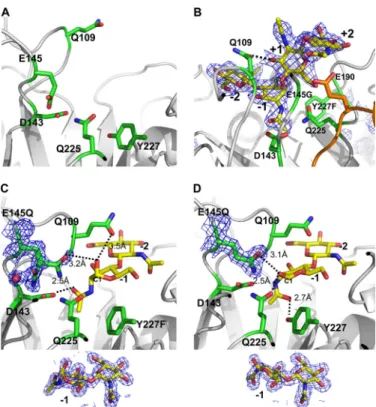
Structures—As two zinc atoms are located at the CaD in the native structure that might effect the activity of ChiNCTU2 (later in text), we removed zinc acetate dehydrate from the crys-FIGURE 1. The crystal structure of ChiNCTU2. A, overall structure (top view) of ChiNCTU2 consists of 10-strains (in green) and 12 ␣-helixes (in blue) with a TIM barrel shown in a ribbon drawing. B, the topology presentation shows the secondary structure arrangements with sequence numbers. C, the electrostatic surface of wilt-type ChiNCTU2 reveals a negatively charged cleft (in red) on the surface formed by the key residues as labeled. The green color shows the structural features in ChiNCTU2. The loops of L1 (4-␣4), L2 (␣7-␣8), and L3 (␣10-10) and the sheets 7 and 8 are located in the periphery of ChiNCTU2. The helix␣5 is located in the end of the cleft. D, the superimposed structures (side view) of ChiNCTU2 (blue), S. marcescens chitinase A (green, CBD removed), A. fumigatus chitinase B1 (yellow), and H. brasiliensis hevamine (magenta) reveal no CID between␣9⬃␣10 (in the circle) in ChiNCTU2 and their structural differences. The conserved key residues Glu-145 and Tyr-227 are shown in red sticks.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
tallization solution and attempted to obtain co-crystals of the (NAG)n complex. This condition caused, however, protein
molecules to pack into a dimer for wild-type ChiNCTU2 or a tetramer for the mutant E145Q in another P1 crystal form, in which two Glu-145 residues at the active site from each mono-mer face each other at distance 5.4 Å. This molecular packing buried the active site and obstructed the entrance of substrates into ChiNCTU2. As ChiNCTU2 exists as a monomer in the solution based on gel filtration measurements (data not shown), we sought another crystallization condition to generate a monomer structure with space group P212121, in which the active site was exposed for possible co-crystallization with (NAG)n. To identify the chitin-binding site and to probe the
hydrolysis mechanism, we co-crystallized (NAG)4with the sin-gle mutant E145Q and double mutants E145Q/Y227F and E145G/Y227F. X-ray diffraction data of co-crystals of E145Q, E145Q/Y227F, and E145G/Y227F in complex with (NAG)4at resolutions 1.94, 1.2, and 1.6 Å revealed the electron densities of (NAG)2, (NAG)2, and (NAG)4in the active site, respectively (Fig. 3, B–D). The NAG-binding site is located in the cleft on the protein surface revealed by the electrostatic surface plot, named the “saccharide binding cleft” for further discussion. Only (NAG)2fragments were found in the active site of mutants E145Q and E145Q/Y227F even though (NAG)4was added for co-crystallization, indicating that these two mutants main-tained slight activity (Fig. 3, C and D and Table 2).
In the E145Q complex structure (Fig. 3D), the (NAG)2with the chair-form conformation was located in the active site and identified as⫺1 and ⫺2 subsites. The C1-OH of NAG at the ⫺1 subsite (⫺1-(NAG)) forms a hydrogen bond with Gln-225 (dis-tance 2.5 Å). The saccharide N-acetyl group in the⫺1 subsite, which was proposed to mediate the exchange of the chair and boat forms of saccharides (46), is directed toward the OH of Tyr-227 with distance 2.7 Å. The other major interaction to stabilize chitin has the aromatic residue Trp-333 under the⫺1 subsite serving as the stacking force through the-orbital with a distance⬃4 Å. Other residues, such as Asn-45, Gln-109, Asp-143, Gln-145, Asn-228, Ala-287, and Trp-337, also contributed to interactions with the saccharide through hydrogen bonding. Through a comparison with E145Q complex structure, the complex structure of the double mutant E145Q/Y227F shows a similar binding site and hydrogen bonds at the⫺2 subsite for (NAG)2(Fig. 3C). Some distinguished features are, however, found at the⫺1 subsite. First, according to the mechanism proposed by Brameld and Goddard (49), before hydrolysis the conformation of saccharides at the⫺1 subsite is possibly the boat form. A similar boat-form conformation is found in the complex structure of E145Q/Y227F⫹(NAG)2at the ⫺1 subsite. C1-OH of ⫺1-(NAG) interacts with Gln-225 in the E145Q complex structure, whereas C1-OH shifts its direction toward a dynamic loop between3 to ␣3 and forms a hydrogen bond with Gln-109 (distance 3.5 Å) in the E145Q/Y227F complex structure. Second, the N-acetyl group at the⫺1 subsite redirects the orientation from resi-due Y227F to Asp-143 by turning 180° and forms a hydrogen bond with distance⬃2.5 Å.
For the complex structure of E145G/Y227F with (NAG)4, the subsites of (NAG)4are identified to be⫺2, ⫺1, ⫹1, and ⫹2 (Fig. 3B). The structures of NAG at⫺1 and ⫺2 subsites are the same as those in the complex structures of E145Q/Y227F and (NAG)2 with a boat-form conformation at the ⫺1 subsite, whereas⫹1-(NAG) rotates and arises ⬃90° from ⫺1-(NAG),
FIGURE 2. Structural-based multiple-sequence alignment of selected GH-18 chitinases. The sequences of various chitinases with the corresponding PDB codes are aligned with ChiNCTU2 on the similarities of their amino acid sequences (CAZy, available on-line). The endochitinases include Saccharomyces cerevisiae chitinase1 (PDB: 2UY2) (35), H. brasiliensis hevamine (1LLO) (13), Parkia platycephala endochitinase (2GSJ) (36), Pyrococcus furiosus chitinase (2DSK) (37); exochitinases include S. marcescens chitinase A (1CTN) (10), S. marcescens chitinase B (1E15) (38), Homo sapiens chitinase (1GUV) (39), Coccidioides immitis chitinase 1 (1D2K) (40), Bacillus circulans WL-12 chitinase A1 (1ITX) (41), A. fumigatus chitinase B1 (1W9P) (8), Arthrobacter sp. chitinase B (1KFW) (42), A. fumiga-tus YJ-407 chitinase (1WNO) (with both endo- and exo-activities) (43), and Vibrio harveyi chitinase A (3B8S) (44). The complete conserved residues are blocked in gray. The residues of ChiNCTU2 that were selected for mutagenesis in this work were indicated with arrows. Three boxes (A, B, and D) indicate the highly conserved regions. The box C shows the ChiNCTU2 contains a longer amino acid sequence at this region that forms a helix of␣5 (191–199), which exists only in exochitinases, and a loop (200 –207). The box E indicates the location of CID.
FIGURE 3. The active site of ChiNCTU2. A, the active site of the native struc-ture is formed with the key residues shown in stick (C, green; N, blue; O, red). The side chain of Glu-145 exhibits the native conformation before substrate binding. B, the complex structure of mutant E145G/Y227F with (NAG)4reveals four subsites (⫺2, ⫺1, ⫹1, and ⫹2) (yellow) in the active site. The configura-tion of (NAG)4at the⫺1 subsite is a boat form. The loop between5⬃␣5 is shown in orange. C, the complex structure of E145Q/Y227F and (NAG)2shows that there are⫺1 and ⫺2-(NAG) in the active site, of which ⫺1-(NAG) exhibits a boat-form conformation that fits well in the electron density below. The N-acetyl group of⫺1-(NAG) is directed toward Asp-143. The side chain of E145Q with a double conformation might be observed with one interacting with⫺1-(NAG) and the other coordinating a water molecule. D, the complex structure of E145Q and (NAG)2shows that⫺1-(NAG) exhibits a chair-form formation as fits well in the electron density below and directs the N-acetyl group toward Tyr-227 without the Y227F mutation. The electron density 2Fo⫺ Fcmap of the substrates and residues E145Q are shown in blue mesh
(1) in all figures.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
presumably due to the5⬃␣5 loop. Notably, ⫹2-(NAG) fur-ther rotates and bends⬃90° relative to ⫹1-(NAG) and interacts with the side chain of Glu-190 (on5⬃␣5 loop) and the main
chain of Glu-145, differing from exochitinases with a CID that uses the aromatic residue to maintain and to interact with ⫹2-(NAG) (Fig. 3B,supplemental Fig. 1). Glu-190 is the key
residue in coordinating both⫹1- and ⫹2-(NAG). The complex
structure shows that many residues contribute to the binding of (NAG)4, as summarized in Table 4.
We also co-crystallized mutants E145Q, E145Q/Y227F, and
E145G/Y227F with (NAG)5. All structures of (NAG)5
com-plexes were similar to that of the (NAG)4complex, except that
an extra electron density was connected to⫺2-(NAG), which
implies one more NAG moiety to be located after the⫺2
sub-site. However, the electron density is too indistinct to allow
complete modeling⫺3-(NAG) due to its flexible character.
Dynamic (NAG)nBinding Loops—A comparison of wild-type
native and all mutant complex structures (E145Q⫹(NAG)2;
E145Q/Y227F⫹(NAG)2; E145G/
Y227F⫹(NAG)4) shows that overall
structures are similar except for two
dynamic regions from 2 to ␣2
(Gly-67—Thr-69) and3 to ␣3
(Ile-106 –Val-112), which might be identified as the saccharide binding loops (Fig. 4, A and B). An inspec-tion of temperature-factor distribu-tions in structures of the native and
(NAG)4complex shows high
B-fac-tors at the two loops, indicating that
they are structurally dynamic
(supplemental Fig. 2, A and B). The
new finding of dynamic characters of two loops is previously unre-ported among other structurally known chitinases.
The two loops were originally ori-entated away from the catalytic
res-idue Glu-145⬃16.5 and 13.1 Å in
the native structure of wild-type ChiNCTU2 (pre-catalyzing stage),
but upon (NAG)n binding, two
loops shift along the saccharide binding cleft toward the active site
and interacted with (NAG)n
(cata-lyzing stage) (Fig. 4, A and B). The two loops are located side by side
with several hydrogen bonds
between them (the smallest distance ⬃3 Å between main chains of Glu-68 and Asn-110); hence, two loops simultaneously alter their conformations in the same direc-tion. Despite one loop (Gly-67— Thr-69) having no interaction with
(NAG)2 or (NAG)4, it could still
shift⬃4 Å (Fig. 4B).
The other dynamic loop (Ile-106 –Val-112) is notable because
(NAG)nbinding induced its
maxi-mum conformational shift⬃4.6 Å
FIGURE 4. The dynamic loops for chitin binding. A, the electrostatic molecular surface of ChiNCTU2 is shown in gray with the dynamic loop (Ile-106 –Val-112) shown in red and green colors for native and mutant E145G/ Y227F⫹(NAG)4complex structures, respectively, after superimposition. Upon the substrate binding, the loop moved from the native open form (red) to the binding close form (green). B, shown is a close view of the chitin-binding mode. The native (in red sticks) and E145G/Y227F⫹(NAG)4(in green sticks) structures show that the side chain of Glu-68 and Gln-109 alters its position⬃4.0 Å (about the length of the monosaccharide) and 4.6 Å, respectively, to interact with chitin in the active site. The swing of the dynamic loop (Ile-106 –Val-112) between the native and complex structure allows Gln-109 to swing over the⫺2-(NAG). The two dynamic loops in the structure of D143A⫹(NAG)2(blue) are in the position similar to the native open form (pre-catalyzing-stage position). The structure of D143A⫹(NAG)2also showed the main chain of catalytic residue Glu-145 relocated its position (blue). The side chain of Phe43 is fixed among three structures and positioned close to the potential⫺3 subsite. C, shown is a scheme of (NAG)4and residue Gln-109 (red, native open form; green, binding close form). The boat-form⫺1-(NAG) shifts the linear ⫹1 and ⫹2-(NAG) from the horizontal (gray) to the vertical (black) position, which shortens the distance between⫺2 and ⫹1 to the distance corresponding to a mono-(NAG) moiety (⬃4.6 A) related to Gln-109.
TABLE 4
Interactions between residues of mutant E145G/Y227F and bound substrate (NAG)4
Protein residues Subsites for
sugar moieties
Gly-145a, Glu-190 ⫹2
Gln-109, E145Qb, Glu-190, Gln-225 ⫹1
Gln-109a, Asp-143, E145Qb, Gln-225, Asn-228,
Ala-287a, Trp-333 ⫺1
Asn-45, Trp-333, Trp-337 ⫺2
a
The main-chain interaction.
b
The residue E145Q was referred from the complex structure of E145Q/ Y227F⫹(NAG)2.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
from the (NAG)n-free to the (NAG)n-bound state, providing
hydrogen bonds to stabilize (NAG)n (Fig. 4B). This
displace-ment is equal to the size of a NAG moiety, which allows the dynamic loop to swing across⫺2-(NAG). In the structure of the E145G/Y227F⫹(NAG)4complex, Gln-109 provides a hydro-gen bond (2.5 Å) with the oxyhydro-gen atom of C3 of⫹1-(NAG) because the⫺1-(NAG) exhibits a unique boat-form conforma-tion (Fig. 4C). An inspecconforma-tion of the loop (Ile-106 –Val-112) with the native orientation (in red) and (NAG)4shows that Gln-109 is located at the position between⫺2 and ⫺3-(NAG) (Fig. 4B), implying that Gln-109 might interact with the oxygen atom of C3 of⫺2-(NAG). Thus, the conformational change of the loop Ile-106 –Val-112 with a swing distance corresponding to a NAG moiety across-⫺2-(NAG) might enable Gln-109 to bring the NAG moiety from the⫺2 to ⫹1 subsite, which migrates the ⫺1 and ⫺2-(NAG) to new ⫹2 and ⫹1 subsites, respectively. Taking advantage of boat-form ⫺1-(NAG), the shift of di-(NAG) by dynamic loops might explain that chitobiose is the predominant product of ChiNCTU2. We tried also to co-crys-tallize mutant D143A with (NAG)4, but the structure showed only (NAG)2at the⫺1 and ⫺2 subsites in the active site. In this complex form, the loop (Ile-106 –Val-112) exhibits an orienta-tion similar to that in the structure of the wild-type native enzyme (Fig. 4B), implying that two dynamic loops would shift back to the native open (pre-catalyzing stage) position after hydrolysis.
The sequence alignment among various chitinases reveals that all exochitinases are equipped with a residue Trp at the equivalent position of Gln-109 on the dynamic loop (Ile-106 – Val-112) in ChiNCTU2; endochitinases employ smaller resi-dues, Ala or Gly (Fig. 2). This observation might imply separate paths of the substrate entrance for chitinases with and without a CID. The CID-containing exochitinases utilize the Trp on the loop to connect with Trp residues in the CID or CBD to form a continuous tunnel path for saccharide binding and entrance. Endochitinases without a CID utilize smaller amino acids to provide a larger space to allow enzymes to hydrolyze substrates randomly, whereas ChiNCTU2 has a mid-size polar residue Gln-109 to interact with substrates. We performed mutagene-sis of Q109G for an activity assay to examine whether Gln-109 is involved in the catalytic turnover and the short side chain of Gly might change the enzymatic function. The results showed that the mutant Q109G decreased the activity to an undetect-able level rather than just altering its function (Tundetect-able 2), which exhibits the similar essential role of Glu-145. We, thus, propose that 1) Gln-109 is a key residue involved in interacting with NAG moieties for catalysis, and 2) the dynamic loop (Ile-106 – Val-112) is crucial for ChiNCTU2 to shift a di-(NAG) moiety in the catalytic site.
The Residues with Multiple Conformations in the Active Site and the Functional Roles—The dynamic side chains of residues exhibiting multiple conformations in the active site typically play crucial roles for an enzyme function, especially at the inter-mediate state. The double-mutation E145Q/Y227F is unique in generating an intermediate state and maintaining a slight enzy-matic activity to hydrolyze (NAG)4to (NAG)2. However, this mutant could not convert⫺1-(NAG) to the chair-form confor-mation as the boat-form conforconfor-mation was observed in the
complex structure. The native structure of wild-type ChiNCTU2 revealed four residues, Asp-143, E145Q, Glu-190, and Tyr-193, with only a single conformation in the active site. With a substrate bound into the active site, these four residues exhibit multiple conformations in the corresponding complex structures (Fig. 5).
The double conformations of E145Q and Asp-143, as previ-ously reported (46, 50), are observed in the ChiNCTU2 struc-ture. The first side-chain conformation of the mutated residue E145Q is directed to OH of Tyr-193 (similar to that in the native non-bound structure), whereas the second conformation is flipped about 120° to interact with the -OH of C1 of⫺1-(NAG) with a distance 3.2 Å (Figs. 3C and 5B, (II)). A water molecule is coordinated with the first conformation of E145Q with the con-nected electron density at a small distance 1.74 Å. Furthermore, in the same crystal form but with a greater duration of (NAG)4 soaking, the bound water is relocated near E145Q at the second conformation with distances 1.6 and 2.9 Å to E145Q and -OH of C1⫺1-(NAG), respectively. In ChiNCTU2, beside its catalytic role, Glu-145 might carry the water molecule through a struc-tural alteration from the first to the second conformations. The mutant D143A disables the hydrogen bond between Asp-143 and Glu-145, existing in all other wild-type native and mutant complex structures (Fig. 5A), to affect the conformation (in
blue) of the loop (144 –148); hence, Glu-145 is re-orientated away from the active site (Fig. 4B). In addition to stabilizing (NAG)nbinding, Asp-143 might maintain the conformation of
the catalytic residue Glu-145 on the loop (144 –148) for its function.
The residual terminal-COOH group of Glu-190 exists in the triple conformations simultaneously in the complex structure FIGURE 5. Residues with multiple conformations involved in catalysis in
the active site. A, the first conformation (magenta) and the second
confor-mation (green) of residues Asp-143, E145Q, Glu-190, and Tyr-193 are observed in the structure of the mutant E145Q/Y227F complex with (NAG)2(cyan), shown in stereo view. The (NAG)4(yellow) from the structure of E145G/ Y227F⫹(NAG)4complex is superimposed with (NAG)2. B, close views show that the respective residues exhibit double conformations revealed by the electron density 2Fo⫺ Fcmap (blue mesh, 1): (I), Asp-143; (II), E145Q and
Tyr-193; (III), Glu-190. The color assignment is the same as in A. The interac-tions between side chains and substrates (or water) are shown in dashed lines with distances.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
(Fig. 5B, (III)). Glu-190 together with E145Q with the corre-sponding positions might be involved in the regulation of prod-uct release. The side chain of Glu-190 is located near the mid-position of two conformations of E145Q. The smallest distance between multiple conformations of E145Q and Glu-190 is 2.5 Å when the side chain of E145Q is situated in the first conforma-tion. When the side chain of E145Q orientates to the second conformation, the two residues are separate from each other by the largest distance 3.4 Å (Fig. 5A). This shift⬃1 Å provides different hydrogen bonding between residues E145Q and Glu-190, which might trigger the dynamic C terminus of Glu-190. Based on our (NAG)4 complex structure, Glu-190 not only interacts with⫹1- and ⫹2-(NAG) but also provides several hydrogen bonds by the C-terminal group (Fig. 5B, (III)). During hydrolysis, the side-chain switch of Glu-145 mediates Glu-190 with triple conformations, which results in the binding forces different between Glu-190 and⫹1- and ⫹2-(NAG). The kinetic data showed that the mutant E190Q increased Kmslightly and
decreased the activity⬃70% in kcat(Table 2).
The residue Tyr-193 exhibits multiple conformations with a vibration of the side chain of about 1.3 Å (Fig. 5, B(II)). We explained the function of multiple conformations of Tyr-193 only after the water molecule between E145Q and Tyr-193 was located. Tyr-193 is located at the midpoint of the shortest path for water molecules from the protein surface to the first con-formation of E145Q. An additional water molecule on the pro-tein surface is found to coordinate with Tyr-193. Tyr-193, hence, switches its side chain to bind with separate water molecules.
Binding Sites of Zinc Atoms and the Inhibitor—Metals are reported to be effective inhibitors for GH-18 (51). The asym-metric unit of crystals from crystallization in the presence of zinc acetate dihydrate comprises one ChiNCTU2 molecule in space group P21. Four zinc atoms and one acetate molecule are bound on the protein surface, of which two zinc atoms and the acetate molecule were found at the active site and interacted with five residues, Asp-143, Glu-145, Glu-190, Gln-225, and Tyr-227 (Fig. 6A). The other two zinc atoms, located away from the catalytic site, were coordinated with histidine residues 178 (together with symmetrical Asp-87) and 257, respectively. The structure of the zinc-free wild-type ChiNCTU2 was also deter-mined (Table 3). Superimposed zinc-bound and zinc-free structures of ChiNCTU2 revealed that the zinc atoms provide ionic forces to rearrange the orientations of the side chains, especially of essential residue Glu-145, which was shifted about 6.1 Å from the conformations of the zinc-free to the zinc-bound state (Fig. 6A).
Because zinc atoms could be bound in the active site, we examined the influence of various metal ions on the enzymatic activity (18). Among tested metal ions, Hg2⫹, Zn2⫹, and Cu2⫹ showed a strongly inhibitory effect. The presence of Hg2⫹and Cu2⫹ions (10 mM) resulted in a 95% activity loss, whereas Zn2⫹ decreased activity about 50%. The enzymatic activity was enhanced 100% in the presence of Ca2⫹(10 mM). Other ions, such as Ba2⫹, Cr3⫹, Mn2⫹, and Mg2⫹, had no significant effect on the activity.
Another group of inhibitors for GH-18 contains chitin ana-logues, such as allosamidin (50) and cyclo-(L-His-L-Pro) (52).
To characterize the inhibitory mechanism, we co-crystallized ChiNCTU2 with cyclo-(L-His-L-Pro) and derived the structure of mutant E145G/Y227F in a complex with the inhibitor (Fig. 6B). The complex structure shows that the inhibitor also occu-FIGURE 6. The inhibitors in the active site. A, two zinc atoms (yellow spheres), designed as Zn1 and Zn2 in the structure, are located in the active site and interact with residues Glu-145 and Glu-190. The distance between Zn1 and Zn2 is⬃4 Å. An acetate molecule interacts with Asp-143 and Tyr-227. The interactions are shown in dashed lines labeled with related distances. The 2Fo⫺ Fcdensity map of the acetate (blue mesh, 1.2) and
anomalous-differ-ence Fourier map of zinc atoms (red mesh, 4) are shown respectively. The Glu-145 was orientated from un-bond position (white stick) to the zinc-bond position (green sticks). B, the inhibitor cyclo-(L-His-L-Pro) (lemon yellow) bound with the surrounding residues (green sticks) in the active is site shown in a surface plot (light pink), with the important residue Gln-109 on one dynamic loop labeled in orange. The electron density with the 2Fo⫺ Fccoefficient is
shown (blue mesh, 0.9). C, the zinc atoms and the inhibitor are superim-posed with (NAG)4 from the complex structure to show their related positions.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
pies the entrance of the catalytic domain and interacts with residues Phe-43, Phe-66, Gln-109, Asp-143, Gln-225, Y227F, and Trp-333 (through a-orbital stacking force). Regardless of the inhibitory function of metals and inhibitors, their binding sites in ChiNCTU2 are varied,⫺1, ⫹1, and ⫹2 subsites for zinc atoms and⫺1 and ⫺2 subsites for cyclo-(L-His-L-Pro) (Fig. 6C), implying varied mechanisms of inhibition. The chitin analogue, located at the substrate entrance, likely obstructs substrates from entering the active site, whereas metals, interacting with critical residue Glu-145 to fix its side-chain conformation, might restrict the hydrolysis capabilities of essential residues.
DISCUSSION
ChiNCTU2 as a GH-18 Member Comprising Only a Catalytic Domain—The enzymatic hydrolyzes of colloidal chitin and chi-tooligosaccharides with the degree of polymerization 3– 6 were studied in a time course manner (18). Chitobiose was observed as the predominant product throughout the enzymatic hydro-lysis of colloidal chitin and chitohexaose, suggesting that ChiNCTU2 likely functions as an exochitinase.
A structural comparison of ChiNCTU2 with various chiti-nases in preceding sections showed that all exochitichiti-nases con-sist of CaD⫹CID or CaD⫹CID⫹CBD and utilize exposed aro-matic residues to form a long and deep substrate binding cleft (11, 53, 54). The clefts with aligned aromatic residues are thought to facilitate substrate binding and might slide the poly-mer chain of chitin along the cleft acting like a procession (55– 57). Distinct from exochitinases, endochitinases comprise only a CaD without CID and CBD in the structures and utilize a shallow anchoring substrate binding site to bind and regulate substrates into the catalytic center (6), resulting in nonspecific hydrolysis products from random cleavages at internal points of the chitin chains.
ChiNCTU2 is suggested as an exochitinase containing only a CaD without CID and CBD to hydrolyze chitin into the chito-biose specifically, differing from previously known GH-18 chitinases (6). A structural comparison of wild-type native enzyme and the mutant complexes revealed notable structural changes at the dynamic loops (Gly-67—Thr-69) and (Ile-106 – Val-112) (Fig. 4). In the structure of wild-type native enzyme without (NAG)n, the two loops show an open-form
conforma-tion (shown in red), whereas in the structures of mutants E145Q, E145Q/Y227F and E145G/Y227F complex with (NAG)n, the two loops, especially Ile-106 –Val-112, exhibit a
conformational alteration to a closed form to grab and to slide the substrate into the active site (in green). A “tunnel-like” sub-strate binding site in the active cleft is, thus, formed to lock the substrate into a position for specific production of chitobiose. As exochitinases generally possess long binding clefts for sub-strate sliding, the tunnel length might be another reason for ChiNCTU2 to act more like an exochitinase. The length of the tunnel of ChiNCTU2 is⬃40 Å, a size similar to those in CaD of other exochitinases, whereas the binding cleft is much shorter, ⬃20 Å, in endochitinases (supplemental Fig. 3).
Substrate Binding through Dynamic Loops for Catalysis— The catalytic mechanisms of GH-18 chitinases have been extensively studied (13, 46, 49, 58, 59). Based on our kinetic assessments of mutants and structures of wild-type native
ChiNCTU2 and the mutant complexes with (NAG)n, we
pro-pose a catalytic action through the dynamic loops of ChiNCTU2 containing only a CaD without CBD and CID. The structure of wild-type native ChiNCTU2 might be considered as one enzymatic state before substrate binding (Fig. 3A). When the substrate (NAG)napproaches the active site, chitin is bound into the active site and interacts with the dynamic loop Ile-106 –Val-112 through a hydrogen bond with Gln-109 (Fig. 4). With the conformational switches of the dynamic loops, ChiNCTU2 is enabled to orientate (NAG)4in the active site and
further imposes a structural change of⫺1-(NAG), which was
achieved with Asp-143 to interact with the boat-form ⫺1-(NAG), as observed in the complex structure of E145G/ Y227F with (NAG)4(Fig. 3B).
The cleavage of the glycosidic bond between ⫺1 and
⫹1-(NAG) by Glu-145 is a crucial step in hydrolysis, which brings a water molecule to complete a catalytic turnover (49). The intermediate state of catalysis can be sketched with the
(NAG)2-incoorporated complex structure of E145Q/Y227F
(Fig. 3C). The Glu-145 controls not only hydrolysis but also product release. Because double conformations of Glu-145 and Glu-190 could bring two residues together within a hydrogen-bond range, the flexibility of the side chain of Glu-145 might affect the side-chain conformations of Glu-190. The
con-strained interaction between Glu-190 and⫹1- and ⫹2-(NAG)
can be released due to the cleavage of the glycosidic bond and
further allows Glu-190 to draw the catalytic product,⫹1- and
⫹2-(NAG) (a di-NAG), away from the catalytic site, whereas
Gln-109 provides hydrogen bonding to maintain⫺1-(NAG) in
a boat-form conformation.
After hydrolysis,⫺1-(NAG) is thought to alter its conforma-tion from the energetically unfavorable boat form to the
favor-able chair form.⫺1-(NAG) exhibits the chair-form
conforma-tion in the complex structure of E145Q with (NAG)2(Fig. 3D),
whereas it remains as the boat-form geometry in the structure
of E145Q/Y227F with (NAG)2(Fig. 3C). This observation
sug-gests that the -OH group of Tyr-227 contributes to the flip of
the N-acetyl group and the conformation change of⫺1-(NAG).
Our kinetic data showed that the mutant Y227F significantly decreased the catalytic activity (kcat) ⬃93%, confirming the important role of Tyr-227 in ChiNCTU2. In addition, the mutant Q225G decreased activity (kcat) 99% (Table 2),
indicat-ing that Gln-225 is essential in the catalytic turnover by
retriev-ing⫺1-(NAG) from the boat-form to the chair-form
confor-mation (46).
To complete a catalytic turnover, a new (NAG)2moiety at the
reducing end of chitin (or chitooligosaccharides) should slide
into the active site. Based on the structures of (NAG)4 and
(NAG)5complexes, the active site of ChiNCTU2 can rigidly
accommodate four NAG moieties, whereas the (NAG)nafter
the⫺2 subsite is flexible. The flexible end of the long-chain substrate with a dynamic motion might induce a conforma-tional change of the loop Gly-67—Thr-69 back to the open (pre-catalyzing-stage) position through the interaction with ⫺4-(NAG). The loop Ile-106–Val-112, hence, might be simul-taneously drawn back to the pre-catalyzing-stage position because of the hydrogen bonds between two associated loops. Concurrently, Gln-109 of the dynamic loop (Ile-106 –Val-112),
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
positioned between ⫺1 and ⫺2-(NAG), might shift cross ⫺2-(NAG) and back to the position between ⫺2 and ⫺3-(NAG), as we observed in the complex structure of D143A⫹(NAG)2(Fig. 4B, in blue) and interact with⫺2-(NAG). Such structural alternations are energetically unfavorable, which then triggers the loop to move forward 4.6 Å from the open (pre-catalyzing stage) form to the close (catalyzing stage) form again. As Gln-109 could interact with the C3 atom of the boat-form⫹1-(NAG) at this orientation, Gln-109 might act to bring NAG from the⫺2 to the ⫹1 subsite to begin another hydrolysis cycle. Thus, by the consecutive actions of the two dynamic loops, two sugar moieties can be processed in the cat-alytic site for the next turnover reaction. These observations provide a mechanistic explanation for ChiNCTU2 having an exochitinase activity.
The aromatic residue Trp167 interacting with⫺3-(NAG) in
S. marcescenschitinase A was been shown to be involved in processivity (57). The position of Trp167 in chitinase A corre-sponds structurally to that of Phe-43 in the catalytic center of ChiNCTU2, suggesting that Phe-43 might interact with ⫺3-(NAG), which agrees with the observation of the complex structure of E145G/Y227F with (NAG)4(Fig. 4B), to play a role in the processive action in ChiNCTU2. The conserved aromatic residue Trp-333 under the⫺1 subsite exhibiting the stacking force to stabilize chitin might also be involved in processivity.
Some mutations that are related to the catalytic activity, sub-strate binding, and product release based on our structural analysis have been examined with the activity assay and kinetics study to confirm their essential roles of key residues. Further investigation of mutagenesis and structures of the mutant com-plexes with chitooligosaccharides of an extended chain is in progress.
Acknowledgments—We are indebted to Yuch-Cheng Jean and the supporting staffs at beamlines BL13B1 and BL13C1 at the National Synchrotron Radiation Research Center and Jeyaraman Jeyakan-than, Masato Yoshimura, and Hirofumi Ishii at the Taiwan con-tracted beamline BL12B2 at SPring-8 for technical assistance. Por-tions of this research were carried out at the NSRRC-NCKU Protein Crystallography Laboratory at National Cheng Kung University.
REFERENCES
1. Cohen-Kupiec, R., and Chet, I. (1998) Curr. Opin. Biotechnol. 9, 270 –277 2. Flach, J., Pilet, P. E., and Jolle`s, P. (1992) Experientia 48, 701–716 3. Graham, L. S., and Sticklen, M. B. (1994) Can. J. Bot. 72, 1057–1083 4. Felse, P. A., and Panda, T. (1999) Appl. Microbiol. Biotechnol. 51, 141–151 5. Takaya, N., Yamazaki, D., Horiuchi, H., Ohta, A., and Takagi, M. (1998)
Microbiology 144,2647–2654
6. Li, H., and Greene, L. H. (2010) PLoS One 5, 1–11
7. Watanabe, T., Kobori, K., Miyashita, K., Fujii, T., Sakai, H., Uchida, M., and Tanaka, H. (1993) J. Biol. Chem. 268, 18567–18572
8. Jaques, A. K., Fukamizo, T., Hall, D., Barton, R. C., Escott, G. M., Parkin-son, T., Hitchcock, C. A., and Adams, D. J. (2003) Microbiology 149, 2931–2939
9. Horn, S. J., Sørbotten, A., Synstad, B., Sikorski, P., Sørlie, M., Vårum, K. M., and Eijsink, V. G. H. (2006) FEBS J. 273, 491–503
10. Perrakis, A., Tews, I., Dauter, Z., Oppenheim, A. B., Chet, I., Wilson, K. S., and Vorgias, C. E. (1994) Structure 2, 1169 –1180
11. Uchiyama, T., Katouno, F., Nikaidou, N., Nonaka, T., Sugiyama, J., and Watanabe, T. (2001) J. Biol. Chem. 276, 41343– 41349
12. Terwisscha van Scheltinga, A. C., Kalk, K. H., Beintema, J. J., and Dijkstra, B. W. (1994) Structure 2, 1181–1189
13. Terwisscha van Scheltinga, A. C., Armand, S., Kalk, K. H., Isogai, A., Hen-rissat, B., and Dijkstra, B. W. (1995) Biochemistry 34, 15619 –15623 14. Brunel, B., Perissol, C., Fernandez, M., Boeufgras, J. M., and Le Petit, J.
(1994) FEMS Microbiol. Ecol. 14, 331–342
15. Silo-Suh, L. A., Lethbridge, B. J., Raffel, S. J., He, H., Clardy, J., and Han-delsman, J. (1994) Appl. Environ. Microbiol. 60, 2023–2030
16. Emmert, E. A., and Handelsman, J. (1999) FEMS Microbiol. Lett. 171, 1–9 17. Pleban, S., Chernin, L., and Chet, I. (1997) Lett. Appl. Microbiol. 25,
284 –288
18. Wen, C. M., Tseng, C. S., Cheng, C. Y., and Li, Y. K. (2002) Biotechnol. Appl. Biochem. 35,213–219
19. Hofmann, K., Bucher, P., Falquet, L., and Bairoch, A. (1999) Nucleic Acids Res. 27,215–219
20. Henrissat, B., and Bairoch, A. (1996) Biochem. J. 316, 695– 696 21. Kuo, C. Y., Wu, Y. J., Hsieh, Y. C., Guan, H. H., Tsai, H. J., Lin, Y. H., Huang,
Y. C., Liu, M. Y., Li, Y. K., and Chen, C. J. (2006) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62,916 –919
22. Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276, 307–326 23. Terwilliger, T. C., and Berendzen, J. (1999) Acta Crystallogr. D Biol.
Crys-tallogr. 55,849 – 861
24. Terwilliger, T. C. (2000) Acta Crystallogr. D Biol. Crystallogr. 56, 965–972
25. Lamzin, V. S., and Wilson, K. S. (1993) Acta Crystallogr. D Biol. Crystal-logr. 49,129 –147
26. Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. A 47,110 –119
27. Bru¨nger, A. T. (1992) Nature 355, 472– 475
28. Bru¨nger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54,905–921
29. Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126 –2132
30. Winn, M. D., Isupov, M. N., and Murshudov, G. N. (2001) Acta Crystal-logr. D Biol. CrystalCrystal-logr. 57,122–133
31. Vagin, A., and Teplyakov, A. (1997) J. Appl. Crystallogr. 30, 1022–1025 32. Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M.
(1993) J. Appl. Crystallogr. 26, 283–291
33. Engh, R. A., and Huber, R. (1991) Acta Crystallogr. A 47, 392– 400 34. Ramachandran, G. N., and Sasisekharan, V. (1968) Adv. Protein Chem. 23,
283– 438
35. Hurtado-Guerrero, R., and van Aalten, D. M. F. (2007) Chem. Biol. 14, 589 –599
36. Cavada, B. S., Moreno, F. B., da Rocha, B. A., de Azevedo, W. F., Jr., Castello´n, R. E., Goersch, G. V., Nagano, C. S., de Souza, E. P., Nascimento, K. S., Radis-Baptista, G., Delatorre, P., Leroy, Y., Toyama, M. H., Pinto, V. P., Sampaio, A. H., Barettino, D., Debray, H., Calvete, J. J., and Sanz, L. (2006) FEBS J. 273, 3962–3974
37. Nakamura, T., Mine, S., Hagihara, Y., Ishikawa, K., Ikegami, T., and Ue-gaki, K. (2008) J. Mol. Biol. 381, 670 – 680
38. van Aalten, D. M., Synstad, B., Brurberg, M. B., Hough, E., Riise, B. W., Eijsink, V. G., and Wierenga, R. K. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5842–5847
39. Rao, F. V., Houston, D. R., Boot, R. G., Aerts, J. M., Sakuda, S., and van Aalten, D. M. (2003) J. Biol. Chem. 278, 20110 –20116
40. Hollis, T., Monzingo, A. F., Bortone, K., Ernst, S., Cox, R., and Robertus, J. D. (2000) Protein Sci. 9, 544 –551
41. Ikegami, T., Okada, T., Hashimoto, M., Seino, S., Watanabe, T., and Shirakawa, M. (2000) J. Biol. Chem. 275, 13654 –13661
42. Lonhienne, T., Mavromatis, K., Vorgias, C. E., Buchon, L., Gerday, C., and Bouriotis, V. (2001) J. Bacteriol. 183, 1773–1779
43. Hu, H., Wang, G., Yang, H., Zhou, J., Mo, L., Yang, K., Jin, C., Jin, C., and Rao, Z. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 939 –940 44. Songsiriritthigul, C., Pantoom, S., Aguda, A. H., Robinson, R. C., and
Sug-inta, W. (2008) J. Struct. Biol. 162, 491– 499
45. Vaaje-Kolstad, G., Vasella, A., Peter, M. G., Netter, C., Houston, D. R.,
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
Westereng, B., Synstad, B., Eijsink, V. G., and van Aalten, D. M. F. (2004) J. Biol. Chem. 279,3612–3619
46. Synstad, B., Gåseidnes, S., Van Aalten, D. M., Vriend, G., Nielsen, J. E., and Eijsink, V. G. H. (2004) Eur. J. Biochem. 271, 253–262
47. Vocadlo, D. J., Davies, G. J., Laine, R., and Withers, S. G. (2001) Nature
412,835– 838
48. Tews, I., Terwisscha van Scheltinga, A. C., Perrakis, A., Wilson, K. S., and Dijkstra, B. W. (1997) J. Am. Chem. Soc. 119, 7954 –7959
49. Brameld, K. A., and Goddard, W. A. (1998) J. Am. Chem. Soc. 120, 3571–3580
50. Vaaje-Kolstad, G., Houston, D. R., Rao, F. V., Peter, M. G., Synstad, B., van Aalten, D. M., and Eijsink, V. G. (2004) Biochim. Biophys. Acta 1696, 103–111
51. Dahiya, N., Tewari, R., and Hoondal, G. S. (2006) Appl. Microbiol. Biotech-nol. 71,773–782
52. Houston, D. R., Synstad, B., Eijsink, V. G., Stark, M. J., Eggleston, I. M., and van Aalten, D. M. (2004) J. Med. Chem. 47, 5713–5720
53. Katouno, F., Taguchi, M., Sakurai, K., Uchiyama, T., Nikaidou, N., Nonaka, T., Sugiyama, J., and Watanabe, T. (2004) J. Biochem. 136, 163–168
54. Watanabe, T., Ariga, Y., Sato, U., Toratani, T., Hashimoto, M., Nikaidou, N., Kezuka, Y., Nonaka, T., and Sugiyama, J. (2003) Biochem. J. 376, 237–244
55. Horn, S. J., Sikorski, P., Cederkvist, J. B., Vaaje-Kolstad, G., Sørlie, M., Synstad, B., Vriend, G., Vårum, K. M., and Eijsink, V. G. H. (2006) Proc. Natl. Acad. Sci. U.S.A. 103,18089 –18094
56. Sikorski, P., Sørbotten, A., Horn, S. J., Eijsink, V. G., and Vårum, K. M. (2006) Biochemistry 45, 9566 –9574
57. Zakariassen, H., Aam, B. B., Horn, S. J., Vårum, K. M., Sørlie, M., and Eijsink, V. G. H. (2009) J. Biol. Chem. 284, 10610 –10617
58. Knapp, S., Vocadle, D., Gao, Z., Kirk, B., Lou, J., and Withers, S. G. (1996) J. Am. Chem. Soc. 118,6804 – 6805
59. van Aalten, D. M., Komander, D., Synstad, B., Gåseidnes, S., Peter, M. G., and Eijsink, V. G. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 8979 – 8984
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/