Ab Initio Chemical Kinetics for H + NCN: Prediction of NCN Heat of
Formation and Reaction Product Branching via Doublet and Quartet
Surfaces
Wen-Shuang Teng,
†Lyudmila V. Moskaleva,
‡Hui-Lung Chen,*
,†and M. C. Lin*
,⊥,§†Department of Chemistry and Institute of Applied Chemistry, Chinese Culture University, Taipei 111, Taiwan ‡Institut für Angewandte und Physikalische Chemie, Universität Bremen, 28359 Bremen, Germany
⊥Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States
§Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung University, Hsinchu 30010, Taiwan
*
S Supporting InformationABSTRACT: The reaction of NCN with H atoms has been investigated by ab initio MO and RRKM theory calculations. The mechanisms for formation of major products on the doublet and quartet potential energy surfaces have been predicted at the CCSD(T) level of theory with the complete basis set limit. In addition, the heat of formation for NCN predicted at this rigorous level and those fromfive isogyric reactions are in close agreement with the best value based on the isodesmic process,3CCO + N2= 3NCN + CO, 109.4 kcal/mol, which lies within the two existing experimental values. The rate constants for the three
possible reaction channels, H + NCN→ CH + N2(kP1), HCN +4N (kQP1), and HNC +4N (kQP2), have been calculated in the
temperature range 298−3000 K. The results show that kP1 is significantly higher than kQP1 and kQP2and that the total rate constant agrees well with available experimental values in the whole temperature range studied. The kinetics of the reverse CH + N2reaction has also been revisited at the CCSD(T)/CBS level; the predicted total rate constants at 760 Torr Ar pressure can be represented by kr= 4.01× 10−15T0.90exp(−17.42 kcal mol−1/RT) cm3molecule−1s−1at T = 800−4000 K. The result agrees
closely with the most recent experimental data and the best theoretical result of Harding et al. (J. Phys. Chem. A 2008, 112, 522) as well as that of Moskaleva and Lin (Proc. Combust. Inst. 2000, 28, 2393) evaluated with a steady-state approximation after a coding error correction made in this study.
1. INTRODUCTION
The spin-forbidden process, CH(2Π) + N2→ HCN + N(4S),
first proposed by Fenimore in 1971 to be one of kinetically feasible processes which could be fast enough to account for prompt NO formation in the primary zone of hydrocarbon−air flames,1
was shown to be at least 2 orders of magnitude slower than the experimental kinetic data,2−5 based on high-level quantum-chemical and statistical-theory calculations by Cui et al. in 1999.6 The difficulty was soon resolved by Lin and co-workers7,8 who predicted the existence of the spin-allowed doublet low-energy path giving rise to H + NCN radical products via a multistep isomerization from the initial ring-adduct, -C(H)NN-, to the HNCN radical intermediate. Their predicted rate constants for the production of NCN and its
potential ensuing reactions involving H and CHx (x = 1−3)
radicals, based on the energetics computed at the G2M level of theory,7,8were shown to be able to account reasonably for the decay rates of CH radicals measured by Hanson and co-workers4 and the formation rates of N atoms detected by Lindackers et al.5 in their respective high-temperature shock tube studies. Since the publication in 2000 of the new prompt NO mechanism,7,8 which will be referred to as the FML (Fenimore−Moskaleva−Lin) mechanism hereafter, NCN has been detected in shock-heated mixtures of N2 and C2H6 (or
Received: March 25, 2013
Revised: June 7, 2013
Published: June 11, 2013
acetic anhydride) above 2000 K9and in several hydrocarbon− airflame experiments10,11attributable to CH + N2; the validity of our potential energy surface for NCN formation from the reaction has also been confirmed by other theoretical calculations.12,13
In the high-temperature shock tube studies of CH + N2 kinetics, hydrocarbon molecules such as C2H6 were typically employed as CH radical precursors through their successive thermal decomposition reactions, producing mixtures of H atoms and CHx (x = 1−3) radicals. The presence of these
reactive species unavoidably affects the quantitative interpreta-tion of experimentally measured CH decay rates and/or N and NCN radical production rates. In the modeling of measured CH and NCN concentration profiles by Hanson and co-workers9 in shock-heated mixtures of C2H6 and Ar, and by Lamoureux et al.11 in a fuel rich CH4−O2−N2 flame and a
stoichiometric C2H2−O2−N2flame, the effects of H + NCN
have been reported. The former group determined the rate constant for H + NCN to be 4.22× 10−11∼ 5.73 × 10−11cm3 mol−1s−1 in the narrow temperature range of 2378−2492 K with an estimated error of±100%. The values are consistent with the rate constant predicted by Moskaleva.7,14In the latter flame studies, a similar but temperature independent value, 4.2 × 10−11cm3mol−1s−1, was reported. Very recently, Lamoureux
et al.15 also reported the first absorption spectrum at high temperature (1660 K) measured by CRDS between 329 and 329.3 nm in a rich low-pressure C2H2−O2−N2 flame. The experimental absorption spectrum, compared to the LIF and simulated spectra, demonstrates that the principal intense feature at 329.13 nm is indubitably assignable to NCN. El Bakali et al.16had earlier simulated CH and NO concentration profiles in several hydrocarbon flames and analyzed the effects of NCN reactions inflames with H and other reactive species, whose kinetics had been predicted by Zhu et al.17−19
The emergence of NCN as one of the key intermediates of prompt NO formation by the FML mechanism has motivated recent experimental work on NCN chemistry. Several high-temperature studies of NCN related kinetics were carried out by Friedrichs and associates.20−22 Their advancement in quantitative detection of NCN behind shock waves in the parts per million (ppm) range opens a straightforward way to NCN detection inflames and to direct high-temperature rate constant measurements of NCN reactions much needed for implementation in the detailed combustion mechanisms.
Theoretically, Moskaleva14 noted that in the H + NCN reaction, unlike its reverse CH + N2process, a quartet surface can be accessed without the need for kinetically rate-limiting surface crossing. Thus, what is the contribution of the quartet surface reaction to the overall rate constant of H + NCN in addition to the known doublet surface,7,8,12,13 and what is the relative rate for H attacking a terminal N atom vs H attacking the central C atom respectively producing the HNC and HCN isomers? Another factor which affects strongly the rate of H + NCN production from CH + N2 is the heat of formation of
NCN. As the process is endothermic without an intrinsic barrier, the rate constant for NCN production is mainly enthalpically controlled. The heat of formation of NCN determined experimentally has a large deviation between the two reported values: 111.38 ± 0.6923and 107.4 ± 3.2 kcal/ mol.24 In this work, we shall attempt to utilize a series of isogyric and isodesmic reactions to help determine the best value which can rationalize existing experimental kinetic data
for H + NCN9,11 and those of the reverse CH + N2 reactions.2−5,9,11
2. COMPUTATIONAL METHOD
With the Gaussian 09 computational chemistry software25we performed molecular-orbital calculations involving density-functional theory with Becke’s three-parameter (B3) exchange functional and the Lee−Yang−Parr (LYP) correlation func-tional (B3LYP)26,27 using a 6-311++G(3df,2p) basis set. Frequency calculations were performed at the same level to check whether the obtained equilibrium structures are local minima or saddle points, among which the reactants, intermediates, and products possess all real frequencies, whereas transition states have only one imaginary frequency. Zero-point vibrational energy (ZPE) corrections are also computed at the B3LYP/6-311++G(3df,2p) level. Intrinsic reaction coordinate (IRC)28calculations were also performed to confirm the connection between the transition state and the designated intermediates. To obtain reliable energies, we performed single-point calculations employing a coupled-cluster technique with single and double excitations and evaluations by perturbation theory of triple contributions CCSD(T)29,30 based on those geometries optimized at the B3LYP/6-311++G(3df,2p) level. Further calculations were made to extrapolate the CCSD(T) results to the complete basis set limit byfitting the results obtained from different aug-cc-PVnZ basis sets and extrapolating to n → ∞. For this purpose, the“cardinal number” X was introduced, where X = 2, 3, 4, and 5 for n = D, T, Q, and 5, respectively. For the extrapolation to the complete basis set limit, we used the two-parameter exponential model,31E = a + b(X + 1)−4, tofit the data points. The highest level of theory used in this work is thus denoted as CCSD(T)/CBS//B3LYP/6-311++G(3df,2p). Un-less otherwise specified, the CCSD(T)/CBS single-point energies are used in the following discussion. The rate constants and branching ratios for the key product channels were computed with the variational transition-state theory (TST) and Rice−Ramsperger−Kassel−Marcus (RRKM) theory32,33 using the VariFlex program.34
3. RESULTS AND DISCUSSION
3.1. Heat of Formation of NCN. The best experimental values for the heat of formation of3NCN existing today, 111.38
± 0.69 kcal/mol23
and 107.4 ± 3.2 kcal/mol,24 differ by as much as 4 kcal/mol. The difference, as will be illustrated later, significantly affects the rate of NCN production from CH + N2.
To ascertain the present computational approach, we calculated the geometrical parameters and thermochemical data of the NCN radical. The C−N bond length and singlet−triplet splitting energy of NCN radical predicted by the hybrid density-functional B3LYP method with a modest 6-311+ +G(3df,2p) basis set are 1.224 Å and 1.02 eV, respectively; they are in satisfactory agreement with experimental values (1.232 Å and 1.01 eV).24 For the evaluation of heats of formation from heats of reaction, it is desirable to use well-balanced (isodesmic or at least isogyric)35−38 reactions whenever possible, so as to achieve the cancellation of errors inherent in the calculation for different types of molecular structures. The current state-of-the-art procedure for obtaining highly accurate heats of formation is the application of basis set extrapolation techniques combined with coupled-cluster methodology.
In Table 1, we present the predicted heats of formation of
3NCN radical from several model reaction energies computed
at the CCSD(T) level of theory using large basis sets (including aug-cc-PVTZ, aug-cc-PVQZ, and the complete basis set limit), along with specific experimental data from the literature. A good test for correlation balance is provided by the comparison of the results obtained at different correlation levels. For an ideally balanced reaction, all errors should cancel out; thus, a less scattered set of values should indicate a better balance. According to this criterion, the values of theΔfH° predicted for
reactions 1, 5, and 6 (Table 1) show the lowest scatter of values with respect to increasing the basis set size, indicative of the good balance (however, for reaction 1, the agreement seems to be coincidental because, as shown earlier, the calculated enthalpy of this reaction is quite sensitive to the level of theory employed).8 As shown in Table 1, the values of ΔfH° predicted for reactions 1, 2, 3, 4, 5, and 6 with the CBS limit are respectively 109.18, 108.94, 108.16, 108.75, 109.64, and 109.44 kcal/mol, which are close to the reported experimental values 111.38 ± 0.69 kcal/mol20 and 107.4 ± 3.2 kcal/mol21 and other values computed from experimental thermodynamic results 112.98 kcal/mol39 and 108.1 ± 0.7 kcal/mol.40 The average of our computed values using model reactions given in Table 1 is 109.01± 0.61 kcal/mol, which is very close to the value of 109.44 kcal/mol obtained by the isodesmic calculation from reaction 6, considered by the authors as most reliable, and
also close to the computed result 109.18 kcal/mol by Harding and Klippenstein,12who estimated the heat of reaction for H +
3NCN → CH + N
2to be −19.54 kcal/mol by multireference
second-order perturbation theory, CASPT2, combined with CCSD(T)/CBS single-point calculations. The value agrees exactly with our result presented in Table 1 (reaction 1). Accordingly, the heat of formation for 3NCN predicted with the isodesmic reaction, 109.4 kcal/mol, may be considered as quite reliable. We mention that in an earlier work of Moskaleva and Lin on the energetics of NCN isomers8a very similar value, 109.1 kcal/mol, was computed at the G2M level, using a somewhat different set of model reactions. Note that G2M is a modified G2 model chemistry providing an approximation to the RCCSD(T)/6-311++G(3df,2p) electronic energy.41 That earlier estimation had nevertheless a relatively large uncertainty of ±1.5 kcal/mol. Significantly, from reaction 1, the G2M method yields the NCN heat of formation as high as 111.1 kcal/mol, even outside the upper value of the given interval. In contrast, the current CCSD(T)/CBS approach gives a much smaller scatter of the calculated values ofΔfH° (NCN) from
the different model reactions, ca. 1.5 kcal/mol (Table 1). It should be noted that theΔfH° values predicted with the AVTZ and AVQZ basis sets for the same set of reactions in the table yield a broader range of values, ca. 4.5 and 2.0 kcal/mol, respectively. Thus, the extrapolation to the CBS limit seems to be the key to the success of our CCSD(T)/CBS methodology. Table 1. Isogyric Reactions Used To Calculate the Heats of Formation of the3NCN Moleculea
ΔrxnH° (kcal mol−1) ΔfH° of NCN (kcal mol−1)
reaction AVTZc AVQZc CBS limitd AVTZc AVQZc CBS limitd lit.
+ → + H 3NCN CH N2 (1) −19.33 −19.46 −19.54 108.97 109.10 109.18 111.0e + → + NCN OH HCN NO 3 (2) −61.21 −63.16 −64.22 105.93 107.88 108.94 109.18f + → + NCN O NCO NO 3 2 (3) −53.41 −55.22 −56.18 105.39 107.20 108.16 111.38± 0.69g + → + NCN NO NCO N 3 2 (4) −96.76 −98.68 −99.73 105.78 107.70 108.75 107.4± 3.2h + → + NCN NO NCO N O 3 2 2 (5) −65.00 −66.35 −67.28 107.36 108.71 109.64 112.98i + → + CCO N NCN CO 3 2 3 (6) −9.51 −9.14 −8.89 108.82 109.19 109.44 108.1± 0.7j + → + CCO N NCN CO 1 2 1 (7) 0.95 1.12 1.23 134.34 134.51 134.62 130.69± 3.2 ∼ 136.27k
aHeats of reactions are evaluated using different basis sets at the CCSD(T) level.bSome experimental data from the literature are also listed.bAll
energies were evaluated using geometries optimized at the B3LYP/6-311++G(3df,2p) level.cAVTZ = aug-cc-PVTZ and AVQZ = aug-cc-PVQZ.
dThe two-parameter exponential model, E = a + b(X + 1)−4, was used tofit the complete basis set limit.eReferences 7 and 8.fReference 12. g
Reference 23.hReference 24.iReference 39.jReference 40.kReference 42.
As the heat of formation of1CCO is also known from the study of Neumark and co-workers,42we have accordingly predicted the heat of formation of 1NCN using the isodesmic reaction
1CCO + N
2→1NCN + CO. A number of different basis sets
are also employed for comparison, including cc-PVTZ, aug-cc-PVQZ, and the CBS limit as listed in Table 1 (reaction 7). The predicted results are quite consistent; the deviation between the CCSD(T)/CBS (134.62 kcal/mol) and exper-imental values (130.69± 3.2∼136.27 kcal/mol) is less than ca. 1.7−3.9 kcal/mol. The CCSD(T)/CBS//B3LYP/6-311++G-(3df,2p) method appears to be quite reliable and is thus suitable for evaluation of the H + NCN kinetics and that of its reverse process.
3.2. Potential Energy Surfaces and Rate Constant Calculations for H + NCN with Doublet and Quartet Configurations. The doublet and quartet schematic diagrams of the key paths for NCN reactions with H atoms are depicted in Figure 1, in which the names and numbers of intermediates and transition states used here are consistent with our previously published work for the same structures using the G2M composite scheme.7,8,14 The profile of the doublet and
quartet PESs calculated at the CCSD(T)/CBS//B3LYP/6-311++G(3df,2p) level is shown in Figure 2, and the calculated energetics for the doublet and quartet species are tabulated in Table 2 (including our previously published data), in which the ZPE corrections and the energies with respect to the reactants (H + NCN) calculated at the CCSD(T)/CBS level, denoted as CRE, are presented. The optimized geometries for the doublet and quartet species involved in the H + NCN reaction at the B3LYP/6-311++G(3df,2p) level are shown in Figures S1 and S2 (Supporting Information).
3.2.1. Doublet Surface. On the doublet surface, shown in Figure 1 by dotted paths, the H + NCN reaction mayfirst form the primary adduct HNCN (INT3), which is energetically more stable than the reactants by 82.99 kcal/mol. After the formation of INT3, there are two distinct paths for formation of the final products CH + N2. Along the energetically most
favorable path, the INT3 can undergo a simple H-shift transition state, TS3, lying 8.82 kcal/mol below the reactants, forming a quasi-cyclic intermediate INT2 with an exothermicity of 43.20 kcal/mol. The reaction may proceed further through isomerization from INT2 to INT1 via TS2 with a barrier of
Figure 2.Calculated profiles on the potential energy surface for possible paths of the2H +3NCN reaction. Dotted and solid lines represent doublet
and quartet subsurfaces at the level of CCSD(T)/CBS//B3LYP/6-311++G(3df,2p). The data in parentheses are from our previously published work.7,8
18.34 kcal/mol by shortening the N−N bond from 1.566 Å in INT2 to 1.244 Å in INT1. Finally, the cyclic intermediate INT1can overcome an activation barrier of 19.19 kcal/mol at TS1by stretching its C−N bonds to ca. 1.59 Å to produce P1 (CH + N2), with an exothermicity of 19.54 kcal/mol. It is
evident from Figure 2 that the G2M relative energies with respect to H + NCN calculated in our earlier study7 are consistently lower by ca. 1.0−2.0 kcal/mol than the present CCSD(T)/CBS values. Along the less favorable reaction path, the INT3 adduct can cyclize via TS4 (lying 7.68 kcal/mol below the reactants) to form a three-membered ring intermediate, INT5, and proceed further by passing over two energy barriers (−4.58 and 0.66 kcal/mol for TS5 and TS6, respectively) to open the ring by breaking a C−N bond, to be followed by a simple H-shift from N atom to the C atom to produce P1 (CH + N2).
3.2.2. Quartet Surface. On the quartet surface, shown in Figure 1 by solid paths, the H + NCN reaction may generate the following possible products with the predicted enthalpy changes: HCN +4N (QP1),−17.14 kcal/mol and HNC +4N
(QP2), −3.55 kcal/mol. The former channel results from H addition to the central C atom, whereas the latter from H addition to the N site of NCN. The most favorable reaction path is R → QTS1 → QINT2 → QTS2 → QP1, with the highest energy barrier at QTS1, 5.51 kcal/mol, having an overall exothermicity of 17.14 kcal/mol. Because QTS2 lies only 0.1 kcal/mol above the reactants, clearly, QTS1 is expected to be rate-controlling. On the other hand, the energetically less competitive product QP2 (HNC +4N) could
be formed via the path R→ QTS3 → QINT3 → QTS4 → QP2, with the highest activation barrier at QTS3, 22.60 kcal/ mol, and an overall exothermicity of 3.55 kcal/mol. Both reaction paths on the quartet PES have been previously addressed by Moskaleva at the G2M level.14 G2M relative energies with respect to H + NCN are consistently somewhat higher than the current CCSD(T)/CBS values, though the differences in most cases do not exceed ca. 1.5 kcal/mol. Only for the energetically high lying QTS3 and QTS4 are the deviations of G2M energies larger, ca. 3.1 and 2.0 kcal/mol, respectively. Even though the relative energies of each stationary point on the quartet surface are mostly higher than those on the doublet surface, the former pathway with the quartet configuration may become competitive at higher temperatures.
3.2.3. Rate Constant Calculations. Variational TST and RRKM calculations have been carried out for this reaction with the VariFlex code34including the doublet and quartet reaction channels (omitting kinetically irrelevant intermediate steps) shown as follows:
In the above scheme we ignored the effect of INT4 and INT2 in our prediction of the rate constants for CH2+ N2→ H + NCN in both directions. The well depths of those intermediates, which appear in paths A and B, respectively, are much shallower than that of HNCN. In addition, under the prompt NO formation condition inflames above 1500 K, the effect of pressure is negligible and the values of rate constants for both forward and reverse directions are controlled mainly by the variational transition states near the dissociation threshold H···NCN. In the original work of Moskaleva and Lin,7 treated by the steady-state method, the INT2 was included in their canonical RRKM calculation, while, in the work of Harding and Klippenstein,12 by solving the master equation, INT2 was also ignored in the rate-constant calculation, similar to the present work. The results of the three calculations, as will be shown later, all agree closely with existing experimental data. The energies used in the calculation are plotted in Figure 2, and the vibrational frequencies and moments of inertia are listed in Table S1. The Lennard-Jones (LJ) parameters employed in the calculation are as follows: for Ar,43σ = 3.465 Å, ε/k = 113.50 K, and for HNCN,14σ = 4.560 Å,ε/k = 48.20 K. For the variational rate constant calculations by the VariFlex code, a statistical treatment of the transitional-mode contributions to the transition-state partition functions is performed variationally. The energy-transfer process was computed on the basis of the exponential down model with a ⟨ΔE⟩down value (the mean energy transferred per collision) of
400 cm−1for Ar. In addition, the estimate of the transitional-mode contribution to the transition-state number of states for a given energy was evaluated via Monte Carlo integration with 10000 configuration numbers. In order to achieve convergence in the integration over the energy range, an energy grain size of 120 cm−1was used. The total angular momentum J covered the Table 2. ZPE (hartree), Total Energies (TE, hartree), and
Relative Energies (RE, kcal/mol) of Reactants,
Intermediates, Transition States, and Products Calculated at the CCSD(T)/CBS//B3LYP/6-311++G(3df,2p) Level (CTE and CRE) for the H + NCN Systema
ZPEb CTE + ZPE CREc Moskalevae
R(H+NCN) 0.008506 −147.806644 00.00 0.00 INT1 0.019301 −147.851259 −28.00 −29.50 INT2 0.019485 −147.875485 −43.20 −45.00 INT3 0.019610 −147.938900 −82.99 −84.01 INT4 0.019409 −147.876131 −43.60 INT5 0.018971 −147.827919 −13.35 QINT3 0.018355 −147.812205 −3.49 −2.24 QINT2 0.017780 −147.840530 −21.26 −19.77 TS1 0.014276 −147.820694 −8.82 −10.70 TS2 0.017292 −147.846258 −24.86 −26.20 TS3 0.015554 −147.820686 −8.81 −9.80 TS4 0.014835 −147.805595 0.66 TS5 0.016798 −147.813942 −4.58 TS6 0.016112 −147.818888 −7.68 QTS1 0.009912 −147.797868 5.51 6.84 QTS2 0.016353 −147.806487 0.10 0.52 QTS3 0.010055 −147.770635 22.60 25.74 QTS4 0.015862 −147.791408 9.56 11.62 P1 (CH + N2) 0.012021 −147.837789 −19.54 −21.50 QP1 (HCN + N) 0.016308 −147.833952 −17.14 −16.34 QP2 (HNC + N) 0.014552 −147.812298 −3.55 −3.96
aThe data of our previously published work are also included.b
Zero-point energy (au) at the B3LYP/6-311++G(3df,2p) level. cRelative energy (kcal/mol) with respect to the reactants.dThe two-parameter exponential model, E = a + b(X + 1)−4, was used tofit the complete basis set limit.eReferences 7 and 14.
range from 1 to 250 in steps of 10 for the E, J-resolved calculation.
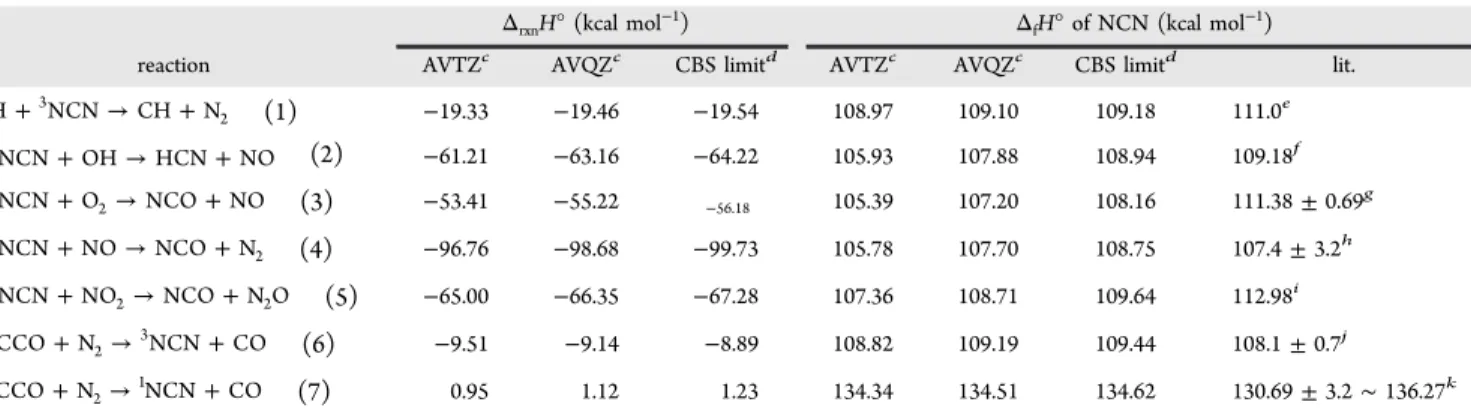
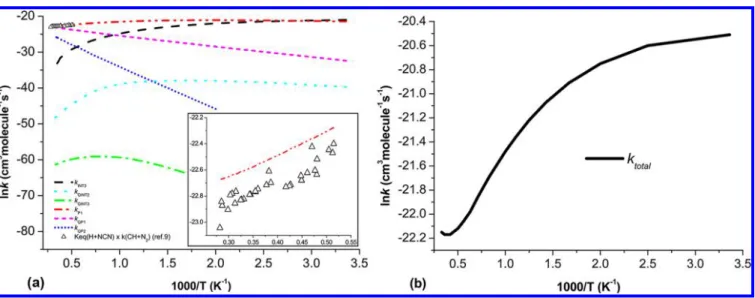
The predicted values for individual rate constants (kINT3,
kQINT2, kQINT3, kP1, kQP1, and kQP2) and total rate constants (ktotal = kINT3+ kQINT2+ kQINT3+ kP1+ kQP1+ kQP2) for reaction of H
+ NCN at 760 Torr Ar pressure in the temperature range of 298−3000 K are shown in parts a and b, respectively, of Figure 3. As we can see from Figure 3a, the values of kINT3and kQINT2 substantially decrease as the temperature increases from 298 to 3000 K, indicating that collisional deactivation giving INT3 and QINT2 becomes increasingly less competitive at higher temperatures. In addition, the calculated results reveal that the rate constant kP1giving CH + N2via the doublet paths is
more favorable than kQP1and kQP2of the quartet paths in the whole temperature range. In order to further ascertain the computational approach used in rate constant predictions, we also separately calculated the equilibrium constant Keqfor H +
NCN→ CH + N2to convert the experimental rate constants of Vasudevan et al.9for CH + N2→ H + NCN to those for H +
NCN→ CH + N2; the results given by open triangles are also
presented in Figure 3a. From the figure it is evident that our predicted values of kP1 are in reasonable agreement with the converted experimental results in the temperature range of 1943−3543 K. It is also clear from the expanded inset of the figure that the H + NCN → CH + N2has negative temperature
dependence, at least in the temperature range studied. Second, there is a small (∼15%) deviation between theory and experiment which may be attributed in part to the theoretical overprediction and in part to the large error (estimated to be ±25% at ∼3350 K and ±35% at ∼2100 K9
) from the modeling of the measured CH profiles involving the complex CHx and
NCN reactions unavoidably present in the complex chemical system, as alluded to before.
The branching ratios for the four primary reaction channels on the doublet and quartet surfaces (kINT3, kP1, kQP1, and kQP2)
at 760 Torr Ar pressure in the temperature range of 298−3000 K are shown in Figure 4. The stabilization rate constant kINT3in
the low-temperature range of 298−350 K clearly has the largest value, accounting for 0.61−0.49 of the total rate. However, at higher temperatures (350−1100 K), CH + N2(P1) formation
becomes dominant, accounting for 0.49−0.95 of the total rate.
Besides, it is also clear that the formation of HCN +4N (QP1)
becomes more competitive as the temperature goes beyond 1100 K.
We summarize the rate expressions for each individual rate constant (kINT3, kQINT2, kQINT3, kP1, kQP1, and kQP2) at seven specific pressures between 5 Torr and 100 atm in the temperature range of 298−3000 K in Table 3. In this temperature range, the values of kINT3, kQINT2, and kQINT3
have strong pressure dependencies, as one would expect; the values of kP1, kQP1, and kQP2, however, are pressure independent
below 100 atm. The predicted total rate constant, ktotal, at 760 Torr Ar pressure can be represented by the following: ktotal =
(7.68× 10−8)T−0.75exp(0.16 kcal mol−1/RT) at T = 298−3000 K, in units of cm3 molecule−1 s−1. Comparison with the previous experimental and computational values has also been made (as shown in Table 4 and Figure 5); the predicted specific rate constants at different temperatures (2378, 2420, 2455, 2491, and 2492 K) with the corresponding pressures 0.421, 0.413, 0.437, 0.401, and 0.447 atm for the H + NCN→ HCN + N reaction (kQP1) vary within the small range of values, 6.60
Figure 3.Predicted rate constants of (a) kINT3, kQINT2, kQINT3, kP1, kQP1, kQP2, and converted experimental values from ref 9; (inset) expanded view of
the high T region of the graph; and (b) the total rate constants (ktotal= kINT3+ kQINT2+ kQINT3+ kP1+ kQP1+ kQP2) at Ar pressure of 760 Torr in the
temperature range of 298−3000 K.
Figure 4.Predicted branching ratios for the principal reaction channels of H + NCN reactions at 760 Torr Ar pressure in the temperature range of 298−3000 K.
× 10−11 ∼ 7.09 × 10−11 cm3 molecule−1 s−1, which are
consistent with our previously predicted values7as well as with the temperature independent result, 4.2 × 10−11 cm3
molecule−1 s−1, by Lamoureux et al.11 in a flame study and with the kinetically modeled values from the experiment of Hanson and co-workers (4.22 × 10−11 ∼ 5.73 × 10−11 cm3 molecule−1s−1, with an uncertainty estimated to be a factor of 2).9
3.3. Rate Constant for CH + N2→ H + NCN Revisited.
From our calculated potential energy surface, it is clear that there are two different paths for the reverse reaction CH + N2 → H + NCN. Path A, CH + N2→ TS1 → INT1 → TS2 →
INT2→ TS3 → INT3 → H + NCN, was proposed in our previous computational work.7 Another potential path, B,
which follows the route CH + N2→ TS6 → INT4 → TS5 →
INT5→ TS4 → INT3 → H + NCN, forming the same final products, has been proposed by Berman et al.13and Harding and Klippenstein.12We compare the potential energy surface with that of Berman et al.,13 in which the CH + N2reaction
starts with a barrierless association to form HCNN and then proceeds through a simple H-shift transition state with a very high energy barrier, 83.6 kcal/mol, to form INT4. In our calculation, however, we found another more attainable transition state, TS6, connecting directly to the INT4, with a much lower activation barrier of 20.20 kcal/mol, which is very close to the 19.5 kcal/mol barrier reported by Harding and Klippenstein,12 who indicated that the contribution from this path might increase at higher temperatures.
The predicted rate constants for the CH + N2reaction at an
Ar pressure of 760 Torr in the temperature range of 800−4000 K are shown in Figure 6, and the rate expressions for the two paths and their sum can be represented as follows:
Table 3. Predicted Rate EquationsaofkINT3, kQINT3, kQINT2,
kP1, kQP1, andkQP2at the Ar Pressures of 5 Torr, 100 Torr,
380 Torr, 760 Torr, 10 atm, 50 atm, and 100 atm in the Temperature Range of 298−3000 K for the H + NCN Reaction reaction P A n B kINT3 5 Torr 7.07× 1012 −9.03 −4.42 100 Torr 2.95× 1017 −9.58 −5.25 380 Torr 3.86× 1018 −9.50 −6.19 760 Torr 2.98× 1018 −9.28 −6.56 10 atm 5.89× 1015 −7.99 −6.95 50 atm 5.35× 1012 −6.86 −6.60 100 atm 1.52× 1011 −6.32 −6.30 kQINT2 5 Torr 6.52× 1018 −11.85 −13.38 100 Torr 2.29× 1020 −11.90 −13.22 380 Torr 2.22× 1021 −12.00 −13.10 760 Torr 1.04× 1022 −12.08 −13.07 10 atm 2.78× 1025 −12.61 −13.56 50 atm 1.92× 1028 −13.02 −14.89 100 atm 1.14× 1029 −13.05 −15.56 kQINT3 5 Torr 2.49× 109 −10.44 −28.42 100 Torr 5.41× 1010 −10.45 −28.36 380 Torr 2.80× 1011 −10.48 −28.28 760 Torr 8.32× 1011 −10.53 −28.24 10 atm 1.37× 1014 −10.82 −28.06 50 atm 4.30× 1016 −11.23 −27.94 100 atm 1.25× 1018 −11.50 −27.95 kP1 all pressures 6.27× 10−05 −1.61 −1.49 kQP1 all pressures 8.24× 10−12 0.41 −5.46 kQP2 all pressures 4.75× 10−13 0.79 −22.24
aRate constants are represented by k = ATnexp(B kcal mol−1/RT) in
units of cm3molecule−1s−1.
Table 4. Predicted Rate Constantsafor the H + NCN→ HCN + N Reaction (kQP1) at the He Pressures of 0.421,
0.413, 0.437, 0.401, and 0.447 atm in the Temperature Range of 2378, 2420, 2455, 2491, and 2492 K and the Corresponding Experimental Data from the Literature
T(K) P(atm) kQP1 expb 2378 0.421 6.60× 10−11 4.22× 10−11 2420 0.413 6.78× 10−11 5.45× 10−11 2455 0.437 6.93× 10−11 5.58× 10−11 2491 0.401 7.08× 10−11 5.73× 10−11 2492 0.447 7.09× 10−11 5.73× 10−11
aRate constants are in units of cm3molecule−1 s−1.bUncertainty of
this data was estimated to be a factor of 2 (ref 9).
Figure 5.Rate coefficient data for H + NCN → HCN + N: solid squares, this work; red triangle, Vasudevan et al.,9uncertainty in their data was estimated to be a factor of 2; solid line, Moskaleva et al.;7and dashed line, Lamoureux et al.11
Figure 6. Predicted rate constants of the contributions from two different pathways (paths A and B) for the CH + N2→ H + NCN
reaction. The pressure is 760 Torr, and the temperature range is 800− 4000 K.
• kA = (1.93 × 10−14)T0.69 exp(−17.72 kcal mol−1/RT)
cm3molecule−1s−1
• kB= (2.53× 10−18)T1.61exp(−18.56 kcal mol−1/RT) cm3
molecule−1s−1
• kA&B= (4.01 × 10−15)T0.90 exp(−17.42 kcal mol−1/RT)
cm3molecule−1s−1
in which kA and kBrepresent the individual rate constants for
the formation of H + NCN via paths A and B, respectively, and kA&Bis the total rate constant (kA+ kB). The computed results
clearly show that, under the conditions studied, path A, proposed in 2000 by Moskaleva et al.,7,8 predominates. The calculated total rate constant for CH + N2 is compared in Figure 7 with experimental and theoretical data available in the
literature3−5,9,12,16,44 including our previously predicted value (M&L, ref 7) and the present results (this work and M&L corrected). In the course of preparation of this article, we found an error in the original RRKM code used for the calculation of the rate constant of CH + N2,7which unfortunately originated
from a programming typo. The old result based on the PES calculated at the G2M level of theory, which had noticeably underpredicted the NCN production rate, has now been corrected (M&L corrected), and the good agreement with the present calculation on the basis of CCSD(T)/CBS PES is evident as shown in Figure 7. Our newly predicted results based on the CCSD(T)/CBS energies with ME solution in the RRKM calculation (this work; black solid line) and the coding-error corrected based on G2M energies with the steady-state approximation in the RRKM calculation (M&L corrected; green dash line) both agree well with the most recent experimental work of Hanson and co-workers9 and the best theoretical result from Harding and Klippenstein12 in the temperature range of 800−4000 K. The reduction of the reaction endothermicity from 21.5 to 19.5 kcal/mol in the present work gives rise to an especially good agreement with the best experimental and theoretical results to date.
4. CONCLUSION
The potential energy surfaces and mechanisms for the reaction of NCN with H and its reverse CH + N2reaction have been
computed at the CCSD(T)/CBS level of theory; the rate constants for the possible reaction channels have been predicted. The predicted total rate constant for the H + NCN reaction, ktotal, at 760 Torr Ar pressure can be
represented by the following: ktotal = (7.68 × 10−8)T−0.75 exp(0.16 kcal mol−1/RT) cm3 molecule−1 s−1 at T = 298− 3000. The result agrees well with available experimental data (4.22× 10−11∼ 5.73 × 10−11cm3mol−1s−1). In addition, our newly predicted rate constants for the CH + N2reaction at 760 Torr Ar pressure in the temperature range of 800−4000 K are also given; the computed result agrees well with the most recent experimental data of Hanson and co-workers9 and the theoretical prediction of Harding and Klippenstein,12as well as with that of the (corrected) earlier prediction of Moskaleva and Lin7using the best heat of formation of NCN obtained with the isodesmic reaction, CCO + N2→ NCN + CO, 109.4 kcal/mol
(which was shown to be in close agreement with the results predicted withfive different isogyric processes).
■
ASSOCIATED CONTENT*
S Supporting InformationFrequencies and moments of inertia for the species involved in the reaction of H + NCN, calculated at the B3LYP/6-311+ +G(3df,2p) level; and calculated structural parameters of all involved stationary geometries of the H + NCN reaction at the B3LYP/6-311++G(3df,2p) level. This material is available free of charge via the Internet at http://pubs.acs.org.
■
AUTHOR INFORMATIONCorresponding Author
*E-mail: [email protected] and [email protected]. Tel: +886-2-28610511, ext 25313. Fax: +886-2-28617006.
Notes
The authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSWe would like to acknowledge support from (1) Taiwan’s National Science Council for H.-L.C. (under Grant Number NSC 100-2113-M-034-002-MY2) and M.C.L. (the support from Taiwan’s MOE ATU program as well as the National Science Council and the Taiwan Semiconductor Manufacturing Co. for the NSC-distinguished visiting professorship and the TSMC distinguished professorship, respectively, at the Center for Interdisciplinary Molecular Science, National Chiao Tung University), (2) the financial support by Chinese Culture University, and (3) Taiwan’s National Center for High-performance Computing, for the use of computer time.
■
REFERENCES(1) Fenimore, C. P. Formation of Nitric Oxide in Premixed Hydrocarbon Flames. Proc. Combust. Inst. 1977, 13, 373−380.
(2) Blauwens, J.; Smets, B.; Peeters, J. Mechanism of‘‘Prompt” NO Formation in Hydrocarbon Flames. Proc. Combust. Inst. 1977, 16, 1055−1064.
(3) Matsui, Y.; Nomaguchi, T. Spectroscopic Study of Prompt Nitrogen Oxide Formation Mechanism in Hydrocarbon-Air Flames. Combust. Flame 1978, 32, 205−214.
(4) Dean, A. J.; Hanson, R. K.; Bowman, C. T. A Shock Tube Study of Reactions of Carbon Atoms and Methylidyne with Nitric Oxide Including Product Channel Measurements. J. Phys. Chem. 1991, 95, 3180−3189.
(5) Lindackers, D.; Burmeister, M.; Roth, P. Perturbation Studies of High Temperature C and CH Reactions with N2 and NO. Proc.
Combust. Inst. 1991, 23, 251−257. Figure 7. Plot of the theoretical predictions and experimental
measurements for the CH + N2 → H + NCN rate constant. The
(6) Cui, Q.; Morokuma, K.; Bowman, J. M.; Klippenstein, S. The Spin-Forbidden Reaction CH(2Π) + N
2→ HCN + N(4S) Revisited.
II. Nonadiabatic Transition State Theory and Application. J. Chem. Phys. 1999, 110, 9469−9482.
(7) Moskaleva, L. V.; Lin, M. C. The Spin-Conserved Reaction CH + N2→ H + NCN: A Major Pathway to Prompt NO Studied by
Quantum/Statistical Theory Calculations and Kinetic Modeling of Rate Constant. Proc. Combust. Inst. 2000, 28, 2393−2401.
(8) (a) Moskaleva, L. V.; Xia, W.-S.; Lin, M. C. The CH + N2
Reaction over the Ground Electronic Doublet Potential Energy Surface: A Detailed Transition State Search. Chem. Phys. Lett. 2000, 331, 269−277. (b) Moskaleva, L. V.; Lin, M. C. Computational Study on the Energetics of NCN Isomers and the Kinetics of the C + N2= N
+ CN Reaction. J. Phys. Chem. A 2001, 105, 4156−4163.
(9) Vasudevan, V.; Hanson, R. K.; Bowman, C. T.; Golden, D. M.; Davidson, D. F. Shock Tube Study of the Reaction of CH with N2:
Overall Rate and Branching Ratio. J. Phys. Chem. A 2007, 111, 11818− 11830.
(10) Smith, G. P. Evidence of NCN as a Flame Intermediate for Prompt NO. Chem. Phys. Lett. 2003, 367, 541.
(11) Lamoureux, N.; Desgroux, P.; El Bakli, A.; Pauwels, J. F. Experimental and Numerical Study of the Role of NCN in Prompt-NO Formation in Low-Pressure CH4−O2−N2 and C2H2−O2−N2
Flames. Combust. Flame 2010, 157, 1929−1941.
(12) Harding, L. B.; Klippenstein, S. J. Kinetics of CH + N2Revisited
with Multireference Methods. J. Phys. Chem. A 2008, 112, 522−532. (13) Berman, M. R.; Tsuchiya, T.; Gregusova, A.; Perera, A.; Bartlett, R. J. HNNC Radical and Its Role in the CH + N2Reaction. J. Phys.
Chem. A 2007, 111, 6894−6899.
(14) Moskaleva, L. V. Ph.D. Dissertation, Emory University, 2001. (15) Lamoureux, N.; Western, C. M.; Mercier, X.; Desgroux, P. Reinvestigation of the Spectroscopy of the A3Π
uX3Σg−Transition of
the NCN Radical at High Temperature: Application to Quantitative NCN Measurement in Flames. Combust. Flame 2013, 160, 755−765. (16) El Bakali, A.; Pillier, L.; Desgroux, P.; Lefort, B.; Gasnot, L.; Pauwels, J. F.; da Costa, I. NO Prediction in Natural Gas Flames Using GDF-Kin®3.0 Mechanism NCN and HCN Contribution to Prompt-NO Formation. Fuel 2006, 85, 896−909.
(17) Zhu, R. S.; Lin, M. C. Ab Initio Study of the Oxidation of NCN by O2. Int. J. Chem. Kinet. 2005, 37, 593−598.
(18) Zhu, R. S.; Lin, M. C. Ab Initio Study on the Oxidation of NCN by O (3P): Prediction of the Total Rate Constant and Product
Branching Ratios. J. Phys. Chem. A 2007, 111, 6766−6771.
(19) Zhu, R. S.; Nguyen, H. M. T. Ab Initio Study on the Oxidation of NCN by OH: Prediction of the Individual and Total Rate Constants. J. Phys. Chem. A 2009, 113, 298−304.
(20) Dammeier, J.; Friedrichs, G. Direct Measurements of the Rate Constants of the Reactions NCN + NO and NCN + NO2 Behind
Shock Waves. J. Phys. Chem. A 2011, 115, 14382−14390.
(21) Dammeier, J.; Faßheber, N.; Friedrichs, G. Direct Measure-ments of the High Temperature Rate Constants of the Reactions NCN + O, NCN + NCN, and NCN + M. Phys. Chem. Chem. Phys. 2012, 14, 1030−1037.
(22) Friedrichs, G.; Dammeier, J.; Faßheber, N. Shock Tube Study on the Reactions NCN + H and NCN + O. 7th International Conference on Chemical Kinetics; MIT, Cambridge, USA, July 2011.
(23) Bise, R. T.; Choi, H.; Neumark, D. M. Photodissociation Dynamics of the Singlet and Triplet States of the NCN Radical. J. Chem. Phys. 1999, 111, 4923−4933.
(24) Clifford, E. P.; Wenthold, P. G.; Lineberger, W. C.; Petersson, G. A.; Ellison, G. B. Photoelectron Spectroscopy of the NCN−and HNCN−Ions. J. Phys. Chem. A 1997, 101, 4338−4345.
(25) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin,
K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N. J.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, 2009.
(26) (a) Becke, A. D. Density-Functional Thermochemistry. I. The Effect of the Exchange-Only Gradient Correction. J. Chem. Phys. 1992, 96, 2155−2160. (b) Becke, A. D. Density-Functional Thermochem-istry. II. The Effect of the Perdew−Wang Generalized-Gradient Correlation Correction. J. Chem. Phys. 1992, 97, 9173−9177. (c) Becke, A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648−5652.
(27) Lee, C.; Yang, W.; Parr, R. G. Development of the Colle− Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. 1988, B37, 785−789.
(28) Gonzalez, C.; Schlegel, H. B. An Improved Algorithm for Reaction Path Following. J. Phys. Chem. 1989, 90, 2154−2161.
(29) Lee, T. J.; Scuseria, G. In Quantum-mechanical Electronic Structure Calculations with Chemical Accuracy; Langhoff, S. F., Ed.; Kluwer: Dordrecht, Netherlands, 1995.
(30) Knowles, P. J.; Hampel, C.; Werner, H.-J. Coupled Cluster Theory for High Spin, Open Shell Reference Wave Functions. J. Chem. Phys. 1993, 99, 5219−5227.
(31) Martin, J. M. L.; Uzan, O. Basis Set Convergence in Second-Row Compounds. The Importance of Core Polarization Functions. Chem. Phys. Lett. 1998, 282, 16−24.
(32) (a) Klippenstein, S. J.; Marcus, R. A. High Pressure Rate Constants for Unimolecular Dissociation/Free Radical Recombina-tion: Determination of the Quantum Correction via Quantum Monte Carlo Path Integration. J. Chem. Phys. 1987, 87, 3410−3417. (b) Klippenstein, S. J. Implementation of RRKM Theory for Highly Flexible Transition States with a Bond Length as the Reaction Coordinate. Chem. Phys. Lett. 1990, 170, 71. (c) Klippenstein, S. J. Variational Optimizations in the Rice−Ramsperger−Kassel−Marcus Theory Calculations for Unimolecular Dissociations with No Reverse Barrier. J. Chem. Phys. 1992, 96, 367−371. (d) Klippenstein, S. J. An Efficient Procedure for Evaluating the Number of Available States within a Variably Defined Reaction Coordinate Framework. J. Phys. Chem. 1994, 98, 11459−11464.
(33) (a) Wardlaw, D. M.; Marcus, R. A. RRKM Reaction Rate Theory for Transition States of Any Looseness. Chem. Phys. Lett. 1984, 110, 230−234. (b) Wardlaw, D. M.; Marcus, R. A. Unimolecular Reaction Rate Theory for Transition States of Partial Looseness. II. Implementation and Analysis with Applications to NO2 and C2H6
Dissociations. J. Chem. Phys. 1985, 83, 3462−3480.
(34) Klippenstein, S. J.; Wagner, A. F.; Dunbar, R. C.; Wardlaw, D. M.; Robertson, S. H. VARIFLEX, version 1.00; Argonne National Laboratory: Argonne, IL, 1999.
(35) Hehre, W. J.; Ditchfield, R.; Radom, L.; Pople, J. A. Molecular Orbital Theory of the Electronic Structure of Organic Compounds. V. Molecular Theory of Bond Separation. J. Am. Chem. Soc. 1970, 92, 4796−4801.
(36) Radom, L.; Hehre, W. J.; Pople, J. A. Molecular Orbital Theory of the Electronic Structure of Organic Compounds. VII. Systematic Study of Energies, Conformations, and Bond Interactions. J. Am. Chem. Soc. 1971, 93, 289−300.
(37) Hehre, W. J.; Radom, L.; Schleyer, P. v. R.; Pople, J. A. Ab Initio Molecular Orbital Theory; John Wiley & Sons: New York, 1986.
(38) Minkin, V. I. Glossary of Terms Used in Theoretical Organic Chemistry. Pure Appl. Chem. 1999, 71, 1919−1981.
(39) Chase, M. W., Jr. NIST-JANAF Thermochemical Tables, 4th ed.; American Chemical Society: Washington, DC, American Institute of Physics for the National Institute of Standards and Technology: Woodbury, New York, 1998.
(40) Clifford, E. P.; Wenthold, P. G.; Lineberger, W. C.; Petersson, G. A.; Broadus, K. M.; Kass, S. R.; Kato, S.; DePuy, C. H.; Bierbaum, V. M.; Ellison, G. B. Properties of Diazocarbene [CNN] and the Diazomethyl Radical [HCNN] via Ion Chemistry and Spectroscopy. J. Phys. Chem. A 1998, 102, 7100−7112.
(41) Mebel, A. M.; Morokuma, K.; Lin, M. C. Modification of the
GAUSSIAN-2 Theoretical Model: The Use of Coupled-Cluster Energies,
Density-Functional Geometries, and Frequencies. J. Chem. Phys. 1995, 103, 7414−7421.
(42) Choi, H.; Mordaunt, D. H.; Bise, R. T.; Taylor, T. R.; Neumark, D. M. Photodissociation of Triplet and Singlet States of the CCO Radical. J. Chem. Phys. 1998, 108, 4070−4078.
(43) Mourits, F. M.; Rummens, F. H. A. A Critical Evaluation of Lennard−Jones and Stockmayer Potential Parameters and of Some Correlation Methods. Can. J. Chem. 1977, 55, 3007−3020.
(44) Baulch, D. L.; Cobos, C. J.; Cox, R. A.; Frank, P.; Hayman, G.; Just, Th.; Kerr, J. A.; Murrells, T.; Pilling, M. J.; Troe, J.; Walker, R. W.; Warnatz, J. Evaluated Kinetic Data for Combustion Modeling. Supplement I. J. Phys. Chem. Ref. Data 1994, 23, 847−848.