Resource
Epigenomic Analysis of
Multilineage Differentiation of
Human Embryonic Stem Cells
Wei Xie,
1Matthew D. Schultz,
2Ryan Lister,
2,14Zhonggang Hou,
3Nisha Rajagopal,
1Pradipta Ray,
11John W. Whitaker,
4Shulan Tian,
3R. David Hawkins,
1,15Danny Leung,
1Hongbo Yang,
7Tao Wang,
4Ah Young Lee,
1Scott A. Swanson,
3Jiuchun Zhang,
3,8Yun Zhu,
4Audrey Kim,
1Joseph R. Nery,
2Mark A. Urich,
2Samantha Kuan,
1Chia-an Yen,
1Sarit Klugman,
1Pengzhi Yu,
3Kran Suknuntha,
12Nicholas E. Propson,
3Huaming Chen,
2Lee E. Edsall,
1Ulrich Wagner,
1Yan Li,
1Zhen Ye,
1Ashwinikumar Kulkarni,
11Zhenyu Xuan,
11Wen-Yu Chung,
11,16Neil C. Chi,
7Jessica E. Antosiewicz-Bourget,
3Igor Slukvin,
8,9,12Ron Stewart,
3Michael Q. Zhang,
11,13Wei Wang,
4,6James A. Thomson,
3,9,10,*
Joseph R. Ecker,
2,*
and Bing Ren
1,5,*
1Ludwig Institute for Cancer Research, La Jolla, CA 92093, USA
2Genomic Analysis Laboratory, Howard Hughes Medical Institute, The Salk Institute for Biological Studies, La Jolla, CA 92037, USA 3Morgridge Institute for Research, Madison, WI 53707, USA
4Department of Chemistry and Biochemistry
5Department of Cellular and Molecular Medicine, Institute of Genomic Medicine and Moores Cancer Center 6Department of Cellular and Molecular Medicine
7Department of Medicine, Division of Cardiology
University of California, San Diego, La Jolla, CA 92093, USA
8Wisconsin National Primate Research Center 9Department of Cell and Regenerative Biology
University of Wisconsin-Madison, Madison, WI 53715, USA
10Department of Molecular, Cellular, and Developmental Biology, University of California, Santa Barbara, Santa Barbara, CA 93106, USA 11Department of Molecular and Cell Biology, Center for Systems Biology, The University of Texas at Dallas, Richardson, TX 75080, USA 12Department of Pathology and Laboratory Medicine, University of Wisconsin Medical School, Madison, WI 53792, USA
13Bioinformatics Division, Center for Synthetic and Systems Biology, Tsinghua National Laboratory for Information Science and Technology,
Tsinghua University, Beijing 100084, China
14Present address: Plant Energy Biology (ARC CoE) and Computational Systems Biology (WA CoE), School of Chemistry and Biochemistry,
The University of Western Australia, Perth, WA 6009, Australia
15Present address: Division of Medical Genetics, Department of Medicine, Department of Genome Sciences, University of Washington,
Seattle, WA 98195, USA
16Present address: Department of Computer Science and Information Engineering, National Kaohsiung University of Applied Sciences,
Kaohsiung 807, Taiwan
*Correspondence:jthomson@morgridgeinstitute.org(J.A.T.),ecker@salk.edu(J.R.E.),biren@ucsd.edu(B.R.) http://dx.doi.org/10.1016/j.cell.2013.04.022
SUMMARY
Epigenetic mechanisms have been proposed to
play crucial roles in mammalian development, but
their precise functions are only partially understood.
To investigate epigenetic regulation of embryonic
development, we differentiated human embryonic
stem cells into mesendoderm, neural progenitor
cells, trophoblast-like cells, and mesenchymal stem
cells
and
systematically
characterized
DNA
methylation, chromatin modifications, and the
tran-scriptome in each lineage. We found that promoters
that are active in early developmental stages tend to
be CG rich and mainly engage H3K27me3 upon
silencing in nonexpressing lineages. By contrast,
promoters for genes expressed preferentially at later
stages are often CG poor and primarily employ DNA
methylation upon repression. Interestingly, the early
developmental regulatory genes are often located
in large genomic domains that are generally devoid
of DNA methylation in most lineages, which we
termed DNA methylation valleys (DMVs). Our results
suggest that distinct epigenetic mechanisms
regu-late early and regu-late stages of ES cell differentiation.
INTRODUCTION
Embryonic development is a complex process that remains to be
understood despite knowledge of the complete genome
se-quences of many species and rapid advances in genomic
tech-nologies. A fundamental question is how the unique gene
expression pattern in each cell type is established and
main-tained during embryogenesis. It is well accepted that the gene
expression program encoded in the genome is executed by
tran-scription factors that bind to cis-regulatory sequences and
modulate gene expression in response to environmental cues
(
Young, 2011
). Growing evidence now shows that maintenance
of such cellular memory depends on epigenetic marks such as
DNA methylation and chromatin modifications (
Bird, 2002
;
Kou-zarides, 2007
).
DNA methylation at promoters has been shown to silence
gene expression and thus has been proposed to be necessary
for lineage-specific expression of developmental regulatory
genes, genomic imprinting, and X chromosome inactivation
(
Bird, 2002
). Indeed, the DNA methyltransferases DNMT1 or
DNMT3a/3b double-knockout mice exhibit severe defects in
embryogenesis and die before midgestation, supporting an
essential role for DNA methylation in embryonic development
(
Li et al., 1992
;
Okano et al., 1999
). On the other hand, mouse
embryonic stem cells (mESCs) lacking all three DNMTs can
sur-vive and self-renew and can even begin to differentiate to some
germ layers (
Jackson et al., 2004
;
Tsumura et al., 2006
), raising
the possibility that DNA methylation is dispensable for at least
initial lineage specification in early embryos. Thus, the role of
DNA methylation in animal development needs to be more
pre-cisely defined. Like DNA methylation, chromatin modifications
have also been shown to play a key role in animal development.
Enzymes responsible for methylation of histone H3 at lysine 4, 9,
and 27, in particular, are essential for embryogenesis (
Kouzar-ides, 2007
;
Vastenhouw and Schier, 2012
). Additionally,
deple-tion of the histone acetyltransferase p300 or CBP also leads to
early embryonic lethality (
Yao et al., 1998
). Although both DNA
methylation and chromatin modifications are critical for
mamma-lian development, the exact role of each epigenetic mark in the
maintenance of lineage-specific gene expression patterns
re-mains to be defined.
In humans, studying the epigenetic mechanisms regulating
early embryonic development often requires access to
embry-onic cell types that are currently difficult or impractical to obtain.
Human embryonic stem cells (hESCs) (
Thomson et al., 1998
) can
be differentiated into a variety of precursor cell types, providing
an in vitro model system for studying early human developmental
decisions. We have established protocols for differentiation of
hESCs to various cell states, including trophoblast-like cells
(TBL) (
Xu et al., 2002
), mesendoderm (ME) (
Yu et al., 2011
),
neu-ral progenitor cells (NPCs) (
Chambers et al., 2009
;
Chen et al.,
2011
), and mesenchymal stem cells (MSCs) (
Vodyanik et al.,
2010
). The first three states represent developmental events
that mirror critical developmental decisions in the embryo (the
decision to become embryonic or extraembryonic, the decision
to become mesendoderm or ectoderm, and the decision to
become surface ectoderm or neuroectoderm, respectively).
MSCs are fibroblastoid cells that are capable of expansion and
multilineage differentiation to bone, cartilage, adipose, muscle,
and connective tissues (
Vodyanik et al., 2010
). The specific
hESC derivatives chosen thus reflect key lineages in the human
embryo and also represent those lineages that currently can be
produced in sufficient quantity and purity for epigenomic
studies. These lineages will complement other cells from more
mature sources, many of which have had their epigenomes
well characterized (
Hawkins et al., 2010
;
Lister et al., 2009
;
Zhu
et al., 2013
). Importantly, epigenomic analysis of these cell types
allows for investigation of chromatin and transcriptional changes
that drive the initial developmental fate decisions.
Here, we used high-throughput approaches to examine
the differentiation of hESCs into four cell types by generating
in-depth maps of transcriptomes, a large panel of histone
mod-ifications, and base-resolution maps of DNA methylation for
each cell type. Our study provided a full view of the dynamic
epi-genomic changes accompanying cellular differentiation and
line-age specification. As outlined below, an integrative analysis of
these data sets provided us with substantial insights into the
role of DNA methylation and chromatin modifications in animal
development.
RESULTS
Generation of Comprehensive Epigenome Reference
Maps for hESCs and Four hESC-Derived Lineages
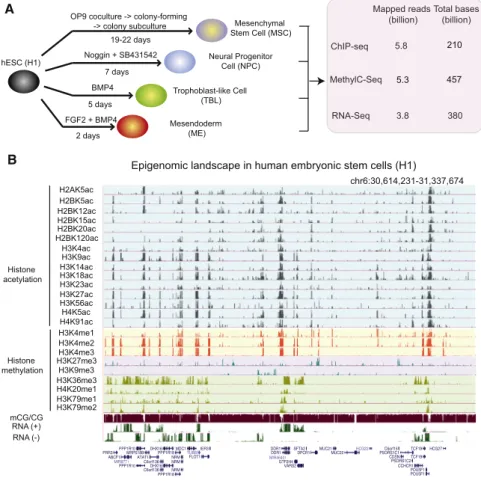
We differentiated the hESC line H1 to ME, TBL, NPCs, and MSCs
(
Figure 1
A) (
Extended Experimental Procedures
). ME, TBL, and
NPC differentiation occurred quickly (2 days, 5 days, and
7 days, respectively) compared to that of MSC (19–22 days).
The expression of various marker genes in these cells was
confirmed using immunofluorescence and
fluorescence-acti-vated cell sorting (FACS), and the purity of each cell population
ranged from 93% to 99% (
Figures S1
A–S1C available online).
ME, NPCs, and MSCs possess further differentiation potentials
as shown in
Figures S1
D and S1E (for ME and NPCs) and our
previous study (for MSCs) (
Vodyanik et al., 2010
). On the other
hand, the nature of TBL is still currently under debate (
Bernardo
et al., 2011
;
Xu et al., 2002
). As a control for terminally
differenti-ated cells, we also cultured and analyzed IMR90, a primary
hu-man fetal lung fibroblast cell line. For each cell type, we mapped
DNA methylation at base resolution using MethylC-seq (
Lister
et al., 2009
) (20–35
3 total genome coverage or 10–17.53
coverage per strand). We also mapped the genomic locations
of 13–24 chromatin modifications by chromatin
immunoprecipi-tation sequencing (ChIP-seq). Additionally, we performed
paired-end (100 bp
3 2) RNA-seq experiments, generating
more than 150 million uniquely mapped reads for every cell
type (
Figures 1
A and 1B). At least two biological replicates
were carried out for each analysis, and the data were publicly
released as part of the NIH Roadmap Epigenome Project
(
http://www.epigenomebrowser.org/
). Selected data are also
available at
http://epigenome.ucsd.edu/differentiation
.
Identification of Differentially Expressed Genes in
hESC-Derived Cells
We first asked how the genome is differentially transcribed when
hESCs are differentiated into each cell type. To do so, we
exam-ined the expression of 19,056 RefSeq coding genes (33,797
iso-forms), among which 76.6% (14,595) were expressed in at least
one cell type (
Figure S2
A). Using an entropy-based method (
Bar-rera et al., 2008
;
Schug et al., 2005
) (
Figure S2
B), we identified
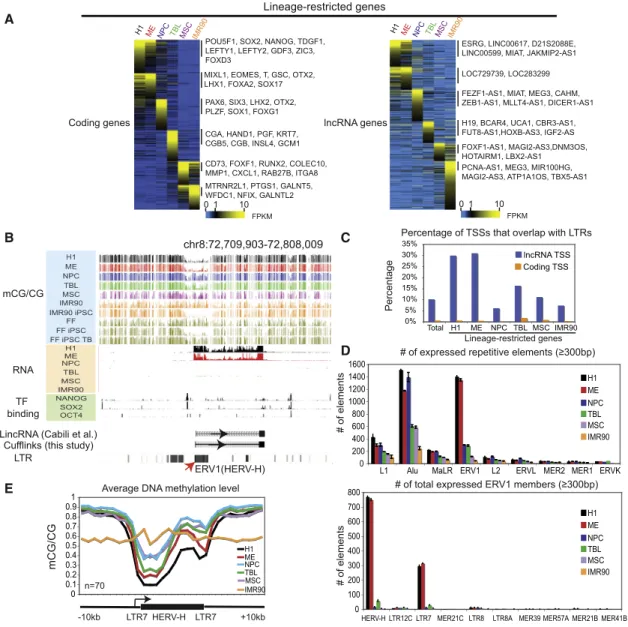
2,408 genes that showed cell-type-specific expression (
Figures
2
A and
S2
A). For convenience, we use ‘‘lineage-restricted
genes’’ to reflect both H1-specific and differentiated
cell-spe-cific genes. As expected, known lineage markers were highly
ex-pressed in the corresponding cell types (
Figure 2
A). It is worth
noting that, in line with a previous report (
Yu et al., 2011
), the
ME cells also express high levels of the hESC regulators
NANOG, POU5F1, and a reduced but significant level of SOX2.
We then investigated a cohort of long noncoding RNA (lncRNA)
genes and detected significant levels of transcripts for 2,175
known and 281 unannotated lncRNA genes in at least one cell
type (
Figures 2
A and
S2
A). Using the same entropy-based
approach, we found 930 lncRNA genes defined as lineage
restricted (
Figure S2
C), which constitute 37.9% of total
ex-pressed lncRNA genes. By contrast, only 16.5% of exex-pressed
coding genes are characterized as lineage restricted (
Fig-ure S2
D). The above analysis defined a large number of coding
and noncoding genes that are differentially expressed in H1
and its derived cells. The lists of all lineage-restricted genes
are included in
Table S1
.
Intriguingly, the promoters of several lncRNA genes highly
expressed in H1 overlap with the long terminal repeat
(LTR)-containing retrotransposons (
Figure 2
B). This appears to be a
general phenomenon as we observed that significant
percent-ages of transcription start sites (TSSs) of lncRNA genes directly
fall into LTRs (
Figure 2
C). The percentages are notably higher for
H1- and ME-enriched lncRNA genes (30% and 31%,
respec-tively), which are in contrast to those of coding genes (<2%).
By quantifying the transcription levels of all major classes of
mappable repetitive elements, we found that the ERV1 (class I
endogenous retrovirus) elements are preferentially expressed
in H1 and ME, but not in other cell types (
Figure 2
D, top).
Strik-ingly, such lineage-specific expression occurs almost
exclu-A
MethylC-Seq ChIP-seq RNA-Seq chr6:30,614,231-31,337,674B
H2AK5ac H2BK5ac H2BK12ac H2BK15ac H2BK20ac H2BK120ac H3K4ac H3K9ac H3K14ac H3K18ac H3K23ac H3K27ac H3K56ac H4K5ac H4K91ac H3K4me1 H3K4me2 H3K4me3 H3K27me3 H3K9me3 H3K36me3 H4K20me1 H3K79me1 H3K79me2 mCG/CG RNA (+) RNA (-)Epigenomic landscape in human embryonic stem cells (H1)
PRR3 ABCF1 MIR877 PPP1R10 PPP1R10 MRPS18B ATAT1 C6orf136 C6orf136 DHX16 DHX16 PPP1R18 PPP1R18 NRM NRM NRM MDC1 TUBB FLOT1 IER3 DDR1 DDR1 MIR4640 GTF2H4 VARS2 SFTA2
DPCR1 MUC21MUC22 HCG22 PSORS1C1C6orf15 CDSN PSORS1C2 CCHCR1 TCF19 TCF19 POU5F1 POU5F1 HCG27 Noggin + SB431542 FGF2 + BMP4 2 days 7 days 19-22 days , , , , Histone acetylation Histone methylation hESC (H1) Mesendoderm (ME) BMP4 5 days Trophoblast-like Cell (TBL) Neural Progenitor Cell (NPC) Mesenchymal Stem Cell (MSC) 5.8 Mapped reads (billion) Total bases (billion) 3.8 380
OP9 coculture -> colony-forming -> colony subculture MethylC-Seq ChIP-seq RNA-Seq 5.8 Mapped reads (billion) Total bases (billion) 3.8 380 210 5.3 457
Figure 1. Generation of Comprehensive Epigenome Reference Maps for hESCs and Four hESC-Derived Lineages
(A) Schematic of hESC differentiation procedures and a summary of the epigenomic data sets pro-duced in this study.
(B) A snapshot of the UCSC genome browser shows the DNA methylation level (mCG/CG),
RNA-seq reads (+, Watson strand; , Crick strand),
and ChIP-seq reads (RPKM) of 24 chromatin marks in H1.
See alsoFigure S1.
sively at the ERV1 subfamily HERV-H
and its flanking LTR elements LTR7 (
Fig-ure 2
D, bottom). Together, HERV-H and
LTR7 account for more than 43% of
LTRs that are present at H1- and
ME-spe-cific lncRNA gene promoters. A gene
ontology analysis of coding genes near
H1-specific HERV-H/LTR7 sites revealed
an
enrichment
of
POU5F1-targeted
genes (p value = 4
3 10
15), which is
consistent with a previous study showing
that NANOG and POU5F1 preferentially
bind to repetitive elements (
Kunarso
et al., 2010
). We did not find significant
enrichment of LTR subclasses for other
lineage-restricted lncRNA genes.
Repeti-tive elements are known to be regulated
by DNA methylation and H3K9me3 in
ESCs (
Leung and Lorincz, 2012
). We do not find significant
enrichment of H3K9me3 around most HERV-H elements (data
not shown). By contrast, a subset of the H1-specific HERV-H
elements (n = 70) show hypomethylation in H1 and ME but
gain DNA methylation in other H1-derived cells (
Figures 2
B and
2E). Notably, the overall low level of DNA methylation in IMR90
reflects its globally hypomethylated genome, likely due to the
presence of partially methylated domains (PMDs) (
Figures S2
E
and S2F) (
Lister et al., 2009
). Additionally, by examining
pub-lished methylomes (
Lister et al., 2011
), we found that DNA
methylation at these regions was depleted upon reprogramming
of IMR90 or foreskin fibroblasts to iPSCs and was then
reestab-lished when the fibroblast-derived iPSCs were differentiated to
trophoblast-like lineage (
Figure 2
B). Together, these data
sug-gest that many noncoding RNA genes may be transcriptionally
regulated by endogenous retroviral sequences. Of particular
in-terest, the expression of HERV-H/LTR7 is closely correlated with
the state of pluripotency and may be regulated by DNA
methylation.
Dynamic DNA Methylation and Chromatin Modifications
at Promoters of Lineage-Restricted Transcripts
Previous studies have shown that the promoters for
somatic-tis-sue-specific genes are often CG poor and lack CpG islands
(CGIs), in contrast to those for housekeeping genes, which
are CG rich and predominantly contain CGIs (
Barrera et al.,
2008
;
Schug et al., 2005
). Therefore, we asked whether early
lineage-restricted promoters also demonstrate similar features
as tissue-specific promoters. We first identified promoters for
each lineage-restricted gene and excluded those with
ambig-uous active promoters (
Extended Experimental Procedures
).
Next, we divided the promoters into three groups based on CG
density (high, medium, and low) (
Figure S3
A). Surprisingly, genes
preferentially expressed in early embryonic lineages H1, ME, and
NPC tend to be CG rich and contain CGIs (
Figure 3
A). The
per-centages of CGI-containing promoters decreased for genes
en-riched in MSCs and IMR90, which are at relatively late
develop-ment stages. By contrast, a much lower percentage of
promoters (23%) contain CGIs for somatic-tissue-specific genes
identified from 18 human tissues (
Zhu et al., 2008
) (
Figure 3
A).
We further verified this using an independent set of
somatic-tis-sue-specific genes (35%) (
Chang et al., 2011
). These data
A
B
E
C
D
Figure 2. Identification of Lineage-Restricted Transcripts in H1 and H1-Derived Cells
(A) Heatmaps showing the expression levels of lineage-restricted coding genes (left) and lncRNA genes (right). Genes are organized by the lineage in which their expression is enriched. Note that certain genes (such as SOX2) can be expressed in more than one cell type.
(B) The levels of DNA methylation and RNA, as well as the binding of NANOG, SOX2, and POU5F1, are shown around an annotated lincRNA gene with the promoter overlapping a HERV-H element.
(C) The percentages of TSSs that overlap with LTRs are shown for coding genes (yellow) and lncRNA genes (blue) for all genes (total) or lineage-restricted genes.
(D) The numbers of expressed (FPKMR 1), mappable repetitive elements are shown in each cell type for various repeat classes (top) or subclasses of ERV1
(bottom). Data are represented as mean± SD based on two replicates of RNA-seq.
(E) The average DNA methylation level in each cell type is shown for a subset of H1-specific HERV-H elements. See alsoFigure S2.
noticed that many DMV genes demonstrate a bivalent state
(H3K4me3 and H3K27me3), which is linked to poised
transcrip-tion that may enable developmental genes to be more flexibly
modulated (
Bernstein et al., 2006
). DNA methylation, on the other
hand, may be required for more stable silencing of genes in
terminally differentiated cells. Another possibility is that the
ge-netic programs regulating embryonic development may actually
evolve separately from, or prior to, the evolution of DNA
methyl-ation machinery. Supporting this hypothesis, DNA methylmethyl-ation is
either absent (such as in Drosophila and C. elegans) or varies
considerably in its pattern relative to gene activity in
inverte-brates (
Feng et al., 2010
;
Zemach et al., 2010
). On the other
hand, the Polycomb family of factors regulates key
develop-mental regulatory genes in both invertebrates and vertebrates
in a more conserved manner. Several mechanisms of DNA
hypo-methylation at DMVs can be envisioned. DMVs may be
recog-nized by proteins, such as the Tet family, that actively remove
DNA methylation (
Wu and Zhang, 2011
). Alternatively, DMVs
may be associated with histone modifications or histone
vari-ants, such as H3K4me3 or H2A.Z, that are incompatible to
DNA methylation (
Cedar and Bergman, 2009
). Future
experi-ments are needed to determine which of the above mechanisms
could be responsible for DMV formation in the mammalian
genome.
EXPERIMENTAL PROCEDURES hESC Differentiation
H1 cells were differentiated according to previously established protocols to mesendoderm (Yu et al., 2011), trophoblast-like cells (Xu et al., 2002), neural progenitor cells (Chambers et al., 2009;Chen et al., 2011), and mesenchymal stem cells (Vodyanik et al., 2010). Details of the differentiation methods can be
found inExtended Experimental Procedures.
MethylC-Seq Library Generation and Sequencing
Genomic DNA from H1 and the H1-derived cells was extracted and sonicated. Sequencing libraries were constructed using NEBNext DNA Sample Prep Re-agent Set 1 (NEB). Methylated adapters were used in place of the standard genomic DNA adapters from Illumina. Ligation products were purified, bisulfite treated, PCR amplified, and sequenced using HiSeq2000 (Illumina).
ChIP-Seq Library Generation and Sequencing
H1 and the H1-derived cells were processed following a ChIP protocol as
pre-viously described (Hawkins et al., 2010). ChIP libraries were prepared and
sequenced using the Illumina instrument per manufacturer’s instructions.
RNA-Seq Library Generation and Sequencing
Total RNA from H1 and the H1-derived cells was extracted and sequencing libraries were constructed using the TruSeq RNA Sample Prep Kit (Illumina) (Poly(A) selected) according to manufacturer’s instructions with modifications to confer strand specificity (seeExtended Experimental Proceduresfor details).
ACCESSION NUMBERS
All data have been deposited to the Sequence Read Archive (SRA) under accession number SRP000941.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures, seven figures, and seven tables and can be found with this article online athttp://dx. doi.org/10.1016/j.cell.2013.04.022.
ACKNOWLEDGMENTS
This work is supported by the NIH Epigenome Roadmap Project (U01 ES017166) and also in part by NSFC 91019016 and NIH R01 HG001696 (M.Q.Z.), NIH P01 GM081629 (J.A.T.), and CIRM RN2-00905-1 (B.R.). J.R.E. is a Howard Hughes Medical Institute and Gordon and Betty Moore Investi-gator. N.C.C. is funded by grants from the NIH/NHLBI. H.Y. is funded by grants from the American Heart Association (12POST12050080). We thank Drs. Tomek Swigut and Joanna Wysocka for sharing the zebrafish enhancer re-porter vector. We thank Dr. John Stamatoyannopoulos for generating and providing access to the DNase-seq data sets. We also thank members of the Ren lab for helpful comments of the manuscript. B.R., J.A.T., J.R.E., W.W., and M.Q.Z. designed and supervised the research. Z.H., J.Z., P.Y., N.E.P., K.S., J.E.A.-B., and I.S. performed/supervised the H1 differentiation experiments. W.X., R.D.H., D.L., A.Y.L., A.K., S. Kuan, C.Y., and S. Klugman performed ChIP-seq experiments. R.L. and J.R.N. performed MethylC-seq ex-periments. M.A.U., Y.L., and Y.Z. performed RNA-seq exex-periments. H.Y. and N.C.C. performed/supervised the enhancer-reporter assay in zebrafish. W.X., M.D.S., N.R., P.R., J.W.W., S.T., T.W., S.A.S., Y.Z., R.L., H.C., L.E.E., U.W., A.K., Z.X., W.Y.C., and R.S. analyzed data. W.X., B.R., and R.S. prepared the manuscript. B.R., J.A.T., J.R.E., W.W., and M.Q.Z. are equally responsible for the analysis results. M.D.S., R.L., Z.H., N.R., P.R., J.W.W., S.T., R.D.H., and D.L. contributed equally to this work.
Received: September 29, 2012 Revised: January 7, 2013 Accepted: April 1, 2013 Published: May 9, 2013
REFERENCES
Anokye-Danso, F., Trivedi, C.M., Juhr, D., Gupta, M., Cui, Z., Tian, Y., Zhang, Y., Yang, W., Gruber, P.J., Epstein, J.A., and Morrisey, E.E. (2011). Highly effi-cient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 8, 376–388.
Barrera, L.O., Li, Z., Smith, A.D., Arden, K.C., Cavenee, W.K., Zhang, M.Q., Green, R.D., and Ren, B. (2008). Genome-wide mapping and analysis of active promoters in mouse embryonic stem cells and adult organs. Genome Res. 18, 46–59.
Berman, B.P., Weisenberger, D.J., Aman, J.F., Hinoue, T., Ramjan, Z., Liu, Y., Noushmehr, H., Lange, C.P., van Dijk, C.M., Tollenaar, R.A., et al. (2012). Re-gions of focal DNA hypermethylation and long-range hypomethylation in colo-rectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 44, 40–46.
Bernardo, A.S., Faial, T., Gardner, L., Niakan, K.K., Ortmann, D., Senner, C.E., Callery, E.M., Trotter, M.W., Hemberger, M., Smith, J.C., et al. (2011). BRACHYURY and CDX2 mediate BMP-induced differentiation of human and mouse pluripotent stem cells into embryonic and extraembryonic lineages. Cell Stem Cell 9, 144–155.
Bernstein, B.E., Mikkelsen, T.S., Xie, X., Kamal, M., Huebert, D.J., Cuff, J., Fry, B., Meissner, A., Wernig, M., Plath, K., et al. (2006). A bivalent chromatin struc-ture marks key developmental genes in embryonic stem cells. Cell 125, 315–326.
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21.
Bock, C., Beerman, I., Lien, W.H., Smith, Z.D., Gu, H., Boyle, P., Gnirke, A., Fuchs, E., Rossi, D.J., and Meissner, A. (2012). DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 47, 633–647.
Boyer, L.A., Plath, K., Zeitlinger, J., Brambrink, T., Medeiros, L.A., Lee, T.I., Levine, S.S., Wernig, M., Tajonar, A., Ray, M.K., et al. (2006). Polycomb com-plexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353.
Bracken, A.P., and Helin, K. (2009). Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 9, 773–784.
Cedar, H., and Bergman, Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304. Chambers, S.M., Fasano, C.A., Papapetrou, E.P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280.
Chang, C.W., Cheng, W.C., Chen, C.R., Shu, W.Y., Tsai, M.L., Huang, C.L., and Hsu, I.C. (2011). Identification of human housekeeping genes and tis-sue-selective genes by microarray meta-analysis. PLoS ONE 6, e22859. Chen, G., Gulbranson, D.R., Hou, Z., Bolin, J.M., Ruotti, V., Probasco, M.D., Smuga-Otto, K., Howden, S.E., Diol, N.R., Propson, N.E., et al. (2011). Chem-ically defined conditions for human iPSC derivation and culture. Nat. Methods 8, 424–429.
Creyghton, M.P., Cheng, A.W., Welstead, G.G., Kooistra, T., Carey, B.W., Steine, E.J., Hanna, J., Lodato, M.A., Frampton, G.M., Sharp, P.A., et al. (2010). Histone H3K27ac separates active from poised enhancers and pre-dicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936. Ernst, J., Kheradpour, P., Mikkelsen, T.S., Shoresh, N., Ward, L.D., Epstein, C.B., Zhang, X., Wang, L., Issner, R., Coyne, M., et al. (2011). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49.
Feng, S., Cokus, S.J., Zhang, X., Chen, P.Y., Bostick, M., Goll, M.G., Hetzel, J., Jain, J., Strauss, S.H., Halpern, M.E., et al. (2010). Conservation and diver-gence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 107, 8689–8694.
Gifford, C.A., Ziller, M.J., Gu, H., Trapnell, C., Donaghey, J., Tsankov, A., Shalek, A.K., Kelley, D.R., Shishkin, A.A., Issner, R., et al. (2013). Transcrip-tional and epigenetic dynamics during specification of human embryonic
stem cells. Cell 153. Published online May 9, 2013. http://dx.doi.org/
10.1016/j.cell.2013.04.037.
Hammoud, S.S., Nix, D.A., Zhang, H., Purwar, J., Carrell, D.T., and Cairns, B.R. (2009). Distinctive chromatin in human sperm packages genes for embryo development. Nature 460, 473–478.
Hawkins, R.D., Hon, G.C., Lee, L.K., Ngo, Q., Lister, R., Pelizzola, M., Edsall, L.E., Kuan, S., Luu, Y., Klugman, S., et al. (2010). Distinct epigenomic land-scapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6, 479–491.
Irizarry, R.A., Wu, H., and Feinberg, A.P. (2009). A species-generalized proba-bilistic model-based definition of CpG islands. Mamm. Genome 20, 674–680. Jackson, M., Krassowska, A., Gilbert, N., Chevassut, T., Forrester, L., Ansell, J., and Ramsahoye, B. (2004). Severe global DNA hypomethylation blocks dif-ferentiation and induces histone hyperacetylation in embryonic stem cells. Mol. Cell. Biol. 24, 8862–8871.
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705.
Kunarso, G., Chia, N.Y., Jeyakani, J., Hwang, C., Lu, X., Chan, Y.S., Ng, H.H., and Bourque, G. (2010). Transposable elements have rewired the core regula-tory network of human embryonic stem cells. Nat. Genet. 42, 631–634. Laurent, L., Wong, E., Li, G., Huynh, T., Tsirigos, A., Ong, C.T., Low, H.M., Kin Sung, K.W., Rigoutsos, I., Loring, J., and Wei, C.L. (2010). Dynamic changes in the human methylome during differentiation. Genome Res. 20, 320–331. Lee, T.I., Jenner, R.G., Boyer, L.A., Guenther, M.G., Levine, S.S., Kumar, R.M., Chevalier, B., Johnstone, S.E., Cole, M.F., Isono, K., et al. (2006). Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125, 301–313.
Leung, D.C., and Lorincz, M.C. (2012). Silencing of endogenous retroviruses: when and why do histone marks predominate? Trends Biochem. Sci. 37, 127–133.
Li, E., Bestor, T.H., and Jaenisch, R. (1992). Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926. Li, Y., Zhu, J., Tian, G., Li, N., Li, Q., Ye, M., Zheng, H., Yu, J., Wu, H., Sun, J., et al. (2010). The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 8, e1000533.
Lister, R., Pelizzola, M., Dowen, R.H., Hawkins, R.D., Hon, G., Tonti-Filippini, J., Nery, J.R., Lee, L., Ye, Z., Ngo, Q.M., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322.
Lister, R., Pelizzola, M., Kida, Y.S., Hawkins, R.D., Nery, J.R., Hon, G., Antosie-wicz-Bourget, J., O’Malley, R., Castanon, R., Klugman, S., et al. (2011). Hot-spots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471, 68–73.
Meissner, A., Mikkelsen, T.S., Gu, H., Wernig, M., Hanna, J., Sivachenko, A., Zhang, X., Bernstein, B.E., Nusbaum, C., Jaffe, D.B., et al. (2008). Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770.
Mendenhall, E.M., Koche, R.P., Truong, T., Zhou, V.W., Issac, B., Chi, A.S., Ku, M., and Bernstein, B.E. (2010). GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 6, e1001244.
Mohn, F., Weber, M., Rebhan, M., Roloff, T.C., Richter, J., Stadler, M.B., Bibel, M., and Schu¨beler, D. (2008). Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 30, 755–766.
Molaro, A., Hodges, E., Fang, F., Song, Q., McCombie, W.R., Hannon, G.J., and Smith, A.D. (2011). Sperm methylation profiles reveal features of epige-netic inheritance and evolution in primates. Cell 146, 1029–1041.
Okano, M., Bell, D.W., Haber, D.A., and Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257.
Ong, C.T., and Corces, V.G. (2011). Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 12, 283–293. Paige, S.L., Thomas, S., Stoick-Cooper, C.L., Wang, H., Maves, L., Sandstrom, R., Pabon, L., Reinecke, H., Pratt, G., Keller, G., et al. (2012). A temporal chromatin signature in human embryonic stem cells identifies regu-lators of cardiac development. Cell 151, 221–232.
Pan, G., Tian, S., Nie, J., Yang, C., Ruotti, V., Wei, H., Jonsdottir, G.A., Stewart, R., and Thomson, J.A. (2007). Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1, 299–312.
Rada-Iglesias, A., Bajpai, R., Swigut, T., Brugmann, S.A., Flynn, R.A., and Wysocka, J. (2011). A unique chromatin signature uncovers early develop-mental enhancers in humans. Nature 470, 279–283.
Rajagopal, N., Xie, W., Li, Y., Wagner, U., Wang, W., Stamatoyannopoulos, J., Ernst, J., Kellis, M., and Ren, B. (2013). RFECS: a Random-Forest based algo-rithm for Enhancer Identification from Chromatin State. PLoS Comput. Biol. 9, e1002968.
Schug, J., Schuller, W.P., Kappen, C., Salbaum, J.M., Bucan, M., and Stoeck-ert, C.J., Jr. (2005). Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 6, R33.
Stadler, M.B., Murr, R., Burger, L., Ivanek, R., Lienert, F., Scho¨ler, A., van Nimwegen, E., Wirbelauer, C., Oakeley, E.J., Gaidatzis, D., et al. (2011). DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495.
Suh, M.R., Lee, Y., Kim, J.Y., Kim, S.K., Moon, S.H., Lee, J.Y., Cha, K.Y., Chung, H.M., Yoon, H.S., Moon, S.Y., et al. (2004). Human embryonic stem cells express a unique set of microRNAs. Dev. Biol. 270, 488–498. Thomson, J.A., Itskovitz-Eldor, J., Shapiro, S.S., Waknitz, M.A., Swiergiel, J.J., Marshall, V.S., and Jones, J.M. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147.
Tsumura, A., Hayakawa, T., Kumaki, Y., Takebayashi, S., Sakaue, M., Matsuoka, C., Shimotohno, K., Ishikawa, F., Li, E., Ueda, H.R., et al. (2006). Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 11, 805–814.
Vastenhouw, N.L., and Schier, A.F. (2012). Bivalent histone modifications in early embryogenesis. Curr. Opin. Cell Biol. 24, 374–386.
Vodyanik, M.A., Yu, J., Zhang, X., Tian, S., Stewart, R., Thomson, J.A., and Slukvin, I.I. (2010). A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell 7, 718–729.
Wu, H., and Zhang, Y. (2011). Mechanisms and functions of Tet protein-medi-ated 5-methylcytosine oxidation. Genes Dev. 25, 2436–2452.
Xie, W., Barr, C.L., Kim, A., Yue, F., Lee, A.Y., Eubanks, J., Dempster, E.L., and Ren, B. (2012). Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148, 816–831. Xu, R.H., Chen, X., Li, D.S., Li, R., Addicks, G.C., Glennon, C., Zwaka, T.P., and Thomson, J.A. (2002). BMP4 initiates human embryonic stem cell differentia-tion to trophoblast. Nat. Biotechnol. 20, 1261–1264.
Yao, T.P., Oh, S.P., Fuchs, M., Zhou, N.D., Ch’ng, L.E., Newsome, D., Bronson, R.T., Li, E., Livingston, D.M., and Eckner, R. (1998). Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 93, 361–372.
Young, R.A. (2011). Control of the embryonic stem cell state. Cell 144, 940–954.
Yu, P., Pan, G., Yu, J., and Thomson, J.A. (2011). FGF2 sustains NANOG and switches the outcome of BMP4-induced human embryonic stem cell differen-tiation. Cell Stem Cell 8, 326–334.
Zemach, A., McDaniel, I.E., Silva, P., and Zilberman, D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919.
Zhao, X.D., Han, X., Chew, J.L., Liu, J., Chiu, K.P., Choo, A., Orlov, Y.L., Sung, W.K., Shahab, A., Kuznetsov, V.A., et al. (2007). Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compart-ments in human embryonic stem cells. Cell Stem Cell 1, 286–298.
Zhu, J., He, F., Hu, S., and Yu, J. (2008). On the nature of human housekeeping genes. Trends Genet. 24, 481–484.
Zhu, J., Adli, M., Zou, J.Y., Verstappen, G., Coyne, M., Zhang, X., Durham, T., Miri, M., Deshpande, V., De Jager, P.L., et al. (2013). Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152, 642–654.
Note Added in Proof
While this manuscript was in revision, the two following related papers describing hESC-specific expression of the HERV-H retrotransposable ments were published. Kelley, D.R., and Rinn, J.L. (2012). Transposable ele-ments reveal a stem cell specific class of long noncoding RNAs. Genome Biol. 13, R107. Santoni, F.A., Guerra, J., and Luban, J. (2012). HERV-H RNA is abundant in human embryonic stem cells and a precise marker for pluripo-tency. Retrovirology 9, 111.