行政院國家科學委員會專題研究計畫 成果報告
催化不對稱合成(I)
計畫類別: 個別型計畫
計畫編號: NSC93-2113-M-006-007-
執行期間: 93 年 08 月 01 日至 94 年 07 月 31 日 執行單位: 國立成功大學化學系(所)
計畫主持人: 宋光生
報告類型: 精簡報告
報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢
中 華 民 國 94 年 10 月 31 日
行政院國家科學委員會專題研究計畫成果報告 計畫編號: NSC 93-2113-M-006-007
執行期限: 92/08/01 ~ 93/07/31 主持人 : 宋光生 執行機構: 成大化學系 中文摘要 :
茲附上三篇已發表文章的研究成果.
關鍵詞: push-pull olefin, stereoselective reaction, ketenimine, amination
Abstract: The research results in three published papers are enclosed.
Key words: push-pull olefin, stereoselective reaction, ketenimine, amination
壹.Ethyl (2-cyano-3-ethoxyacryloyl)carbamate: irreversibly thermal
isomerization of a push-pull olefins
Sung, Kuangsen; Lin, Ming-Chi; Huang, Pin-Mei; Zhuang, Bo-Ren; Sung, Robert;
Wu, Ru-Rong;Arkive of Org. Chem. (ARKIVOC), 2005, xiii, 131~140. (SCI)
Introduction
5-Fluorouracil is an effective antitumor agent1 and we are trying to prepare its derivatives by multi-component reaction (MCR).2 To prepare an intermediate to the uracil derivatives, ethyl (2-cyanoacetyl)carbamate, 1, was treated with ethyl orthoformate in the presence of acetic anhydride in our laboratory, and only E-ethyl (2-cyano-3-ethoxyacryloyl)carbamate, E-2, was found. In addition, we found that isomerization of E-2 to Z-2 is driven only by ultraviolet light while the backward isomerization can be driven by heat. On the other hand, when we treated ethyl acetoacetate with ethyl orthoformate and acetic anhydride, both E-3 and Z-3 were obtained at a ratio of 1:1. Even though both 2 and 3 are push-pull olefins, Z-E isomerization equilibrium of 2 is completely different from that of 3.
Z-E isomerization of some alkenes by light or chiral light may function like
molecular switch or motor and has applications in optical data storage system, image processor, laser-addressable device, erasable optical recording support, and so on.3 It turns out that 2 can be a prototype of molecular switch. Dynamic NMR spectroscopy has been used to determine rotational barriers of push-pull olefins, but the rotational barriers of push-pull olefins with only one geometric isomer detected, such as 2, cannot be measured by the DNMR.4 Therefore, both UV and NMR spectrometers were used to study the Z-E isomerization of 2 in this article.
HC(OC
2H
5)
3+
NH O
O O
N
NH O
O O
N
H O
NH O
O O
N
O H
h
acetic anhydride
1
E-2 Z-2
O
O O
HC(OCH
2CH
3)
3acetic anhydride +
O
O O
EtO H
O
O O
H OEt
+
E-3 Z-3
1 : 1
Results and Discussion
Reaction of 1 with ethyl orthoformate in the presence of acetic anhydride in chloroform was carried out under reflux for 2 hours, and only E-2 was isolated in 75%
yield. No trace of Z-2 has been found. The E-2 is well characterized by its 1H and
13C NMR spectra. Its vinyl proton has characteristic resonance absorption at 8.17.
The 3J coupling constant between the vinyl proton and nitrile carbon is 11 Hz while the one between the vinyl proton and amide carbon is 2 Hz. Electrons delocalize preferentially through trans configuration, and this is confirmed by much larger vicinal spin-spin coupling/interaction for trans than for cis configurations in NMR spectra.5 It indicates that the vinyl proton of the isolated product is trans to the nitrile group and its structure is E-2, instead of Z-2. NOESY (Fig. 1) of the isolated product provides another evidence to support that its configuration is E-2. In the NOESY, vinyl proton at 8.17 correlates with both imide proton at 9.12 and methylene protons ( 4.39) of ester group, while methylene protons ( 4.12) of vinyl ether don’t correlate with both vinyl proton at 8.17 and imide proton at 9.12.
The configuration assignment for E-2 is consistent with the one for E-4 Ceder and Stenhede did.6 Compounds 5~8 were also prepared highly stereoselectively with one geometric isomer isolated, but their configurations were not assigned.4a
C
NH O
O
O H
Some correlations in NOESY of E-2 N
O H2 C
CH3
H2 C H3C
O O
EtO H
E-4 N
Ar S
O N
O
5 : Ar = 2-Furyl 6 : Ar = 2-Thiophenyl 7 : Ar = Phenyl 8 : Ar = p-Cl-Phenyl
After E-2 in CD3CN has been subjected to 2 hours of irradiation at = 254 nm, intensity of the vinyl proton resonance absorption at 8.17 decreases and another resonance absorption at 7.80 appears. After stopping the irradiation, the peak at
7.80 disappears slowly while intensity of the original peak at 8.17 increases, indicating that the resonance absorption at 7.80 belongs to the vinyl proton of Z-2.
(Fig. 2)
Fig. 1. NOESY of E-2 in CD3CN
Around 40% of E-2 was isomerized to Z-2 after 2 hours of ultraviolet irradiation at a wavelength of 254 nm. Ultraviolet spectrum of the whole sample in acetonitrile displayed a blue shift. Two days after stopping the irradiation, most of Z-2 was isomerized back to E-2 at room temperature, and ultraviolet spectrum of the sample shifted back to original position (red shift). The isomerization is faster at higher temperature.
Fig. 2. Part of 1H NMR spectra of E-2 in CD3CN measured (a) before ultraviolet irradiation, (b) 5 min, (c) 10hrs, and (d) 1 day after 2 hrs irradiation at 254 nm.
After 2 hours of photo-isomerization at =254 nm, the mixture of E-2 and Z-2 was used for kinetic study of the isomerization of Z-2 back to E-2. The isomerization in acetonitrile at 25°C was monitored by UV spectrophotometer at =
272 nm, where both isomers have the biggest absorption difference, at six different short time intervals distributed evenly throughout the whole isomerization. The plots of absorption vs. time are fitted with a first-order exponential rise very well and they are shown in Fig. 3 and 4. The kinetic studies were carried out at 25, 30, 35, 40, and 45℃, respectively, and the corresponding rate constants are shown in Table 1.
The Ea, H‡, S‡, and G‡ for the isomerization of Z-2 to E-2 were determined to be 19.6 kcal mol-1, 19.0 kcal mol-1, -17.5 cal K-1mol-1, and 24.4 kcal mol-1 according to the Eyring equation.7
Table 1. Observed Rate Constants for Isomerization of Z-2 to E-2
Temp. (°C) 25 30 35 40 45
kobs (s-1) 1.05 ×10-5 1.71 ×10-5 3.15 ×10-5 5.03 ×10-5 8.28 ×10-5
It was reported that reactions generating electric charge exhibit negative entropies of activation.8 For example, solvolysis of t-butyl chloride in 80% aqueous ethanol displays negative entropy of activation (-6.6 cal K-1mol-1) even though two particles are being generated from one in the transition state.9 Similarly, negative entropy of activation (-17.5 cal K-1mol-1) for the isomerization of Z-2 to E-2 indicates that the transition state has much more charge separation than the ground state.
Because of more polar character, the transition state requires a greater degree of ordering of solvent molecules than the ground state,9 leading to the negative entropy of activation.
A'
A
D'
D
A'
A
D'
D
push-pull olefin
Rotational barriers in alkyl-substituted ethylenes are >55 kcal mol-1,10 while those of push-pull olefins, whose -bonds are dramatically polarized,11 are significantly lowered and shown in Table 2.4a Rotational barrier (G‡ = 24.4 kcal mol-1) of the isomerization of Z-2 to E-2 is much lower than those of the alkyl-substituted ethylenes, indicating that Z-2 is a push-pull olefin. However, the rotational barrier of Z-2 is around 9.4 kcal mol-1 higher than those (G‡ = 9.9 ~ 15.0 kcal mol-1) of push-pull olefins 9~12. It was reported that the sequence of-donating ability is N > S > O.4a Compounds 9~12 all have two strong -donors (N) while Z-2 has only one less strong -donor (O). Therefore, it is reasonable that the rotational barrier of Z-2 is much higher than those of 9~12.
Table 2. Barrier of Rotation, G‡ (kcal/mol), about the Central C=C Partial Double Bond of Push-pull Olefins in Acetone
Ph HN
N H
(CH
2)n
N X
9 : X = O, n = 1 10 : X = O, n = 2 11 : X = O, n = 3 12 : X = S, n = 1
Compound 9 10 11 12
G‡ 15.0 12.3 12.7 9.9
N H O
OEt
EtO H
13C NMR of E-2
N
O
175.13 ppm 89.30 ppm
Push-pull effect of olefins can be recognized by both rotational barrier and chemical shift difference (C=C) of two sp2-hybridized carbons of C=C partial double bond.4 Due to dramatically polarized -bonds,11 push-pull olefins display a deshielding effect on the alkenyl carbon of donor side and a shielding effect on the alkenyl carbon of acceptor side, causing C=C being around 82 ~ 100 ppm.4 The
C=C of E-2 is 85.83 ppm in CD3CN, and that confirms 2 is a push-pull olefin.
N
O Me Me
NO2
N Me Me
O O2N
light on light off
13 14
E-2 Z-2
h
It was reported that 13/14 are good molecular switches, whose isomerization equilibrium can be driven by light for the forward equilibrium and by heat for the
backward isomerization.3c Z-E isomerization equilibrium of 2 can be driven by the same way, so 2 is a prototype of molecular switch. Next challenge of this issue is to modify structure of 2 in order to make response time of the system as short as possible.
Conclusion
The highly stereoselective reaction of 1 with ethyl orthoformate in the presence of acetic anhydride produces E-2 only. 2 is a push-pull olefin and E-2 is much more stable than Z-2. The Ea, H‡, S‡, and G‡ for the isomerization of Z-2 to E-2 were determined to be 19.6 kcal mol-1, 19.0 kcal mol-1, -17.5 cal K-1 mol-1, and 24.4 kcal mol-1. The negative entropy of activation for this isomerization indicates that the transition state has much more charge separation than the ground state. E-2 cannot be isomerized to Z-2 thermally but photochemically. On the other hand, Z-2 can be irreversibly isomerized back to E-2 thermally.
Experimental Section
General. Unless otherwise stated reagents were obtained from commercial suppliers
and used as received. Ethyl (2-cyanoacetyl)carbamate, 1, was prepared according the literature method.12
E-Ethyl (2-cyano-3-ethoxyacryloyl)carbamate (E-2).
To a solution of 1 (0.156 g, 1 mmol) and acetic anhydride (1 mL) in 2 mL of chloroform was ethyl orthoformate (0.296 g, 2 mmol) added. The mixture was refluxed at 80°C under nitrogen atmosphere for 2 h. After the reaction was complete, the reaction mixture was cooled down and concentrated by rotary evaporator. Ether was poured into the reaction mixture and the mixture stayed in refrigerator for 12 hrs.
After filtration of the mixture, white powder was collected and recrystallized from
chloroform/ether. Yield: 75%;1H NMR (CD3CN) 1.23 (3H, t, CH3), 1.34 (3H, t, CH3), 4.12 (2H, q, CH2), 4.39 (2H, q, CH2), 8.17 (1H, s, CH), 9.12 (1H, s, NH);13C NMR (CD3CN) 14.80, 15.87, 63.13, 75.69, 89.30, 114.69, 152.22, 162.01, 175.13;
IR (thin film) 2227 (CN), 1774, 1689 (C=O) cm-1; MS (EI) m/z 212 (4, M+), 118 (100), 88 (32), 74 (24), 57 (28); HRMS (EI) m/z calcd for C9H12N2O4 212.0797, found 212.0801.
NMR Study of Isomerization between E-2 and Z-2.
Proton NMR spectra of a CD3CN solution (0.5 mL) of E-2 (0.01 mmol) were measured before ultraviolet irradiation, 5 min, 10hrs, and 1 day after 2 hrs irradiation at 254 nm. The sample solution was transferred to a quartz flask with a sealed cap when it was subject to the ultraviolet irradiation.
Kinetic Studies for Isomerization of Z-2 to E-2.
Kinetics for isomerization of Z-2 was carried out by ultraviolet absorption measurement of a sample solution at
=272 nm, which was approximately 0.1 mM E-2 solution in CH3CN and was subjected to 2 hrs of ultraviolet irradiation at 254 nm. Six different short periods of time, which is spaced evenly throughout the whole isomerization, was chosen for each of kinetic studies. The kinetic studies were carried out at 25, 30, 35, 40, and 45°C, respectively, in the thermostatic UV cell with Perkin-Elmer Lambda 12 spectrophotometer. The plot of ultraviolet absorption vs.
time was fitted with a first-order exponential rise by the SigmaPlot software to get a first-order rate constant. All rate constants were measured at least in duplicate with maximum deviations of ±5%.
Reference
1. Ozaki, S. Medicinal Research Reviews 1996, 16, 51-86.
2. Dömling, A.; Ugi, I. Angew. Chem. Int. Ed. 2000, 39, 3168-3210.
3. (a) Dugave, C.; Demange, L. Chem. Rev. 2003 103, 2475-2532. (b) Feringa, B. L.
Acc. Chem. Res. 2001, 34, 504-513. (c) Tomasulo, M.;
Sortino, S.; Raymo, F. M.Org. Lett. 2005, 7, 1109-1112. (d) Pina, F.;
Lima, J. C.; Parola, A. J.; Afonso, C.A. M. Angew. Chem. Int. Ed. 2004, 43, 1525-1527. (e) Giordani, S.; Raymo, F. M.
Org. Lett. 2003, 5, 3559-3562. (f) van Delden, R. A.;
Hurenkamp, J. H.; Feringa, B. L. Chem. Eur. J. 2003, 9, 2845-2853.4. (a) Kleinpeter, E.; Klod, S.; Rudorf, W.-D. J. Org. Chem. 2004, 69, 4317-4329. (b) Fischer, G.; Rudorf, W.-D.; Kleinpeter, E. Magn. Reson. Chem. 1991, 29, 212. (c) Sandström, J. In Topics in Stereochemistry; Allinger, N. L.; Eliel, E. L.; Wilen, S.
H.; Eds. John Wiley and Sons: New York, 1983; Vol. 14. (d) Markovic, R.;
Baranac, M.; Jovanovic, V.; Dzambaski, Z. J. Chem. Edu. 2004, 81, 1026-1029. (e) Sin, H. S.; Holler, M.; Burger, A.; Biellmann, J. F. Tetrahedron Lett. 1997, 38, 3585-3586. (f) Zhu, S. Z.; Qin, C. Y.; Xu, G. L.; Chu, Q. L. Tetrahedron Lett.
1998, 39, 5265-5268. (g) Abbotto, A.; Bradamante, S.; Capri, N.; Rzepa, H.;
Williams, D. J.; White, A. J. Org. Chem. 1996, 61, 1770-1778.
5. Silverstein, R.M. Webster FX. Spectrometric Identification of Organic
Compounds, 6
th Ed. John Wiley & Sons: New York, 1998.6. Ceder, O.; Stenhede, U. Tetrahedron 1973 29, 1585-1590.
7. Connors, K. A. Chemical Kinetics. VCH Publishers: New York, 1990.
8. Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry, Part A: Structure and
Mechanism, 2
nd ed. Plenum Press: New York, 1984.9. Grunwald, E.; Winstein, S. J. Am. Chem. Soc. 1948, 70, 846.
10. (a) Douglas, J. E.; Rabinovitch, B. S.; Looney, F. S. J. Chem. Phys. 1955, 23, 315. (b) Rabinovitch, B. S.; Michel, K.-W. J. Am. Chem. Soc. 1959, 81, 5056. (c) Kristiakovsky, G. B.; Smith, W. R. J. Am. Chem. Soc. 1934, 56, 638.
11. Forni, A.; Destro, R. Chem. Eur. J. 2003, 9, 5528.
12. Atkinson, M. R.; Shaw, G.; Warrener, R. N. J. Chem. Soc. 1956, 4118-4123.
貳. Why is reaction of ethyl (2-cyanoacetyl)carbamate with ethyl orthoformate highly stereoselective ?
Sung, Kuangsen; Lin, Ming-Chi; Huang, Pin-Mei; Zhuang, Bo-Ren; Sung, Robert;
Wu, Ru-Rong;J. Phys. Org. Chem. 2005, 18, 1183-1189. (SCI)
INTRODUCTION
One of popular methods to prepare vinyl ethers is to treat active methylene compounds with ethyl orthoformate.1 (Eq. 1) Some of reactions of this type are highly stereoselective. Not until 1973, Ceder and Stenhede figured out configuration of the highly stereoselective product by NMR studies using a chemical-shift reagent Eu(fod)3-d27.2 However, no literature reports why the type of reactions is highly stereoselective.
CH2 + HC(OC2H5)3 CHOC2H5+ 2 C2H5OH Y
X X
Y (Eq. 1)
To prepare an intermediate to uracil derivatives, ethyl (2-cyanoacetyl)carbamate, 1, was treated with ethyl orthoformate in the presence of acetic anhydride in our laboratory, and only E-ethyl (2-cyano-3-ethoxyacryloyl)carbamate, E-2, was found.
(Eq. 2) The reaction is a good model to study why this type of the reactions is highly stereoselective.
HC(OC
2H
5)
3+
NH O
O O
N
C(O)NHC(O)OEt
NH O
C(O)NHC(O)OEt
NO H
h
acetic anhydride
1
E-2 Z-2
(Eq. 2)
COMPUTATION
All the calculations reported here were performed with Gaussian98 program.3 Geometry optimizations of Z-2, Z-2a, E-2, E-2a, Z-3, E-3, Z-4, E-4, Z-5, E-5, Z-6, E-6,
E-7, 8, 9, and 10 at level of B3LYP/6-31+G* and geometry optimizations of 11a~d
and 12a~d at level of HF/6-31+G* were carried out without any symmetry restriction.
Optimized structures of Z-2, Z-2a, E-2, E-2a, Z-3, E-3, Z-4, E-4, Z-5, E-5, Z-6, E-6,
E-7, 8, 9, and 10 at level of B3LYP/6-31+G* are shown in Figure 1 and 2. After all
the geometry optimizations were performed, analytical vibration frequencies were calculated at the same level to determine the nature of the located stationary points.
Thus all the stationary points found were properly characterized by evaluation of the harmonic frequencies. The single point energies of the optimized structures of Z-2 and E-2 were calculated at levels of B3LYP/6-31+G*, HF/6-311++G (3df,3pd), or B3LYP/6-311++G(3df,3pd) with scale zero-point vibration energies included. The single point energies of the optimized structures of 11a~d and 12a~d were calculated at levels of MP2/6-31+G* with scale zero-point vibration energies included. The scale
factor of 0.9804 for zero-point vibration energies is used according to the literatures.4 Calculated energies of all the above structures are shown in Table 1.
Z-3 E-3 Z-4 E-4
Z-5 E-5 Z-6 E-6
E-7 8 9 10
Fig. 1. Optimized structures of Z-3, E-3, Z-4, E-4, Z-5, E-5, Z-6, E-6, E-7, 8, 9, and 10 at level of B3LYP/6-31+G*.
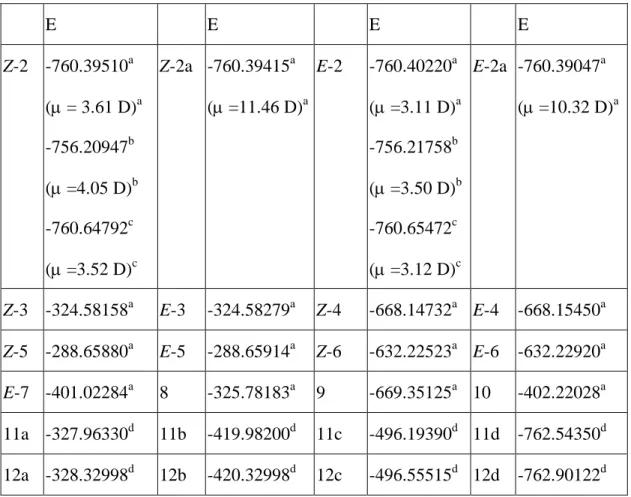
Table 1. Calculated energies E (hartrees) of Z-2, Z-2a, E-2, E-2a, Z-3, E-3, Z-4, E-4,
Z-5, E-5, Z-6, E-6, E-7, 8, 9, 10, 11a~d and 12a~d
E E E E
Z-2
-760.39510a ( = 3.61 D)a -756.20947b (=4.05 D)b -760.64792c (=3.52 D)cZ-2a -760.39415
a ( =11.46 D)aE-2
-760.40220a ( =3.11 D)a -756.21758b (=3.50 D)b -760.65472c (=3.12 D)cE-2a -760.39047
a ( =10.32 D)aZ-3
-324.58158aE-3
-324.58279aZ-4
-668.14732aE-4
-668.15450aZ-5
-288.65880aE-5
-288.65914aZ-6
-632.22523aE-6
-632.22920aE-7
-401.02284a 8 -325.78183a 9 -669.35125a 10 -402.22028a 11a -327.96330d 11b -419.98200d 11c -496.19390d 11d -762.54350d 12a -328.32998d 12b -420.32998d 12c -496.55515d 12d -762.90122da B3LYP/6-31+G*//B3LYP/6-31+G*. At this level, E(Z-2) - E(E-2) = 4.5 kcal/mol.
b. HF/6-311++G(3df,3pd)//B3LYP/6-31+G*. At this level, E(Z-2) - E(E-2) = 5.1 kcal/mol.
c. B3LYP/6-311++G(3df,3pd)//B3LYP/6-31+G*. At this level, E(Z-2) - E(E-2) = 4.3 kcal/mol.
d. MP2/6-31+G*//HF/6-31+G*.
RESULTS AND DISCUSSION
Reaction of 1 with ethyl orthoformate in the presence of acetic anhydride in chloroform was carried out under reflux for 2 hours, and only E-2 was isolated in 75%
yield. No trace of Z-2 has been found. In E-2, the 3J coupling constant between vinyl proton and nitrile carbon is 11 Hz while the one between the vinyl proton and amide carbon is 2 Hz, indicating that the vinyl proton is trans to the nitrile group.5 The configuration assignment for E-2 is consistent with the one Ceder and Stenhede did.2
After 2 hours of ultraviolet irradiation at =254 nm, 40% of E-2 was isomerized to Z-2. Two days after stopping the irradiation, most of Z-2 was isomerized back to
E-2 at room temperature. E-2 cannot be isomerized to Z-2 thermally but
photochemically, while Z-2 can be isomerized back to E-2 thermally. It implies that the reaction of 1 with ethyl orthoformate in the presence of acetic anhydride is very likely to be thermodynamic control.
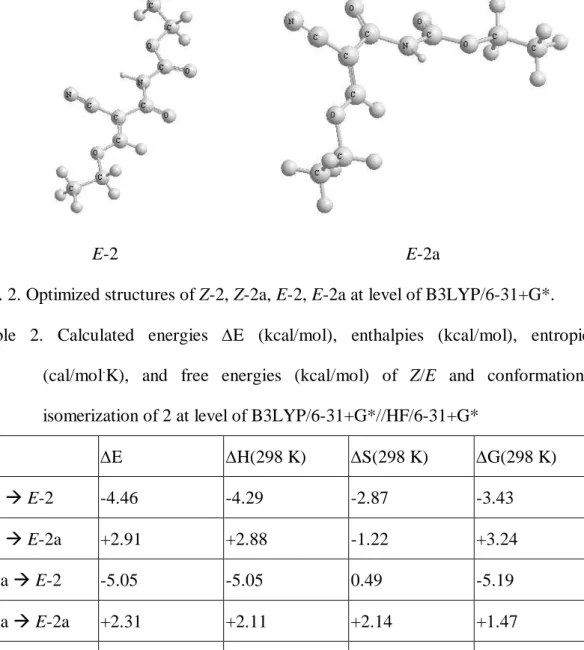
Whether the -ketoester part of E-2 stays as keto-form or enol-form is important for configuration assignment. A NMR technique of 1H-15N HSQC was applied and an off-diagonal cross peak shows a correlation between hydrogen at 9.20 and urethane nitrogen at 138.6 by means of their spin-spin coupling, indicating that E-2 stays as keto-form. The results have been found with solvents like d6-acetone, d3-acetonitrile, and CDCl3. Based on the NMR results, Z/E-isomers of 2 were optimized at level of B3LYP/6-31+G* and at least two interesting conformers were located for each of the geometric isomers. (Fig. 2) Conformers Z-2 and Z-2a were located for Z-isomer of 2, and Z-2 is 0.60 kcal/mol more stable than Z-2a.
Conformers E-2 and E-2a were located for E-isomer of 2, and E-2 is 7.36 kcal/mol more stable than E-2a. Backbones of Z-2, Z-2a, and E-2 stay in the same plane while C(O)NHC(O)OEt group is twisted away from the plane of the rest of structure in E-2a.
The major structure difference of the conformers is dihedral angle of C=C_C=O.
The more stable conformers (Z-2 and E-2) have s-cis conformation of C=C_C=O, so they can pull bulky C(O)NHC(O)OEt group away from vinyl hydrogen or ethoxy group to avoid steric hindrance. The less stable conformers (Z-2a and E-2a) have s-trans conformation of C=C_C=O, causing significant steric hindrance between bulky C(O)NHC(O)OEt group and vinyl hydrogen or ethoxy group. To reduce the steric hindrance, E-2a twists C(O)NHC(O)OEt away from the C=C plane (dihedral angle of C=C_C=O = 145.4°) at expense of lose of resonance stabilization along ethoxy, C=C, and C(O)NHC(O)OEt groups, and that makes it 2.31, 2.91, 7.36 kcal/mol less stable than Z-2a, Z-2, and E-2, respectively, implying that there is an intramolecular hydrogen bonding between N-H group and oxygen of ethoxy group in Z-2a. In Z-2a, C(O)NHC(O)OEt group stays in the same plane with the moiety of ethyl vinyl ether in spite of steric hindrance between C(O)NHC(O)OEt and ethoxy groups, and distance between hydrogen of N-H group and oxygen of ethoxy group is 1.97 Å, indicating that it has an intramolecular hydrogen bonding again and that makes it only 0.6 kcal/mol less stable than Z-2.
Z-2 Z-2a
E-2 E-2a
Fig. 2. Optimized structures of Z-2, Z-2a, E-2, E-2a at level of B3LYP/6-31+G*.
Table 2. Calculated energies E (kcal/mol), enthalpies (kcal/mol), entropies (cal/mol.K), and free energies (kcal/mol) of Z/E and conformational isomerization of 2 at level of B3LYP/6-31+G*//HF/6-31+G*
E H(298 K) S(298 K) G(298 K)
Z-2 E-2
-4.46 -4.29 -2.87 -3.43Z-2 E-2a
+2.91 +2.88 -1.22 +3.24Z-2a E-2
-5.05 -5.05 0.49 -5.19Z-2a E-2a
+2.31 +2.11 +2.14 +1.47Z-2a Z-2
-0.60 -0.77 +3.36 -1.77E-2a E-2
-7.36 -7.16 -1.65 -6.67Calculated thermodynamic data of conformational isomerization for Z/E isomers of 2 are shown in Table 2. According to the relationship of G = -RT(lnK),6 G(298 K) of –1.77 kcal/mol indicates that around 95% of Z-isomer of 2 stay as Z-2, and around 99.99% of E-isomer of 2 stay as E-2 due to G(298 K) of –6.67 kcal/mol.
Calculated thermodynamic data of Z/E isomerization of 2 are shown in Table 2.
According to experimental results, isomerization of 2 from Z-isomer to E-isomer is
spontaneous at room temperature. However, calculated free energies of Z/E isomerization from Z-2 to E-2a or from Z-2a to E-2a are 3.24 and 1.47 kcal/mol, respectively, indicating these two processes are not thermodynamically favorable and
E-2a is much less stable than both Z-2 and Z-2a. On the other hand, calculated free
energies of Z/E isomerization from Z-2 to E-2 or from Z-2a to E-2 are –3.43 and –5.19 kcal/mol, respectively, which are consistent with experimental results and much more negative than –0.62 kcal/mol for Z/E isomerization of 2-butene.7 Since around 95%
of Z-isomer of 2 stay as Z-2, the process from Z-2 to E-2 is considered. Negative entropy is not favorable for this isomerization from Z-2 to E-2, but favorable enthalpy dominates this isomerization. Therefore, to answer the question why the reaction of 1 with ethyl orthoformate is highly stereoselective, one needs to look further into relative stability between Z-2 and E-2.
Single point energies of B3LYP/6-31+G*-optimized structures of Z-2 and E-2 at three different levels are shown in Table 1. E-2 is 4.3~5.1 kcal/mol more stable than
Z-2. Why is E-2 much more stable than Z-2? Relative stability of geometric
isomers may be controlled by several factors such as intramolecular hydrogen bonding, dipole moment, steric hindrance, and electron delocalization. These factors will be discussed as follows in order to explain relative stability between Z-2 and E-2.
Based on the optimized structure of Z-2, it is unlikely that Z-2 has an intramolecular hydrogen bonding. On the other hand, Z-2a does form intramolecular hydrogen bonding, but this stabilization effect is offset by steric hindrance between C(O)NHC(O)OEt and ethoxy groups, making Z-2a even 0.6 kcal/mol less stable than
Z-2. In E-2, distance between vinyl hydrogen and oxygen of amide moiety is 2.40 Å
and electronegativity of vinyl carbon is not strong enough, so it is unlikely that E-2 has an intramolecular hydrogen bonding. Therefore, the factor of intramolecularhydrogen bonding is not important when we explain why E-2 is much more stable than Z-2.
As shown in Table 1, dipole moments of Z-2 and E-2 are 3.52 and 3.12 debye, respectively, at level of B3LYP/6-311++G(3df,3pd)//B3LYP/6-31+G*. Dipole moments of chloroform and acetonitrile are 1.04 and 3.92 debye, respectively.8 Dielectric constants of chloroform and acetonitrile are 4.81 and 36.6, respectively.8 Chloroform is a nonpolar aprotic solvent while acetonitrile is a dipolar aprotic solvent.
Based on a useful rule of thumb -“like dissolves like”, 9 chloroform may stabilize E-2 better than Z-2, while acetonitrile may stabilize Z-2 better than E-2. However, the reaction of 1 with ethyl orthoformate is highly stereoselective in both chloroform and acetonitrile. It indicates that dipole moment is not a major factor to make E-2 much more stable than Z-2.
To investigate contribution of resonance effects and steric hindrance to the stability of both E-2 and Z-2, E-2 is divided into two systems, Z-3 and E-4, and Z-2 is divided into another two systems, E-3 and Z-4. Isodesmic reactions of Eq. 3~6 were designed to predict resonance stabilization in Z-3, E-4, E-3 and Z-4, respectively.
The isodesmic reaction, in which the total number of each type of bond is identical in the reactants and products,4b successfully predicts heat of formation4b and substituent effects on stability of functional groups.10 To consider steric hindrance in Z-2, E-2,
Z-3, and Z-4, n-propyl group replaces ethoxy substituent and Z-5, E-5, Z-6 and E-6
were designed in order to significantly reduce resonance effect along two substituents across C=C. Therefore, steric hindrance in Z-3 is close to that in Z-5, which is 0.21 kcal/mol according to Eq. 7, and resonance stabilization in Z-3 is ca. 10.69 kcal/mol (= 10.48 + 0.21) based on Eq. 3. Similarly, steric hindrance in Z-4 is close to that in
Z-6, which is equal to 2.49 kcal/mol based on Eq. 8, and resonance stabilization in Z-4
is ca. 10.66 kcal/mol (= 2.49 + 8.17) according to Eq. 6.OEt NC
H H
H H
CH3CH3 H2C CH2
+ +
E = 10.48 kcal/mol
Z-3 8
H H
OEt NC
(Eq. 3)
Z-3 - steric hindrance: ca. 0.21 kcal/mol;
resonance stabilization: ca. 10.69 kcal/molH
OEt H
O EtO(O)CHN
E-4
H OEt H
H H
CH3CH3 H2C CH2
+ +
E = 12.68 kcal/mol
9 EtO(O)CHN
O
(Eq. 4)
E-4 - steric hindrance: ca. 0.0 kcal/mol;
resonance stabilization: ca. 12.68 kcal/molE-3
OEt NC
H H
H H
CH3CH3 H2C CH2
+ +
E = 11.24 kcal/mol
8
NC H
OEt H
(Eq. 5)
E-3 - steric hindrance: ca. 0.0 kcal/mol;
resonance stabilization: ca. 11.24 kcal/molH OEt H
H H
9 EtO(O)CHN
O
Z-4
CH3CH3 H2C CH2
+ E = 8.17 kcal/mol +
H H
OEt EtO(O)CHN
O
(Eq. 6)
Z-4 - steric hindrance: ca. 2.49 kcal/mol;
resonance stabilization: ca. 10.66 kcal/molE=0.21 kcal/mol
E-5 Z-5
NC H
CH
2CH
2CH
3H
NC CH
2CH
2CH
3H H
(Eq. 7)
Z-5 - steric hindrance: ca. 0.21 kcal/mol
E=2.49 kcal/mol
E-6 Z-6
H
CH2CH2CH3 H
O EtO(O)CHN
CH2CH2CH3
H H
O EtO(O)CHN
(Eq. 8)
Z-6 - steric hindrance: ca. 2.49 kcal/mol;
E-7
OEt H2N(O)C
H H
H H
CH3CH3 H2C CH2
+ +
E = 12.24 kcal/mol
10
H2N(O)C H
OEt H
(Eq. 9)
E-7 - steric hindrance: ca. 0.0 kcal/mol;
resonance stabilization: ca. 12.24 kcal/molContribution of both resonance stabilization and steric hindrance in E-2 can be estimated to be sum of stabilization energies in both Eq. 3 and Eq. 4, which is equal to 23.16 kcal/mol. Similarly, contribution of both resonance stabilization and steric hindrance in Z-2 presumably equals to sum of stabilization energies in both Eq. 5 and Eq. 6, which is equal to 19.41 kcal/mol. Then, energy difference between E-2 and Z-2, which is calculated from these four isodesmic reactions, is 3.75 kcal/mol, which is close to energy difference (4.5 kcal/mol) of these two molecules calculated at level of B3LYP/6-31+G*.
To investigate the contribution of resonance effects and steric hindrance to the stability of E-2 and Z-2, isodesmic reactions of Eq. 3~8 are considered. Eq. 7 and 8 show a small steric interaction (0.21 kcal/mol) with the cyano group group cis to an n-propyl group, whereas there is a significant steric interaction of 2.49 kcal/mol with the C(O)NHC(O)OEt group cis to an n-propyl group. The difference (2.28 kcal/mol) is taken as the steric contribution to the energy difference between E-2 and Z-2. The total energy difference between E-2 and Z-2 may be estimated as the stabilization
energy difference Eq. 3 + Eq. 4 – Eq. 5 – Eq. 6 = 3.75 kcal/mol, so the resonance stabilization for E-2 relative to Z-2 is estimated as 3.75 – 2.28 = 1.47 kcal/mol.
Trans delocalization energy from ethoxy to C(O)NHC(O)OEt group in E-4 is 12.68 kcal/mol based on Eq. 4, while trans delocalization energy from ethoxy to nitrile group in E-3 is 11.24 kcal/mol according to Eq. 5, indicating that C(O)NHC(O)OEt group is a better -acceptor than nitrile group by 1.44 kcal/mol.
Based on Eq. 9, trans delocalization energy from ethoxy to C(O)NH2 group in E-7 is 12.24 kcal/mol, indicating that C(O)NH2 group is a better -acceptor than nitrile group by 1.00 kcal/mol but a worse -acceptor than C(O)NHC(O)OEt group by 0.44 kcal/mol. However, reported resonance substituent constants R (or R) of nitrile and C(O)NH2 are 0.15, 0.10 by Hansch, et al.,11a and 0.08, 0.08 by Charton,11b indicating that the resonance substituent constants vary with different models or solvents.
-accepting ability: C(O)NHC(O)OEt > C(O)NH2 > CN
-accepting (inductive) ability: CN > C(O)NHC(O)OEt > C(O)NH2
We successfully obtained field substituent constants from relative deprotonation Gibbs free energies of 4-substituted quinuclidinium ions 11 (Eq. 10) by ab initio calculations at level of CBS-4M.11 The relative deprotonation Gibbs free energies were rescaled by a factor of –1/17.53 to become FG,12 which is well correlated with Taft’s F and Charton’s I.11 Now we get the field substituent constants of CN, C(O)NH2, and C(O)NHC(O)OEt by the same way at level of MP2/6-31+G*//HF/6-31+G*. As shown in Table 3, both calculation levels, CBS-4M and MP2/6-31+G*//HF/6-31+G*, give similar and coherent results. The FG of CN, C(O)NH2, and C(O)NHC(O)OEt is 0.66, 0.27, and 0.35, respectively, indicating the sequence of inductive effect is CN > C(O)NHC(O)OEt > C(O)NH2. It was reported
that acetonitrile is 3 order magnitude more acidic than N,N-dimethylacetamide,6 indicating that more acidity of acetonitrile is dominated by the inductive effect because amide group is a better -acceptor than nitrile group.
N
N NH+
NH+ R
R
+ +
G(298 K)
11a~11d 12a 11a 12a~12d
(Eq. 10)11a,12a: R=H; 11b,12b:R=CN; 11c,12c: R=C(O)NH2; 11d,12d: R=C(O)NHC(O)OEt
Table 3. Relative deprotonation Gibbs free energies (G(298 K)) (kcal/mol) of 4-substituted quinuclidinium ions 11a~11d (Eq. 10), the calculated field substituent constants (FG) at levels of MP2/6-31+G*//HF/6-31+G* (and CBS-4M) in gas phase, and Charton’s inductive substituent constant (I) 11b R -G(298 K) FG I
H 0.00 (0.00) 0.00 (0.00) 0.00 CN 11.61 (11.65) 0.66 (0.66) 0.57 C(O)NH2 4.72 (4.21) 0.27 (0.24) 0.28 C(O)NHC(O)OEt 6.16 0.35
Conclusion:
The highly stereoselective reaction of 1 with ethyl orthoformate in the presence of acetic anhydride produces E-2 only. E-2 cannot be isomerized to Z-2 thermally but photochemically while Z-2 can be isomerized back to E-2 thermally, indicating the reaction of 1 with ethyl orthoformate is thermodynamic control. Calculated free energy of Z/E isomerization from Z-2 to E-2 is –3.43 kcal/mol, which is thermodynamically favorable and consistent with the experimental results. Negative
entropy is not favorable for this isomerization, but favorable enthalpy dominates this isomerization. Both resonance stabilization of 1.47 kcal/mol and steric hindrance of 2.28 kcal/mol in favor of E-2 contribute to the energy difference (3.75 kcal/mol) between Z-2 and E-2 calculated from the four isodesmic reactions of Eq. 3~6, and that causes the reaction of 1 with ethyl orthoformate highly stereoselective. Nitrile group is a better -acceptor than C(O)NHC(O)OEt group while C(O)NHC(O)OEt group is a better -acceptor than nitrile group.
EXPERIMENTAL
General. Unless otherwise stated reagents were obtained from commercial suppliers and used as received. Ethyl (2-cyanoacetyl)carbamate, 1, was prepared according the literature method.13
E-Ethyl (2-Cyano-3-ethoxyacryloyl)carbamate (E-2): To a solution of 1 (0.156 g,
1 mmol) and acetic anhydride (1 mL) in 2 mL of chloroform was ethyl orthoformate (0.296 g, 2 mmol) added. The mixture was refluxed at 80°C under nitrogen atmosphere for 2 h. After the reaction was complete, the reaction mixture was cooled down and concentrated by rotary evaporator. Ether was poured into the reaction mixture and the mixture stayed in fridge for 12 hrs. After filtration of the mixture, white powder was collected and recrystallized by chloroform/ether. Yield:
75%;1H NMR (CD3CN) 1.23 (3H, t, CH3), 1.34 (3H, t, CH3), 4.12 (2H, q, CH2), 4.39 (2H, q, CH2), 8.17 (1H, s, CH), 9.12 (1H, s, NH);13C NMR (CDCl3) 14.80, 15.87, 63.13, 75.69, 89.30, 114.69, 152.22, 162.01, 175.13; IR (thin film) 2227 (CN), 1774, 1689 (C=O) cm-1; MS (EI) m/z 212 (4, M+), 118 (100), 88 (32), 74 (24), 57 (28);
HRMS (EI) m/z calcd for C9H12N2O4 212.0797, found 212.0801.
Acknowledgment. Both financial supports by the National Science Council of Taiwan (NSC 92-2113-M-006-010) and computer time from National Center for High-performance Computing of Taiwan are gratefully acknowledged.
REFERENCES
1. (a) Leeming P, Ray CA, Simpson SJ, Wallace TW, Ward RA. Tetrahedron. 2003;
59: 341-352. (b) Volle JN, Schlosser M. Eur. J. Org. Chem. 2002; 9: 1490-1492.
(c) Wang JH, Shen YQ, Yu CX, Si JH. J. Chem. Soc., Perkin Trans. 1. 2000;
1455-1460. (d) Palanki MSS, Erdman PE, Gayo-Fung LM, Shevlin GI, Sullivan RW, Suto MJ, Goldman ME, Ransone LJ, Bennett BL, Manning AM. J. Med.
Chem. 2000, 43: 3995-4004. (e) Hanack M, Hackenberg J, Menke O,
Subramanian LR, Schlichenmaier R. Synthesis. 1994; 249-251. (f) Phillips JG, Chu D, Seif L, Spanton S, Henry R, Plattner JJ. Tetrahedron Lett. 1993; 34:
3259-3262. (g) McFadden HG, Huppatz JL, Aust. J. Chem. 1991;44: 1263-1273.
(h) Chu DTW, Lico IM, Claiborne AK, Faubl H. Can. J. Chem. 1991; 25:
1521-1527. (i) Hosmane RS, Bertha CM. J. Org. Chem. 1990; 55: 5206-5216. (j) Doleschall G, Seres P. J. Chem. Soc., Perkin Trans. 1. 1988; 1875-1879. (k) Moriya O, Urata Y, Ikeda Y, Ueno Y, Endot T. J. Org. Chem. 1986; 51:
4708-4709. (l) Perez MA, Soto JL, Guzman F, Diaz A. Synthesis. 1984;
1045-1047. (m) Arys M, Christensen TB, Eriksen J. Tetrahedron Lett. 1984; 25:
1521-1524.
2. Ceder O, Stenhede U. Tetrahedron. 1973; 29: 1585-1590.
3. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Jr., Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V,
Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, AlLaham MA, Peng CY, Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson B, Chen W, Wong M W, Andres JL, Gonzales C, Head-Gordon M, Replogle ES, Pople JA. Gaussian98. Revision A9. Gaussian, Inc.: Pittsburgh, PA, 1998.
4. (a) Sung K, Wu SH, Wu RR, Sun SY. J. Org. Chem. 2002; 67: 4298-4303.
(b) Foresman JB, Frisch A. Exploring Chemistry with Electronic
Structure Methods, 2
nd Ed. Gaussian Inc.: Pittsburgh, 1996.5. Silverstein RM, Webster FX. Spectrometric Identification of Organic Compounds, 6th Ed. John Wiley & Sons: New York, 1998.
6. Carey FA, Sundberg RJ. Advanced Organic Chemistry, Part A: Structure and
Mechanism, 2
nd ed. Plenum Press: New York, 1984.7. Isaacs N. Physical Organic Chemistry, 2nd Ed. Longman Scientific & Technical:
Essex, 1995.
8. Lide DR. Handbook of Chemistry and Physics, 85th ed. CRC Press: New York, 2004.
9. Morrison RT, Boyd RN. Organic Chemistry, 6th ed. Prentice Hall International Limited: London, 1992.
10. (a) Sung K. J. Org. Chem. 1999; 64: 8984-8989. (b) Sung, K. J. Chem. Soc.,
Perkin Trans. 2 1999;
1169-1173. (c) Sung, K. J. Chem. Soc., Perkin Trans. 2 2000; 847-852.11. (a) Hansch C, Leo A, Taft RW. Chem. Rev. 1991; 91: 165-195. (b) Charton M,
Progress in Physical Organic Chemistry 1981;
13: 119-251.12. Sung, K. J. Chem. Soc., Perkin Trans. 2 2002; 1658-1661.
13. Atkinson MR, Shaw G, Warrener RN. J. Chem. Soc. 1956; 4118-4123.
參. Mechanistic studies of amination of ketenimines: change of rate- determining step by N-substituents through electronic effects
Sung, K.; Chen, F.-L.; Huang, P.-M.; Chiang, S.-M. Tetrahedron 2005, in press. (SCI)
Introduction:
Chemistry of ketenes and ketenimines has been intensely studied for a century.1 They are important reactive intermediates, which may occur as transients in many thermal and photochemical reactions.1,2 There has been intense interest in their addition reactions, including cycloadditions,3 nucleophilic additions,2b,f,4 electrophilic additions,5 and radical additions.6 Both amination of ketenes2f,4c-f and hydration of ketenimines2b,4h,i generate the amide functional group, which is a repeating unit in peptides and other synthetic polymers like nylon.
There was a mechanistic controversy regarding where an initial addition occurs on ketenes and ketenimines when they reacts with amines.2f,4c-f,7,8
Recent results2f,4c-f,7 confirm that they are two-step reactions and involve an initial addition of an amine to C=O or C=N, followed by tautomerization. In the recent literatures regarding amination reactions of ketenimines7 or ketenes,2f,4d,e intermediates of vinylidenediamines or enol amides were found, indicating that the rate-determining step is the second step involving tautomerization.
In our previous research, we found that reaction of N-phenylphenylketenimine 1a with n-butylamine involves two steps including an initial addition to C=N,
followed by tautomerization to give amidine 2a.7 The reaction runs fast at room
temperature, so the metastable intermediate of vinylidenediamine was caught and identified by means of low-temperature proton NMR. In the low-temperature proton NMR experiment, it was found that the second step involving tautomerization is much slower than the first addition step.7 On the other hand, when we changed N-substituent of the ketenimine from phenyl group to i-propyl group 3a and did the same amination reaction, surprisingly no intermediate was found at all by means of the low-temperature proton NMR. In this article, we designed model reactions and used ab initio calculations to inspect how and why the rate-determining step of these amination reactions is changed. To our knowledge, this is the first example to demonstrate change of rate-determining step in the reactions of ketenimines and ketenes.
H
2N(n-Bu)
+
+
H
2N(n-Bu)
3a invisible by
1
H NMR 4aPhCH
2C N
NH(n-Bu)
C C
H
Ph
NH
NH(n-Bu) H
C C N
Ph
observable by
1
H NMR 2a 1aC C N
Ph
Ph H
C C
H
Ph
NHPh
NH(n-Bu)
PhCH
2C NPh
NH(n-Bu) slow
Computational Details
All the calculations reported here were performed with Gaussian98 program.9 The Onsager self-consistent reaction field (SCRF) model has been used to monitor systems in a solvent with dielectric constant of 35.9 which is close to that of acetonitrile. The model treats the solvent as a continuum of uniform dielectric constant (the reaction field) and the solute is placed into a fixed spherical cavity of radius a0 within the solvent. The radius a0 of the cavity for each solute was
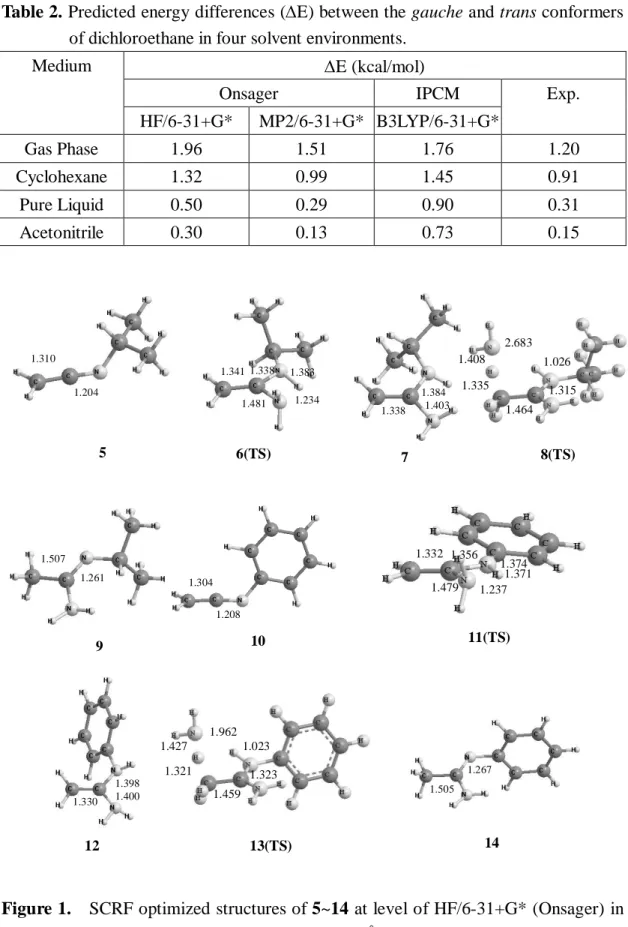
evaluated based on its optimized structure in the gas phase. SCRF geometry optimizations of 5 ~ 14 were carried out at level of HF/6-31+G* in solvent (= 35.9) without any symmetry restriction. Many possible conformations have been optimized for each of these configurations, and the conformation with the lowest energy was chosen for each configuration. Their optimized structures are shown in Fig. 1. An SCRF frequency calculation at each of SCRF optimized structures was run at the same level and analytical vibration frequencies were calculated to determine the nature of the located stationary points. Thus, all the stationary points found were properly characterized by evaluation of the harmonic frequencies. The energies of all the stationary points were calculated at MP2/6-31+G* with scale zero-point vibration energies included, and population analyses of 5 and 10 were carried out at the same level. (Table 1) The scale factor of 0.9135 for zero-point vibration energies is used according to the literatures.10 As shown in Table 2, 10 it was reported that the Onsager model at MP2/6-31+G* level gives the best prediction results for the energy difference between the gauche and trans conformers of dichloroethane in several solvents.10 Therefore, the Onsager model at MP2/6-31+G* level was used in this research to predict the reaction mechanism of the amination of ketenimines.
Table 1. Calculated energies (hartree), imaginary frequencies (cm-1), and spherical cavity of radius a0 (Å) of stationary points along amination of ketenimines in solvent (= 35.9) at MP2/6-31+G*//HF/6-31+G* Level (Onsager model)
Stationary Point
a0 Energy Imaginary Frequency
Stationary Point
a0 Energy Imaginary Frequency
5 3.86 -249.66817 10 4.04 -362.47773
6(TS) 4.29 -305.95331 -2109 11(TS) 4.66 -418.77114 -2115
7 4.06 -306.02672 12 4.41 -418.83987
8(TS) 4.23 -362.31219 -1880 13(TS) 4.60 -475.12451 -1825
9 4.22 -306.05061 14 4.46 -418.86874
NH3 2.77 -56.33085
Table 2. Predicted energy differences (E) between the gauche and trans conformers of dichloroethane in four solvent environments.
E (kcal/mol)
Onsager IPCM
Medium
HF/6-31+G* MP2/6-31+G* B3LYP/6-31+G*
Exp.
Gas Phase 1.96 1.51 1.76 1.20
Cyclohexane 1.32 0.99 1.45 0.91
Pure Liquid 0.50 0.29 0.90 0.31
Acetonitrile 0.30 0.13 0.73 0.15
6(TS) 7 8(TS)
5
9 10
12 13(TS) 14
1.310
1.204
1.383
1.234 1.481
1.338 1.341
1.338 1.384
1.403
1.261 1.507
1.304
1.208
1.330 1.398
1.400 1.505
1.267
11(TS) 1.374
1.371 1.237 1.479 1.332 1.356
1.459 1.321 1.427
1.962 1.023
1.323
1.464 1.335 1.408
2.683 1.026
1.315
Figure 1. SCRF optimized structures of 5~14 at level of HF/6-31+G* (Onsager) in a solvent (=35.9). Bond lengths are in Å.
Results and Discussion:
When we studied the amination of N-phenylphenylketenimine 1a with n-butylamine in CD3CN at –10°C by proton NMR spectrometer, the vinylidenediamine intermediate was caught and we found that it involves two steps, C=N addition and tautomerization, and the second step is much slower than the first step.7 The same reaction mechanism was found for amination of other N-p-substituted-phenyl-phenylketenimines 1b and 1c with n-BuNH2 by monitoring the reactions with proton NMR spectrometer in CD3CN at –10°C. One representative example is shown in Fig. 2.7 On the other hand, reactions of N-i-propyl-p-substituted-phenylketenimines 3a~3e with n-BuNH2 in CD3CN are much slower than those of 1a~1c with n-butylamine, so they were monitored by proton NMR spectrometer at 10°C. Surprisingly, there is no intermediate found for each of the reactions, and one representative example is shown in Fig. 3. To find out this strange result, reactions of ketenimines 5 and 10 with NH3 were monitored by ab
initio calculations in the solvent with = 35.9.
+
H2N(n-Bu)observable by
1H NMR 2a ~ 2c 1a ~ 1c
C C N
Ph
X
H
C C H
Ph
NH
NH(n-Bu) X
PhCH2 C N
NH(n-Bu) slow X
1a, 2a : X = H ; 1b, 2b : X = OMe ; 1c, 2c : X = Br
H2N(n-Bu)
+
invisible by
1H NMR
CH2 C N
NH(n-Bu)
X C C
H NH
NH(n-Bu)
X H
3a, 4a : X = H ; 3b, 4b : X = CH
3; 3c, 4c : X = OMe ; 3d, 4d : X = Cl ; 3e, 4e : X = NO
23a ~ 3e 4a ~ 4e
C C N
X
Figure 2. Part of 1H NMR (CD3CN) spectra for the reaction of 1a with n-butylamine in the presence of an internal
standard (benzyl phenyl ether) at –10°C (a) before adding n-butylamine, (b) at 10 min after mixing the solution,