Calvatia lilacina Protein-Extract Induces Apoptosis through Glutathione Depletion in Human Colorectal
Carcinoma Cells
JWU-GUHTSAY,† KING-THOMCHUNG,‡ CHUNG-HUNGYEH,§ WAN-LINGCHEN,† CHI-HUNG CHEN,| MARTIN HSIU-CHU LIN,⊥ FUNG-JOULU,#
JENG-FONG CHIOU,*,∇ ANDCHING-HSEINCHEN*,O
Department of Nutrition and Health Science, Toko University, Puzih City, Chiayi County 61363, Taiwan, Department of Biology, The University of Memphis, Memphis, Tennessee 38152, Division of Colon and
Rectal Surgery, Department of Surgery, Chiayi Chang Gung Memorial Hospital, Chiayi 613, Taiwan, Graduate Institute of Food Science and Biopharmaceutics, National Chiayi University, 300 University Road, Chiayi 600, Taiwan, Department of Neurosurgery, Chiayi Chang Gung Memorial Hospital, Chiayi 613, Taiwan, Department of Applied Chemistry, Chung Shan Medical University, Taichung 402, Taiwan, Cancer Center and Department of Radiation Oncology, Taipei Medical University and Hospital Taipei,
Taiwan, Graduate Institute of Biomedical and Biopharmaceutical Sciences, College of Life Sciences, National Chiayi University, Chiayi 60004, Taiwan
This paper reports that a novel protein extract isolated from Calvatia lilacina (CL) can induce cell death against four types of human colorectal cancer cells. Importantly, CL was shown to be free of apoptotic effects against normal rat liver cells. We have also identified that CL-induced glutathione (GSH) depletion is the major contributor responsible for the apoptotic cell death induction of SW 480 cells, as evidencd by the observation that exogenously added N-acetylcysteine (NAC), or GSH, but not vitamin C, could offer a near complete protection of CL-treated cells against apoptotic cell death.
Furthermore, the participation of reactive oxygen species (ROS) evoked a drop in the transmembrane potential (∆Ψm) in the CL-induced apoptotic cell death. This observation can only be deemed as a minor pathway due to the fact that cyclosporine A (CyA) could only partially rescue the CL-treated cells from apoptotic cell death. Likewise, despite the fact that CL could induce the upregulation of Bax, its knockdown via siRNA (48 h) failed to completely mitigate apoptotic cell death, indicating that its role in this apoptotic process was insignificant. To further explore the possible underlying mechanism associated with CL-induced GSH depletion, we proceeded to determine the effect of CL on the cellular γ-glutamylcysteine synthetase (γ-GCS), a rate-limiting enzyme responsible for GSH biosynthesis, and demonstrated that indeed γ-GCS could be repressed by CL. Taken together, we report here for the first time that the anticancer effect of CL on human colorectal cancer cells is mediated through GSH depletion mechanism rather than a ROS-mediated killing process. This functional attribute of CL can thus provide the basis for the strategic design of a treatment of colorectal cancer.
KEYWORDS: Calvatia lilacina; colorectal carcinoma; glutathione epletion; γ-glutamylcysteine synthetase;
apoptosis
INTRODUCTION
CalVatia lilacina (Mont. et Berk.) Lloyd (Fam. Lycoper- daceae) is a medicinal and edible mushroom, which was originally grown in China and Taiwan. It was widely used for
the treatment of throat and nasopharyngeal pains, loss of voice, coughing, hemorrhagic vomiting, nose bleeding, and traumatic bleeding. The growing season of CalVatia lilacina is from July to September. It takes only 4-5 days for the maturation of the carpophores, which is the main portion for medicinal usage (1).
In the past decade, there were many reports that the extracts of medicinal or edible mushrooms have therapeutic effects in various cancer cells. Most of these edible mushroom extracts contain water-soluble proteins or polypeptides. The chemical substances in carpophore of CalVatia lilacina contain leucine, tyrosine, urea, ergosterol, lipoid, gemmaetin, and calvacin (2).
Among them, calvacin is an antitumor agent.
* To whom correspondence should be addressed. For C.-H.C.:
E-mail, chench@mail.ncyu.edu.tw; phone, +886-5-2717837; fax, +886- 5-2717778. For J.-F.C.: E-mail, sjfchiou@ms68.hinet.net; phone, +886- 2-27367117; fax, +886-2-27367014.
†Department of Nutrition and Health Science, Toko University.
‡Department of Biology, The University of Memphis.
§Division of Colon and Rectal Surgery, Department of Surgery, Chiayi Chang Gung Memorial Hospital.
|Graduate Institute of Food Science and Biopharmaceutics, National Chiayi University.
⊥Department of Neurosurgery, Chiayi Chang Gung Memorial Hospital.
#Department of Applied Chemistry, Chung Shan Medical University.
∇Cancer Center and Department of Radiation Oncology, Taipei Medical University and Hospital Taipei.
OGraduate Institute of Biomedical and Biopharmaceutical Sciences, College of Life Sciences, National Chiayi University.
10.1021/jf8030265 CCC: $40.75 2009 American Chemical Society Published on Web 01/30/2009
It is important to clarify the possible functional relation- ships between GSH depletion and apoptosis. In view of the GSH depletion, buthionine sulfoximine (BSO) is a GSH synthase inhibitor, which causes GSH depletion. In the BSO- induced apoptosis, mitochondrial dysfunction and Bax over- expression have close functional relations (3). Proapoptotic Bax is a requisite gateway for cell death. Enforced dimer- ization of Bax results in its translocation, mitochondrial dysfunction, and apoptosis. The mitochondrial GSH could prevent cardiolipin peroxidation. Once GSH depletion occurs, cardiolipin peroxidation will cooperate with oligomerized Bax to cause mitochondrial outer membrane permeabilization through destabilization of the lipid bilayer of mitochondria.
The translocation and integration of Bax into mitochondria would induce cytochrome c to release that occurs before mitochondrial dysfunction (4).
Several studies have reported that some mushrooms are cancer fighting. It was reported that FA-2-b-β extracted from Agaricus blazei in combination with 3′-azido-3′-deoxythymidine produced a synergistic effect against the gastric cancer cells MKN45 (5).
Antrodia camphorata crude extract inhibited androgen-respon- sive LNCaP and -independent PC-3 prostate cancer cells through G1/S phase arrest and G2/M phase arrest, respectively (6). The extracts of Grifola frondosa and Phellinus linteus induced a significant growth reduction in bladder cancer cells (7). The water-soluble proteins or polypeptides extracted from Pleurotus ostreatus produced a significant cytotoxicity and apoptosis of PC-3 cells (8). The aqueous polysaccharide extract from Pleurotus ostreatus also induced antiproliferative and pro- apoptotic effects of HT-29 colon cancer cells (9). Coprinus comatus, Coprinellus sp., and Flammulina Velutipes contain potent antitumor compounds against breast cancer (10). Polyo- zellus multiplex was reported as a potent chemopreventive agent against stomach cancer (11). However, the study on the anticancer effects of CalVatia lilacina is lacking. In this paper, we report the anticancer effects of CalVatia lilacina protein- extract (CL) on human colorectal SW480 cancer cells by examining the induction of the glutathione (GSH) depletion and the mitochondrial dysfunction dependent apoptosis.
MATERIALS AND METHODS
Experimental Material. CalVatia lilacina (Mont. et Berk.) Lloyd (Fam. Lycoperdaceae) was collected from National Chiayi University, Chiayi, Taiwan, July, 2007.
Extraction and Isolation of Proteins. CalVatia lilacina (400-450 g) was washed twice with distilled water and then homogenized in cold extraction buffer (50 mM KH2PO4, 150 mM NaCl, and 1 mM EDTA, pH 7.3) at the highest speed in a Waring blender for 10 min.
The water-soluble protein-extract homogenate was filtrated through two layers of surgical gauze, and the filtrate was centrifuged at 8900g for 20 min. The supernatants were precipitated by addition of ammonium sulfate to 80% saturation and standing at 4 °C overnight and then centrifuged at 8900g for 30 min. The protein-pellets were redissolved in phosphate buffer solution (123 mM NaCl, 10.4 mM Na2HPO4, and 3.16 mM KH2PO4, pH 7.3) (weight/volume ) 1:1) and centrifuged.
The supernatants were dialyzed against 50-fold volume of phosphate buffer solution three times at 4°C and clarified by centrifugation. The supernatants were filtrated through 0.45 µm Millex filter units (Milli- pore) and concentrated by Amicon Ultra-4 (10 kDa cutoff, Millipore) at 4000g for 20 min. The 1 mL concentrate was sterilized by 0.22 µm Millex filter units (Millipore) in a laminar flow chamber and exerted freeze-drying to obtain powder and then stored at -80°C as stocks.
Cell Lines and Reagents. Four human colorectal cancer cell lines SW480, Colo 205, Colo 320 DM, and DLD-1 and one rat normal liver cell line clone 9 were obtained from the Bioresource Collection and Research Center (Hsinchu, Taiwan). The “Apo-BrdU” kit was bought
from BD Pharmingen (San Diego, CA). The chloromethylflourescein diacetate (CMF-DA) and lipofectactamine 2000 were acquired from Invitrogen Corporation (Carlsbad, CA). The Bioxytech GSH/GSSG- 412 assay kit was bought from Oxis International Inc. (Foster City, CA). Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were obtained from Hyclone (South Logan, UT).
Propidium iodide (PI), N-acetylcysteine (NAC), glutathione (GSH), cyclosporin A (CyA), 2′,7′-dichlorodihydrofluorescein-diacetate (DCFH- DA), rhodamine 123, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetra- zolium bromide (MTT), DNase-free RNase A, vitamin C (Vit C), and other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Cell Culture and Teatment. SW480 Cells were cultured in DMEM medium containing 10% fetal bovine serum (FBS). Colo 205, Colo 320 DM, and DLD-1 cells were cultured in RPMI-1640 medium containing 10% FBS. Clone 9 cells were cultured in F-12K medium containing 10% FBS. The stock solution of CL was dissolved in the phosphate buffer saline (PBS) (136.89 mM NaCl, 2.68 mM KCl, 10.14 mM Na2HPO4, and 1.76 mM KH2PO4, pH 7.4) and different concentra- tions (µg/mL) were prepared in the DMEM, RPMI-1640, or F-12K medium.
Cell Cycle and DNA Damage Analysis of Cells. The cell cycle and DNA damage was measured with propidium iodide (PI) staining and flow cytometry. PI is a specific fluorescence dye for double-stranded DNA. In methanol-fixed cells, PI could penetrate into the nuclear region and bind to DNA. The cells in G1and G2/M phases would be in the diploid and the tetraploid regions of the DNA distribution histograms, respectively. The cells in S phase would be located between in the diploid and the tetraploid regions of the DNA distribution histograms.
Once DNA was damaged, the length of DNA in cells would be shorter then that of cells in G1phase. The numbers of molecules of PI bound to DNA-damaged cells would be less than that of the cells bound to G1phase; the PI fluorescence intensity in DNA-damaged cells would be weaker than that of the cells in G1phase. The percentage of cells undergoing DNA damage was defined as the percentage of cells in the subdiploid region (SubG1) of the DNA distribution histograms.
Cells (1× 106) were cultured in 60 mm tissue-culture dishes for 24 h. The culture medium was replaced by new medium, and then cells were exposed with or without various concentrations of CL for 24 h. After treatment, adherent and floating cells were pooled, washed with PBS, fixed in PBS-methanol (1:2, volume/volume) solution, and then maintained at 4°C for at least 18 h. After one wash with PBS, the cell pellets were stained with the fluorescent probe solution containing PBS, 40 µg/mL PI, and 40 µg/mL DNase-free RNase A for 30 min at room temperature in the dark. DNA fluorescence of PI-stained cells was analyzed by excitation at 488 nm and monitored through a 630/22 nm band-pass filter using a Becton-Dickinson FACSan flow cytometer (Franklin Lakes, NJ). A minimum of 10000 cells was counted per sample, and the DNA histograms were evaluated further using Modfit software on a PC workstation to calculate the percentage of cells in various phases of the cell cycle.
Cell Viability Assays. Cell viability was determined by MTT assay.
MTT assay can be used to determine cell death but not cellular DNA damage caused by potential medicinal agents and other toxic materials.
Yellow MTT is reduced to purple formazan in the mitochondria of living cells. A solubilization agent such dimethyl sulfoxide (DMSO) or sodium dodecyl sulfate (SDS) is added to dissolve the insoluble purple formazan product into a colored solution. The absorbance of this colored solution can be quantified by measuring at a certain wavelength (usually between 500 and 600 nm) by a spectrophotometer.
When the amount of purple formazan produced by cells treated with a potential medicinal agent or other toxic material is compared with the amount of formazan produced by untreated cells, the effectiveness of the potential medicinal agents or other toxic materials in causing cell death can be deduced through the production of a dose-response curve.
Cells (1× 106) were cultured in 60 mm tissue-culture dishes for 24 h. The culture medium was replaced with new medium and then exposed to 150 µg/mL of CL for 0.5, 1, 3, 6, 12, or 24 h. After the indicated treatments, cells were incubated for 2 h with 0.5 mg/mL of MTT reagent and lysed with DMSO. Absorbance was measured at 595 nm in a microplate reader (Bio-Rad, Richmond, CA).
Measurement of Intracellular ROS by Flow Cytometry. Produc- tion of intracellular ROS was detected by flow cytometry using DCFH- DA. After treatment, cells were treated with 10 µM DCFH-DA for 30 min in the dark, washed once with PBS, detached by trypsinization, collected by centrifugation, and suspended in PBS. The intracellular ROS, as indicated by the fluorescence of dichlorofluorescein (DCF), was measured with a Becton-Dickinson FACSan flow cytometer.
Measurement of GSH Depletion and GSSG/GSH Ratio. After treatment, the SW480 cells were incubated with 25 µM CMF-DA for 20 min at 37°C in a CO2incubator. Once CMF-DA is inside the cell, cytosolic esterase cleaves their acetates, and then the chloromethyl group reacts with intracellular thiols, changing the probe into a cell- impermeable fluorescent dye-thioether adduct, chloromethylflourescein (CMF). The CMF fluorescence is directly related to intracellular GSH level. After CMF-DA staining, the cells were washed with PBS, collected by centrifugation, and then measured with a Becton-Dickinson FACSan flow cytometer. For GSSG/GSH ratio, the SW480 cells were extracted with protein extraction buffer after CL treatment. Then 100 µg of total proteins was analyzed by Bioxytech GSH/GSSG-412 assay kit with the standard protocol supplied by the manufacturer.
Measurement of ∆Ψmby Flow Cytometry. After treatment, the SW480 cells were trypsinized and then incubated with 5 µM rhodamine 123 for 30 min at 37°C in a CO2incubator. Following the incubation step, ∆Ψm, as indicated by the changed fluorescence level of rhodamine 123, was analyzed using a Becton-Dickinson FACScan flow cytometer.
Measurement of Cytosolic Cytochrome c by Western Blot. After treatment, cells were washed with PBS, resuspended in a homogenized buffer (0.25 M sucrose, 10 mM HEPES and 1 mM ethylene glycol bis(β-aminoethylether)-N,N,N′,N′-tetraacetic acid, pH 7.4), and subjected to 40 strokes of homogenization in a glass homogenizer. The homo- genates were centrifuged at 1000g for 15 min at 4°C to precipitate nuclei and unbroken cells. The supernatant was then centrifuged at 10000g for 15 min at 4 °C to obtain cytosolic fraction. Protein concentrations were measured with a Bio-Rad protein assay reagent (Bio-Rad, Richmond, CA). The cytochorme c in cytosolic fraction was evaluated by Western blot analysis.
Bax siRNA Transfection. For silencing of gene expression of Bax, small interference RNAs (siRNA) oligonucleotides with the following 5′-ACUUUGCCAGCAAACUGGUGCUCAA-3′(sense)and5′-UUGAG- CACCAGUUUGCUGGCAAAGU-3′ (antisense) were designed and purchased from Invitrogen Corporation (Carlsbad, CA). SW480 cells (4× 105) were cultured in 60 mm dishes in 5 mL of DMEM medium complemented with 10% FBS and transfected at 40% of confluency by adding lipofectactamine 2000 transfection agent and 10 µL of 200 pM Bax siRNA. Control cells were treated with lipofectactamine 2000 transfection agent alone. Cells were rinsed with the medium after 25 min of incubation and then maintained in culture for additional 48 h.
The Bax expression was evaluated by Western blot.
Measurement of the Cleavage of Poly ADP Ribose Polymerase (PARP), Bax, andγ-GCS by Western Blot. After treatment, cells were washed with PBS, resuspended in a protein extraction buffer for 10 min, and then centrifuged at 12000g for 10 min at 4°C to obtain total extracted proteins (supernatant). The protein concentrations were measured with a Bio-Rad protein assay reagent (Bio-Rad, Richmond, CA). The Bax, PARP, γ-GCS, and actin expressions were evaluated by Western blot analyses. Briefly, the total extracted proteins were boiled in loading buffer, and an aliquot corresponding to 50 µg of protein was separated by 12% SDS- polyacrylamide gel. After electrophoresis, proteins were electrotransferred onto a polyvinylidene fluoride transfer membrane. After blotting, the membranes were incubated with anti-Bax, anti-PARP, anti-γ-GCS, and antiactin antibod- ies (Laboratory Vision, Santa Cruz, CA) overnight and then washed with PBST solution (0.05% Tween 20 in PBS). Following washing, the second antibody labeled with horseradish-peroxidase was adjacently incubated for 1 h and then washed with PBST solution (0.05% Tween 20 in PBS). The antigen-antibody complexes were detected by the enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscat- away, NJ) with a chemiluminescence analyzer.
TUNEL Assay for Apoptosis. Following incubation of various reagents, TdT-mediated dUTP-biotin nick end labeling (TUNEL) was
performed using the “Apo-BrdU” kits (BD Pharmingen, San Diego, CA), with the standard protocol supplied by the manufacturer. Both floating and adherent cells were harvested and fixed using 1%
paraformaldehyde for 30 min at 4 °C. After fixation, cells were permeated with 70% ethanol for 30 min at 4 °C, followed by two washes with PBS. To label DNA strand breaks, 1-2× 106cells were incubated with 50 µL of TUNEL reaction buffer containing terminal deoxynucleotidyl transferase and fluorescein-BrdUTP, and it was then incubated for 1 h at 37°C in a humidified 5% CO2-in-air atmosphere.
Cells were washed twice with PBS, suspended in PBS containing 50 µg/mL of PI and 50 µg/mL of DNase-free RNase A for 30 min at room temperature in the dark and then analyzed by a Becton-Dickinson FACSan flow cytometer.
Cellular Morphology Examination. SW480 cells were treated with 150 µg/mL of CL for 24 h. After treatment, the cellular morphology was examined with a light microscope.
Statistical Analysis. Data were presented as means ( standard deviation from at least three independent experiments and analyzed using Student’s t test. A P value of less than 0.05 was considered as statistically significant (12).
RESULTS
CL Induced DNA Damage and Cytotoxic Effect in Colorec- tal Cancer Cells. To evaluate whether CL induces apoptosis, we exposed SW480 cells to 50, 75, and 150 µg/mL CL for 24 h. The cell cycle of SW480 cells was evaluated by flow cytometry. The percentage of cell cycle phases (SubG1, G1, G2/M, S) are analyzed by Modfit software. The total cells are separated by DNA damaged group (cells with a subG1
DNA content) and normal cell cycle group. The normal cell cycle group includes G1, G2/M, and S content. Under the definition of Modfit software, the total percentage of G1, G2/ M, and S must equal 100%. As to the SubG1percentage, it is calculated as: (cells in SubG1 × 100)/(total number of modeled cells, including SubG1, G1, G2/M, and S). As shown in Figure 1A, the untreated cells expressed 51.5% of G1, 35.2% of S, 13.3% of G2/M, and 4.6% of SubG1. The percentages of SubG1 dose-dependently increased after treatment of SW480 cells with CL at 50, 75, and 150 µg/mL for 24 h (Figure 1C). The study focused on the main apoptotic mechanisms of CL in SW480 colorectal cells. We first used the propidium iodide (PI) staining and flow cytometry to find an effective dose to induce obvious DNA damage (an apoptotic event) in SW480 cells. In Figure 1A, the dose of 150 µg/mL induced half of the total cells to DNA damage (50.1% of SubG1). The PI staining is a chief method to evaluate the DNA damage but not able to see the cell death; however, the MTT assay is used to analyze the cell death. To evaluate the level of cell death or cytotoxicity of CL in the effective dose to induce cellular DNA damage, the dose of 150 µg/mL was used for MTT assay. As indicated in Figure 1D, SW 480 cells were treated with CL extract (150 µg/mL) for 0.5, 1, 3, 6, 12, and 24 h, resulting in a time-dependent decrease in cellular viability. Three other human colorectal cancer cell lines, Colo-205, Colo-320 DM, and DLD-1, were selected to evaluate the anticancer effect of CL. As shown in Figure 1E, the cell viabilities of Colo- 205, Colo-320 DM, and DLD-1 were less than 30% after 24 h of CL treatment. The flow cytometry analysis also demonstrated that the percentage of Sub G1increased in CL- treated Colo-205, Colo-320 DM, and DLD-1 cells (Figure 1F), indicating the anticancer effect of CL works more than one human colorectal cancer cell line. Because the liver is an important detoxifying or metabolic organ, many drugs or toxic substances would be detoxified or metabolized in liver cells. For this reason, we used the clone 9, a rat liver cell
line, as normal somatic cells to evaluate the effect of DNA damage in CL treatment. As shown in Figure 1B, there are no significant differences in DNA damage (the percentage of SubG1) between CL-treated and untreated clone 9 cells.
The percentages of SubG1are 1.1% and 0.7% in CL-treated and untreated clone 9 cells, respectively, after 24 h of treatment. These results indicate that CL could not induce obvious DNA damage in the normal liver cell line. To study the apoptotic mechanisms of CL, an effective dose to induce apoptosis should be selected. Because the dose of 150 µg/
mL could induce a large number of cells showing DNA damage (50.1% of SubG1), we selected this dose to study the detailed mechanisms of CL-induced apoptosis in SW480 cells.
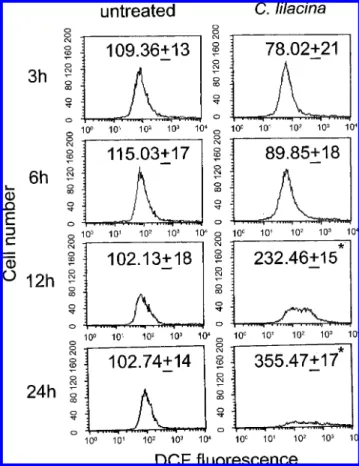
CL Induced ROS in SW480 Cells. Reactive oxygen species (ROS) were known as important mediators of apoptosis induced by various stimuli including many drugs. For this reason, the intracellular ROS production was measured by DCF fluores- cence assay following CL treatment. As indicated in Figure 2, SW480 cells were exposed to CL (150 µg/mL) for different times (3-24 h). The DCF fluorescence increased significantly from 102.13 ( 18 and 102.74 ( 14 to 232.46 ( 15 and 355.47 ( 17 after 12 and 24 h of treatment, respectively.
CL Induced GSH Depletion in SW480 Cells. GSH plays an important role in protection against oxidative stress-induced injury. Intracellular GSH depletion would proceed to apoptosis.
We, therefore, analyzed the changes of GSH level in SW480 cells by a flow cytometer using CMF fluorescence. SW480 cells Figure 1. DNA damage and cell viability analysis in CL-treated SW480 cell line. (A) SW480 and (B) Clone 9 cells were plated in 60 mm cultured dishes at 80% confluence and then treated with 0, 50, 75, or 150 µg/mL of CL for 24 h. Adherent and floating cells were harvested, fixed in PBS-methanol (1:2, volume/volume) solution, and stained with propidium iodide, followed by flow cytometric analysis. (C) The percentage of total cells in SubG1, G1, S, and G2/M phases of cell cycle in CL-treated SW480 cells. (D) SW480 cells were treated with 150 µg/mL of CL for 0.5, 1, 3, 6, 12, or 24 h. (E) SW480, Colo 205, Colo 320 DM, and DLD-1 cells were treated with 150 µg/mL of CL for 24 h. The MTT reagent (0.5 mg/mL) was added to the cells for 2 h at 37
°C and then lysed with DMSO. The absorbance was measured at 595 nm. (F) The percentage of total cells in SubG1phases in CL-treated SW480, Colo 205, Colo 320 DM, and DLD-1 cells. The values shown are mean ( standard deviation (n ) 5-8). Significant differences from the untreated group are P < 0.05 (*).
were treated with 150 µg/mL of CL for different times. Results were shown in Figure 3A. The percentage of intracellular GSH- depletion cells significantly increased to 10.98 ( 0.5, 52.39 ( 3.5, 90.46 ( 3.2, and 99.57 ( 5.4 after 3, 6, 12, and 24 h of treatment, as compared with untreated cells. The GSSG/GSH ratio was evaluated by Bioxytech GSH/GSSG-412 assay kit, which is a colorimetric determination of reduced and oxidized glutathione. When cells exposed to CL for 6 h, the GSSG/GSH ratio in the untreated cells was about 2 and was about 5 in the CL-treated group. The GSSG/GSH ratio increased to 2.5-fold at 6 h in CL-treated SW480 cells as compared with the untreated cells, suggesting large amount of GSH was depleted or changed to GSSG. These results demonstrated that an increased GSSG/
GSH ratio induced an intracellular oxidative stress in CL treatment.
CL Induced Mitochondrial Dysfunction in the SW480 Cells. Mitochondrial dysfunction usually triggers specific cel- lular signaling to induce apoptosis. Cytochrome c releasing to cytosol and ∆Ψm decreasing are two common parameters of mitochondrial dysfunction. To evaluate the effects of CL on cytosolic cytochrome c and ∆Ψm, SW480 cells were exposed to 150 µg/mL of CL for various times. The ∆Ψmand cytosolic cytochrome c were detected by rhodamine123 staining and Western blot, respectively. As indicated in Figure 4A, the percentage of ∆Ψm-decreased cells significantly increased to
20.43 ( 0.8, 26.57 ( 0.9, 54.41 ( 1.7, and 91.57 ( 2.2 after 3, 6, 12, and 24 h of CL treatment as compared to untreated cells, respectively. In Figure 4B, cytosolic cytochrome c initially increased at 3 and 6 h and markedly increased at 12 h of CL treatment. To evaluate the role of GSH depletion and Bax on CL-induced ∆Ψm disruption, NAC (an intracellular GSH complement), GSH, and SiBax (Bax inhibition) were used. As shown in Figure 4C, pretreatment with NAC and GSH markedly prevented the CL-induced ∆Ψm reduction, but the decreased ∆Ψmeffect did not recover by SiBax.
Figure 2. Evaluation of intracellular ROS in CL-treated SW480 cells. Cells were plated in 60 mm cultured dishes. The culture medium was replaced with new medium when the cells were 80% confluence and then exposed to 150 µg/mL of CL for 3, 6, 12, and 24 h. Production of intracellular ROS was detected by flow cytometry using DCFH-DA. The intracellular fluorescence of dichlorofluorescein (DCF) was measured with a Becton- Dickinson FACScan flow cytometer. Data in each panel represent the DCF fluorescence intensity within the cells. The values shown are mean ( standard deviation (n ) 5-8 of individual experiments). Significant differences from the untreated group are P < 0.05 (*).
Figure 3. GSH depletion analysis and GSSG/GSH ratio in CL-treated SW480 cells. Cells were plated in 60 mm cultured dishes. The culture medium was replaced with new medium when the cells were 80%
confluence and then treated with 0 (untreated) and 150 µg/mL of CL for 3, 6, 12, and 24 h. (A) After treatment, the cells were incubated with 25 µM CMF-DA for 30 min in a 37°C CO2incubator and then measured with a flow cytometer. Data show the percentage of cells displaying intracellular GSH-negative cells. (B) After treatment, the cells were extracted with protein extraction buffer. To evaluate the GSSG/GSH ratio, 100 µg of proteins was analyzed by Bioxytech GSH/GSSG-412 assay kit. The values shown in (A) and (B) are mean ( standard deviation (n ) 5-8). Significant differences from the untreated group are P < 0.05 (*).
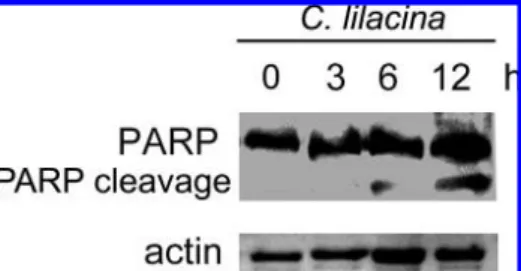
CL Induced PARP Cleavage in SW480 Cells. An important factor in inducing apoptosis is the enzyme PARP, which has been widely studied. To evaluate the effect of CL on PARP cleavage, the Western blot was employed. As shown in Figure 5, PARP cleavage appeared after 6 and 12 h of CL treatment.
Evaluation of Critical Pathway in CL-Induced Apoptosis.
We used the TUNEL analysis to evaluate the percentage of cellular apoptosis in SW480 cells treated with CL and in the meantime studied the critical pathway of apoptosis. As shown in Figure 6A, there were 57.8% of apoptotic cells after treatment Figure 4. The analysis of mitochondrial transmembrane potential (∆Ψm) and cytsolic cytochrome c in CL-treated SW480 cells. Cells were plated in 60 mm cultured dishes at 80% confluence and then treated with 150 µg/mL CL for 3, 6, 12, or 24 h. (A) After drug treatment, the culture medium was replaced with new medium with 5 µM rhodamine 123 for 20 min in the dark. Values in each panel express cellular percentages of decreased ∆Ψm. (B) After drug treatment, the cytosolic fractions were isolated and examined for cytochrome c and actin expressions by Western blot. (C) The SW480 cells were treated with 0 (untreated) or 150 µg/mL CL alone for 6 or 12 h, pretreated with 10 mM N-acetylcysteine (NAC) or 10 mM glutathione (GSH) for 1 h or Bax siRNA (SiBax) for 48 h followed by 150 µg/mL CL treatment for 6 or 12 h and then proceeded to the ∆Ψmassay by rhodamine 123 staining.
Values in each panel express cellular percentages of decreased ∆Ψm. The values shown are mean ( standard deviation (n ) 5-8). Significant differences from the untreated group are P < 0.05 (*).
with CL 150 µg/mL for 24 h as compared with 0.8% of untreated cells. To evaluate the critical pathway, several agents were employed in the study (1): cyclosporin A (Cy A), a mitochondrial permeability transition opening inhibitor, was used to find the ∆Ψmdecreased pathway (2); N-acetylcysteine (NAC), an intracellular GSH synthetic agent, and GSH were used to discover the GSH depletion pathway (3); vitamin C (Vit C), an antioxidant, was used to discover the ROS overproduction
pathway. SW480 cells were treated with these agents for 1 h before the addition of CL. As shown in Figure 6A, NAC and GSH exhibited greatest inhibition to the CL induced apoptosis;
the percentage of apoptotic cells decreased to 5.1 ( 2 and 4.5 ( 1, respectively. Cy A displayed a moderate inhibitory effect;
the percentage of apoptotic cells was 30.9 ( 4.5. The apoptotic effect induced by CL was not prevented by Vit C; the percentage of the apoptotic cells was 61.1 ( 4.3. In Figure 6B, the untreated cells displayed intact cellular morphology, whereas the CL-treated cells were undergoing apoptosis. In NAC and GSH-pretreated cells, the intact cellular morphology was similar to untreated cells, suggesting that the NAC and GSH prevented the cellular morphology damaged by CL.
The Bax Expression of the Cells Treated with the CL. To evaluate the Bax expression of the cells treated with the CL, the Western blot assay was used. SW 480 cells were treated with CL (150 µg/mL) for 0, 3, 6, and 12 h. As shown in Figure 7A, the Bax expression markedly increased 6 h after the treatment of SW 480 cells with CL. As expected, most of CL- treated cells had been dead at 24 h. The decreasing of Bax expression at 24 h treatment might be due to the large number of dead cells induced by CL. In Figure 6, NAC and GSH prevented the apoptosis induced by CL treatment. To evaluate whether NAC or GSH inhibited the Bax expression and then prevented the CL-induced apoptosis, the Western blot assay was Figure 5. PARP cleavage in CL-treated SW480 cell line. Cells were plated
in 60 mm cultured dishes at 80% confluence and then treated with 150 µg/mL CL for 3, 6, and 12 h. After treatment, cells were washed with PBS and extracted with protein extraction buffer. Then 50 µg of proteins were loaded on a 12% SDS-polyacrylamide gel. The expressions of PARP and actin were evaluated by Western blot analysis. These experiments were performed at least three times and a representative experiment is presented.
Figure 6. Evaluation of critical event in CL-induced apoptosis. (A) The SW480 cells were treated with 0 (untreated) or 150 µg/mL CL alone for 24 h, pretreated with 5 µM cyclosporin A (CyA), 10 mM N-acetylcysteine (NAC), 10 mM glutathione (GSH), or 1 mM Vitamin C (Vit C) for 1 h, followed by 150 µg/mL CL treatment for 24 h and then proceeded to the TUNEL assay. The representative plots depict DNA content on the X-axis and BrdU- FITC-labeled apoptotic DNA strand breaks on the Y-axis. Data represent the percentage of cells in the upper box of apoptosis. (B) After treatment, the cellular morphology was evaluated with a light microscope (magnification 200×). These experiments were performed at least three times, and a representative experiment is presented.
used. In Figure 7B, the CL-induced Bax expression was not drastically inhibited by NAC or GSH, suggesting that the prevention of NAC or GSH on CL-induced apoptosis is independent of Bax inhibition. We further evaluated whether the Bax expression was a main pathway in CL-induced SW480 cell death, and the Bax siRNA and MTT assays were conducted.
As shown in Figure 7B, the Bax expression was markedly inhibited by Bax siRNA in CL-treated cells. In Figure 7C, the cell viability was not recovered by Bax siRNA in CL-treated cells. However, the cell viability could be effectively recovered by NAC and GSH treatment. These results suggested that GSH depletion but not Bax expression was a critical event in CL- induced cell death.
Theγ-GCS Expression of the Cells Treated with the CL.
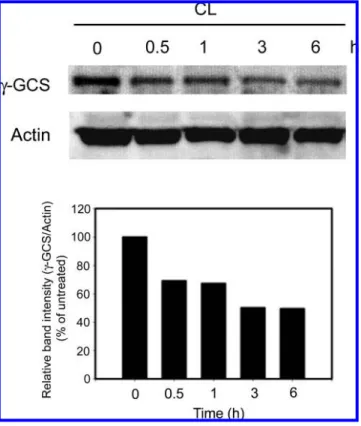
γ-Glutamylcysteine synthetase (γ-GCS) is the rate-limiting enzyme for GSH synthesis. To evaluate the γ-GCS expression of the cells treated with the CL, the Western blot assay was used. As shown in Figure 8, the γ-GCS expression markedly decreased as early as 0.5 h after treatment of SW480 cells with CL. The γ-GCS expression was less than 50% after 3 and 6 h of CL treatment. These results suggested that CL-induced GSH depletion in SW480 cells was related to the γ-GCS inhibition.
DISCUSSION
Many anticancer substances can induce large amounts of intracellular ROS to kill cancer cells (3, 10, 11). The cellular damages of ROS include apoptosis and necrosis. Our results found that CL induced large amounts of intracellular ROS after 12 and 24 h of treatment (Figure 2). The ROS production of CL treatment appeared at a later period and seemed to depend on the intracellular GSH depletion. At 12 and 24 h of treatment of CL, the percentages of GSH-depletion cells were >90%
(Figure 3). The GSH depletion appeared before the intracellular ROS overproduction. Moreover, the antioxidant Vit C could not effectively prevent the cells from CL-induced apoptosis (Figure 6). These results likely explain that the ROS production
is not the major event in CL-induced cell death in SW480 cells.
Changes in the intracellular redox surroundings of cells have been shown to generate two important events (1): the activation of apoptotic enzymes (2) and the progression of programmed cell death (12). In our previous studies in treating cancerous cells, we found that many substances extracted from plants induce the intracellular ROS production immediately and then GSH depletion appeared afterward (13, 14). This phenomenon seems different with the present data. In CL-treated SW480 cells, the GSH depletion occurred as early as 6 h and was retained to 24 h (Figure 3). However, ROS overproduction in CL treatment only appeared at 12 h and lasted until 24 h (Figure 2). Some studies have reported that GSH depletion was an early trademark detected in apoptosis and the GSH loss was independent of the ROS generation (15). Our results are in agreement with the report showing that CL-induced GSH depletion in SW480 cells was independent of the ROS generation. Our data also dem- onstrated that pretreatment with NAC or GSH could significantly decrease the cell population undergoing apoptosis in CL-treated SW480 cells. It was suggested that the GSH depletion was a critical event in CL-induced apoptosis.
GSH is synthesized intracellularly by two GSH synthesizing enzymes, γ-GCS and glutathione synthetase. γ-GCS is the key enzyme for the GSH-synthesis (16). GSH synthesis is also thought to be an important event in cellular defense against stresses such as radiation and drug damage (17). GSH depletion characterizes a potentially important strategy to sensitize tumors to cytotoxic drugs (18). Other studies have reported an increased synthesis of γ-GCS and GSH in drug-resistant malignant cells and suggested that increased GSH synthesis might play a role in drug resistance (19). The inhibition of γ-GCS would result in GSH depletion and be an important screen model for anticancer substances. We are the first to demonstrate that CL treatment markedly decreased the γ-GCS expression as early as 3 h (Figure 3). The effect might result in obvious intracellular GSH depletion at 6 h and might, therefore, provoke the later apoptotic effect after 24 h of treatment.
Figure 7. (A) CL-induced Bax expression; (B) and (C) Bax siRNA (SiBax), NAC, or GSH in CL-induced Bax expression and cellular viability. (A) The SW480 cells were treated with 0 (untreated) or 150 µg/mL CL alone for 3, 6, 12, and 24 h. The expressions of Bax and actin were evaluated by Western blot analysis. (B) Pretreated with siBax for 48 h, 10 mM NAC, or 10 mM GSH for 1 h, followed by 150 µg/mL CL treatment for 6 h, or siBax alone and CL alone for 6 h, and then evaluated the Bax and actin expressions by Western blot analysis. (C) Pretreated with siBax for 48 h, 10 mM NAC, or 10 mM GSH for 1 h, followed by 150 µg/mL CL treatment for 24 h, and then proceeded to the MTT assay. The values shown are mean ( standard deviation (n ) 5-8). Significant differences from the CL group are P < 0.05 (*). These experiments were performed at least three times and a representative experiment is presented.
Mitochondria play a central role in the regulation of apoptotic signaling (20). The decreased ∆Ψm is related to the cellular apoptosis (21, 22). In spite of the fact that ∆Ψmdecrease was time-dependent in the CL-treated SW480 cells, the CyA pretreatment could partially prevent the apoptosis induced by CL treatment (Figure 6A). We presume that the decreased ∆Ψm
may be a secondary pathway in apoptotic process of the CL- induced cell death.
GSH depletion is important in inducing mitochondrial dysfunction. A significant depletion of GSH in the neurons of Parkinson’s disease would result in oxidative stress, mitochon- drial dysfunction, and ultimately cell death (23). The mitochon- drial complex I is believed to be the central player to the mitochondrial dysfunction (23). GSH depletion could impair the activity of mitochondrial complex I. Mitochondrial dysfunc- tion in response to diamide occurs in stages, progressing from oscillations in ∆Ψm to sustained depolarization, which is in association with GSH depletion. The thiol redox status is systematically clamped at GSH/GSSG ratios. In cardiomyocytes, the GSH/GSSG ratio decreasing from 300:1 to 50:1 would irreversibly depolarize ∆Ψm and induce maximal rates of reactive oxygen species (ROS) production and loss of matrix constituents (24). These reports indicate an interdependence of mitochondrial dysfunction with GSH depletion.
Bax is an important pro-apoptotic protein (25, 26). Bax is always overproduction in cancer cells treated with various anticancer drugs (27, 28). In our present studies, the Bax overexpression and mitochondrial dysfunction simultaneously occurred at 3 h of CL treatment (Figures 4A and 7A). However, pretreatment with SiBax for 48 h could not prevent the CL- induced mitochondrial dysfunction (Figure 4C). It explains that Bax overexpression has no relationship with mitochondrial dysfunction in CL-induced apoptosis. Moreover, the SiBax treatment could not recover the cell viability induced by CL (Figure 7C). In addition, NAC or GSH pretreatment also could not inhibit the Bax overexpression in CL-treated cells (Figure 7B). These results implied that the Bax expression was independent of GSH depletion in CL treatment. The Bax overexpression was not the main pathway during CL-induced cell death in SW480 cells. The decreased ∆Ψm could be prevented by the pretreatment of NAC or GSH in CL-treated SW480 cells (Figure 4C). These results indicate that GSH depletion is a critical inducer in CL-induced mitochondrial dysfunction.
CL decreased the γ-GCS expression as early as 0.5 h would suggest that CL induced apoptosis via inhibiting γ-GCS in SW480 cells. The effect of CL is similar to theL-buthionine- S-sulfoximine (L-BSO)-induced apoptosis in other cancer cells.
L-BSO has been studied in both preclinical and early clinical trials for anticancer study (18). It has been discovered that L-BSO strongly and irreversibly inhibits γ-GCS (29) and induces a novel pathway of apoptosis dependent on GSH loss, ROS formation, and independent of Bax levels in neuroblastoma cells (30).
In conclusion, CL decreased the γ-GCS expression and eventually led to GSH depletion and is the critical event to induce apoptosis in SW480 cells. The second important event is mitochondrial dysfunction such as decreased ∆Ψm and cytochorme c released to cytosol. The GSH depletion is independent of Bax expression in the apoptotic event. This proposed model of the CL-induced apoptosis in SW480 cells is shown in Figure 9.
ABBREVIATIONS USED
CL, CalVatia lilacina protein-extract; ROS, reactive oxygen species; NAC, N-acetylcysteine; GSH, glutathione; DCF, 2′,7′- dichlorofluorescein; CMF, chloromethylfluorescein; PARP, poly ADP ribose polymerase; PI, propidium iodide, DMEM, Dul- becco’s modified Eagle’s medium; FBS, fetal bovine serum;
∆Ψm, mitochondrial transmembrane potential; γ-GCS, γ-glutamyl- Figure 8. γ-Glutamylcysteine synthetase (γ-GCS) expression in CL-
treated SW480 cell line. Cells were plated in 60 mm cultured dishes at 80% confluence and then treated with 150 µg/mL CL for 0.5, 1, 3, and 6 h. After treatment, cells were washed with PBS and extracted with protein extraction buffer. Then 50 µg of proteins were loaded on a 12%
SDS-polyacrylamide gel. The expressions of γ-GCS and actin were evaluated by Western blot analysis. A densitometer was used to quantify the expression of γ-GCS. Data are normalized for actin. These experiments were performed at least three times and a representative experiment is presented.
Figure 9. Schematic illustration of a proposed model of CL-induced apoptosis in human colorectal SW480 cancer cells. The “+”and “-” signs represent “increase” and “decrease”, respectively. CL decreased γ-GCS and GSH depletion is the critical pathway. Mitochondrial dysfunction (∆Ψm
decreasing and cytosolic cytochrome c releasing) is the secondary pathway. Bax expression and ROS are the accompanying events but not the main pathways.
cysteine synthetase; CyA, cyclosporine A; Vit C, vitamin C;
siRNA, small interference RNAs; TUNEL, TdT-mediated dUTP-biotin nick end labeling; MTT, 3-(4,5-cimethylthiazol- 2-yl)-2,5-diphenyl tetrazolium bromide.
LITERATURE CITED
(1) Zhao, H. Z.; Xu, Y. Y.; Fu, X. Y.; Fan, L. The progress of food and medical on values of puff-balls. Microbiology (Beijing) 2007, 34, 367–375.
(2) Sternberg, S. S.; Philips, F. S.; Cronin, A. P.; Sodergren, J. E.;
Vidal, P. M. Toxicological studies of calvacin. Cancer Res. 1963, 23, 1036–1044.
(3) Ren, J.; Privratsky, J. R.; Yang, X.; Dong, F.; Carlson, E. C.
Metallothionein alleviates glutathione depletion-induced oxidative cardiomyopathy in murine hearts. Crit. Care Med. 2008, 36, 2106–
2116.
(4) Fernández, A.; Colell, A.; Garcia-Ruiz, C.; Fernández-Checa, J. C.
Cholesterol and sphingolipids in alcohol-induced liver injury. J.
Gastroenterol. Hepatol. 2008, 23, S9-S15.
(5) Sun, Y. Q.; Guo, T. K.; Xi, Y. M.; Chen, C.; Wang, J.; Wang, Z. R. Effects of AZT and RNA-protein complex (FA-2-b-β) extracted from Liang Jin mushroom on apoptosis of gastric cancer cells. World J. Gastroenterol. 2007, 13, 4185–4191.
(6) Chen, K. C.; Peng, C. C.; Peng, R. Y.; Su, C. H.; Chiang, H. S.;
Yan, J. H.; Hsieh-Li, H. M. Unique formosan mushroom Antrodia camphorata differentially inhibits androgen-responsive LNCaP and -independent PC-3 prostate cancer cells. Nutr. Cancer 2007, 57, 111–121.
(7) Konno, S. Effect of various natural products on growth of bladder cancer cells: two promising mushroom extracts. Altern. Med. ReV.
2007, 12, 63–68.
(8) Gu, Y. H.; Sivam, G. Cytotoxic effect of oyster mushroom Pleurotus ostreatus on human androgen-independent prostate cancer PC-3 cells. J. Med. Food 2006, 9, 196–204.
(9) Lavi, I.; Friesem, D.; Geresh, S.; Hadar, Y.; Schwartz, B. An aqueous polysaccharide extract from the edible mushroom Pleu- rotus ostreatus induces antiproliferative and pro-apoptotic effects on HT-29 colon cancer cells. Cancer Lett. 2006, 244, 61–70.
(10) Gu, Y.H.; Leonard, J. In vitro effects on proliferation, apoptosis and colony inhibition in ER-dependent and ER-independent human breast cancer cells by selected mushroom species. Oncol. Rep.
2006, 15, 417–423.
(11) Lee, I. S.; Nishikawa, A. Polyozellus multiplex, a Korean wild mushroom, as a potent chemopreventive agent against stomach cancer. Life Sci. 2003, 73, 3225–3234.
(12) Lucke, J. F. Student’s t Test and the Glasgow Coma Scale. Ann.
Emerg. Med. 1996, 28, 408–413.
(13) Chen, C. Y.; Liu, T. Z.; Liu, Y. W.; Tseng, W. C.; Liu, R. H.;
Lu, F. J.; Lin, Y. S.; Kuo, S. H.; Chen, C. H. 6-Shogaol (alkanone from ginger) induces apoptotic cell death of human hepatoma p53 mutant Mahlavu subline via an oxidative stress-mediated caspase- dependent mechanism. J. Agric. Food Chem. 2007, 55, 948–954.
(14) Ong, P. L.; Weng, B. C.; Lu, F. J.; Lin, M. L.; Chang, T. T.;
Hung, R. P.; Chen, C. H. The anticancer effect of protein-extract from Bidens alba in human colorectal carcinoma SW480 cells via the reactive oxidative species- and glutathione depletion- dependent apoptosis. Food Chem. Toxicol. 2008, 46, 1535–1547.
(15) Franco, R.; Panayiotidis, M. I.; Cidlowski, J. A. Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J. Biol. Chem. 2007, 282, 30452–30465.
(16) Richman, P.; Meister, A. Regulation of γ-glutamylcysteine synthetase by nonallosteric inhibition by glutathione. J. Biol.
Chem. 1975, 250, 1422–1426.
(17) Shimizu, T.; Iwanaga, M.; Yasunaga, A.; Urata, Y.; Goto, S.;
Shibata, S.; Kondo, T. Protective role of glutathione synthesis on radiation-induced DNA damage in rabbit brain. Cell. Mol.
Neurobiol. 1998, 18, 299–310.
(18) Hamilton, D.; Wu, J. H.; Batist, G. Structure-based identification of novel human gamma-glutamylcysteine synthetase inhibitors.
Mol. Pharmacol. 2007, 71, 1140–1147.
(19) Godwin, A. K.; Meister, A.; O’Dwyer, P. J.; Huang, C. S.;
Hamilton, T. C.; Anderson, M. E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 3070–3074.
(20) Lu, G.; Ren, S.; Korge, P.; Choi, J.; Dong, Y.; Weiss, J.; Koehler, C.; Chen, J. N.; Wang, Y. A novel mitochondrial matrix serine/
threonine protein phosphatase regulates the mitochondria perme- ability transition pore and is essential for cellular survival and development. Genes DeV. 2007, 21, 784–796.
(21) Han, J.; Goldstein, L. A.; Gastman, B. R.; Rabinowich, H.
Interrelated roles for Mcl-1 and BIM in regulation of TRAIL- mediated mitochondrial apoptosis. J. Biol. Chem. 2006, 281, 10153–10163.
(22) Neuzil, J.; Wang, X. F.; Dong, L. F.; Low, P.; Ralph, S. J.
Molecular mechanism of “mitocan”-induced apoptosis in cancer cells epitomizes the multiple roles of reactive oxygen species and Bcl-2 family proteins. FEBS Lett. 2006, 580, 5125–5129.
(23) Vali, S.; Mythri, R. B.; Jagatha, B.; Padiadpu, J.; Ramanujan, K. S.; Andersen, J. K.; Gorin, F.; Bharath, M. M. Integrating glutathione metabolism and mitochondrial dysfunction with implications for Parkinson’s disease: a dynamic model. Neuro- science 2007, 149, 917–930.
(24) Aon, M. A.; Cortassa, S.; Maack, C.; O’Rourke, B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 2007, 282, 21889–21900.
(25) Pigneux, A.; Mahon, F. X.; Moreau-Gaudry, F.; Uhalde, M.;
deVerneuil, H.; Lacombe, F.; Reiffers, J.; Milpied, N.; Praloran, V.; Belloc, F. Proteasome inhibition specifically sensitizes leu- kemic cells to anthracyclin-induced apoptosis through the ac- cumulation of Bim and Bax pro-apoptotic proteins. Cancer Biol.
Ther. 2007, 6, 603–611.
(26) Wu, J. Y.; Chung, K. T.; Liu, Y. W.; Lu, F. J.; Tsai, R. S.; Chen, C. H.; Chen, C. H. Synthesis and biological evaluation of novel C(6) modified baicalein derivatives as antioxidative agents. J.
Agric. Food Chem. 2008, 56, 2838–2845.
(27) Ma, L.; Wen, S.; Zhan, Y.; He, Y.; Liu, X.; Jiang, J. Anticancer effects of the Chinese medicine matrine on murine hepatocellular carcinoma cells. Planta Med. 2008, 74, 245–251.
(28) Luo, W.; Liu, J.; Li, J.; Zhang, D.; Liu, M.; Addo, J. K.; Patil, S.; Zhang, L.; Yu, J.; Buolamwini, J. K.; Chen, J.; Huang, C.
Anti-cancer effects of JKA97 are associated with its induction of cell apoptosis via a Bax-dependent and p53-independent pathway.
J. Biol. Chem. 2008, 283, 8624–8633.
(29) Reliene, R.; Schiestl, R. H. Glutathione depletion by buthionine sulfoximine induces DNA deletions in mice. Carcinogenesis 2006, 27, 240–244.
(30) Marengo, B.; DeCiucis, C.; Verzola, D.; Pistoia, V.; Raffaghello, L.; Patriarca, S.; Balbis, E.; Traverso, N.; Cottalasso, D.; Pronzato, M. A.; Marinari, U. M.; Domenicotti, C. Mechanisms of BSO (L-buthionine-S,R-sulfoximine)-induced cytotoxic effects in neu- roblastoma. Free Radical Biol. Med. 2008, 44, 474–482.
Received for review September 28, 2008. Revised manuscript received December 2, 2008. Accepted December 22, 2008. This work was supported by grant NSC 96-2320-B-415-002-MY3 (C.-H. Chen) from the National Science Council, Taiwan, ROC.
JF8030265