I
國 立 交 通 大 學
分子醫學與生物工程研究所
碩 士 論 文
Aspergillus屬中的水平基因轉移及基因體中
的GC變化
The horizontal gene transfer events and alterations
of genomic GC content in the genus Aspergillus.
研 究 生:何宜佩
指導教授:林勇欣 博士
II
Aspergillus 屬中的水平基因轉移及基因體中
的GC變化
The horizontal gene transfer events and alterations
of genomic GC content in the genus Aspergillus
研 究 生:何宜佩
Student:
Yi-Pei
Ho
指導教授:林勇欣 博士 Advisor:Yeong-Shin Lin
國 立 交 通 大 學
分 子 醫 學 與 生 化 工 程 研 究 所
碩 士 論 文
A Thesis Submitted to Institute of molecular medicine and bioengineering National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master in molecular medicine and bioengineering
July 2011
Hsinchu, Taiwan, Republic of China
III X 1
Aspergillus屬中的水平基因轉移及基因體中的GC變化
學生:何宜佩
指導教授:林勇欣
分子醫學與生化工程研究所碩士班
摘 要
水平基因轉移在原核生物及部分的真核生物演化中扮演重要的角色,不同於 一般有性的遺傳物質轉移,水平基因轉移是一個遺傳物質轉移至不同物種的過程, 在原核生物中,水平基因轉移事件發生頻繁且原核生物常經由此過程增加對於環 境的適應力,最廣為被人提及的病人產生抗藥性的事件就是屬於基因轉移發生的 案例。在現在這個基因體大量被解碼的時代中,水平基因轉移事件逐漸被發現, 水平基因轉移的發生可以解釋一些物種所產生的新特徵或人類、動物及植物上的 新疾病。搜尋潛在轉移基因有許多方法,例如找出不同於整個基因體特色的片段 或是發現序列度異於認知中的演化關係,因為許多因素會影響水平基因轉移事件 的判斷,應該利用不同方法及數據來驗證水平基因轉移的案例。在此研究中,發 現一個從真菌轉移至 Stigmatella aurantiaca (屬於黏液菌 myxobacteria 中的一種土 壤細菌)的水平基因轉移案例,從演化關係及基因體特徵分析中推論 Aspergillusclavatus 中的同源基因與水平轉移的基因 STAUR_2131 最相近,但確切的序列來
IV
X 2
The horizontal gene transfer events and alterations of genomic GC
content in the genus Aspergillus
Student: Yi-Pei Ho
Advisor:Yeong-Shin Lin
Institute of molecular medicine and bioengineering
National Chiao Tung University
ABSTRACT
Horizontal Gene Transfer (HGT), also called Lateral Gene Transfer (LGT),
plays an important role in evolution of prokaryotes and part of eukaryotes. HGT is the
nonsexual process of genetic material transfer across species. It is well known that
HGT events occur frequently among prokaryotes that can often increase fitness to
colonize new environment. The common transfer events that people often mentioned
is developing drug resistance. However, as genome sequencing era coming, HGTs
found in eukaryotes increasingly. HGT events may explain some new traits of
organisms and even new disease to human, animals and plants. There are several
approaches, which based on genome-wide features, sequence similarity and
phylogeny incongruence, can screen out potential HGT cases. Since, there are
different possibilities that can contribute to the gene anomalies, HGTs should be
confirmed by different approaches, especially phylogeny analysis. In this study, I find
V
of phylogeny and genome characteristics, the orthologous gene of Aspergillus
clavatus is the most closed gene sequence to the HGT gene, STAUR_2131, but where
VI X 3
謝 誌
兩年的碩士生涯一轉眼就結束了,感謝大家的幫忙與陪伴,首先,感謝林勇 欣老師開啟我進入生資領域的大門,並且耐心與細心指導,除了學業上的指導外, 也非常高興老師能在生活中與我們同樂,感謝俊霖學長的指導,一開始甚麼都不 懂,多虧學長一步一步的講解以及花時間和我討論問題解決問題,才讓我不用在 一個問題上卡太久,也很喜歡再盯著電腦之餘與學長話家常,可以從中得到許多 心得與樂趣,感謝大逵在我進實驗之初教我寫程式還有講解許多電腦的基本觀念 與注意事項,因為你的熱心使我可以早一點融入實驗室,感謝空肉在大小事上的 幫忙,你真是實驗室的支柱,甚麼事找你總是可以迎刃而解,感謝你的細心及貼 心,積極主動幫忙大家,值得我好好的學習,感謝小 Q 的陪伴,不管是在學業 上還是在生活中,都因為有你而豐富許多,學業上的討論及生活中的分享都讓兩 年的生活不枯燥,感謝宗翰在我遇到挫折時的鼓勵,也感謝你在經驗上的分享, 感謝火馬與之杭,常常幫我外帶食物,感謝淑娟常陪我到處逛,每次在宿舍和你 聊天都很開心,也因為你而知道很多訊息還有優惠,很開心生活中有你, 最後,感謝爸媽把我養這麼大,在我每個辛苦或困難的時候給我無限的支持與關 心,讓我可以不用擔心的去選擇我的路。VII
Contents
Abstract ... I List of figures ... VIII
Chapter 1 Introduction ... 1
1.1 Horizontal gene transfer ... 1
Chapter 2 Materials and Methods ... 5
2.1 Genome and sequences ... 5
2.2 Codon usage database ... 5
2.3 HGT candidate screening ... 5 2.4 Phylogenetic analysis ... 6 2.5 GC content ... 6 2.6 Codon usage ... 7 Chapter 3 Results ... 8 3.1 HGT scan ... 8 3.2 GC content ... 9
3.3 Codon usage bias ... 12
Chapter 4 Conclusion ... 13
Chapter 5 Discussion ... 14
VIII
List of figures
Figure 1 The phylogenetic tree of AFUA_5G10930. ... 18
Figure 2 The GC content of the neighbor genes of the HGT gene and Stigmatella aurantiaca genome. ... 19
Figure 3 The GC content of HGT gene compare with the foreign gene and genome. ... 20
Figure 4 The GC content of amino acid code 1,2 and code 3(wobble site) ... 21
Figure 5 The synonymous and nonsynonymous distance distribution. ... 22
Figure 6 Scan the GC content of the transferred sequence region ... 23
Figure 7 The GC content of intergenic region. ... 24
Figure 8 The comparison of Pro codon usage bias. ... 25
Figure 9 The comparison of Phe codon usage bias. ... 26
Figure 10 The comparison of Leu codon usage bias. ... 27
Figure 11 The comparison of Ile codon usage bias. ... 28
Figure 12 The comparison of Ser codon usage bias. ... 29
1
Chapter 1 Introduction
1.1 Horizontal gene transfer
Horizontal gene transfer means genetic materials transfer from one species to
another, and this is a non- sexual process. Different from Darwin inheritance, genetic
materials transmitted generation by generation in the same phylogenetic clade, HGT
events transfer from one clade to a different clade. HGT events could be occurred by
transformation, transduction and conjugation in prokaryotes, while occurred by
phagocytosis and endosymbiont process in eukaryotes[1] . Evidences showed that
HGTs are more frequently found in prokaryotes than in eukaryotes[2]. HGT is a
considerable driving force of prokaryotic evolution. Based on Charles Darwin’s
theory, biologists construct a tree-like phylogenetic relationship. If a wide sequence
region or whole genome is transferred to unrelated lineage, the phylogenetic tree will
become a network-like diagram. It has been obscured that the ancestor of eukaryotes
is the Bacteria or the Archea. It is hard to distinguish the origin of the Eukaryote
depending on one gene. Rivera and Lake suggest that genome fusion or horizontal
gene transfers occur within diverse prokaryotic genomes becoming the origin of the
Eukaryote and convert the phylogenetic trees into rings[3]. As more and more genetic
2
infecting the phylogenetic relationship especially in prokaryotes[4].
Accumulating HGT events suggest that genes transferred particularly. Genes of
genome are divided into operational genes and informational genes. Operational
genes are responsible for metabolic process; in contrast, informational genes are
responsible for transcriptional and translational process. Informational genes are far
less transferred than operational genes in horizontal transfer events. It is summarized
that functional genes are transferred preferentially in HGT process[5]. Because not
every gene transfer event is kept expand in recipient species, it must be profitable
under nature selection. For instance, Dehalococcoides ethenogenes acquir
dehalogenase genes that are important for dehalorespiration process to resist the
polluted environment. Moreover, many studies showed that HGTs occur commonly
for antibiotic-resistance and the frequencies of transfer events must higher than
observed [6]. Through HGT process, host could gain a new function for pathogenicity
or virulent resistance increasing the host fitness and colonizing new environments
[7-8].
There are some genes acquired from HGT events occurred in fungi that can
cause diseases. For instance, A gene encoding ToxA, encoding host-selective toxin, is
transferred from Stagonospora nodorum to Pyrenophora tritici-repentis can cause tan
3
protein in host plant. The recognition of ToxA by Tsn1 in wheat subverts the plant
pathogens resistance mechanism [11]. The ToxA genes are found both in host
Stagonospora nodorum and pathogen Pyrenophora tritici-repentis but not in other
related species of Pyrenophora tritici-repentis. The comparison of ToxA gene in
pathogen fungi shows that the pathogen fungi both contain highly similar region
surrounding ToxA gene, and 57 ToxA gene sequences of Pyrenophora tritici-repentis
are almost identical to ToxA gene sequences of 600 Stagonospora nodorum strain
[11-12]. That suggests the ToxA gene is transferred to Pyrenophora tritici-repentis
and make that fungi cause tan spot on wheat.
Evidences show that not only one gene but also a gene cluster can be lateral
transferred. The ACE1 gene cluster contains 15 genes co-expressed during the process
of the fungi penetrates into host tissues. This horizontal transferred cluster is found by
comparing 26 fungal genomes and search continual orthologs genes. The ACE1
cluster is transferred from Magnapothe grisea to few fungal species. According to the
phylogenetic analysis, the genes of the cluster of Magnapothe grisea are more closed
to the genes in Aspergillus clavatus and the Aspergillus clavatus contains only six
genes in the ACE1 cluster. The observation suggests the ACE1 cluster is transferred
from Magnapothe grisea into an ancestor of Aspergillus clavatus. Comparison of
4
loss after transferred process and become different pattens in the fungi.
Horizontal gene transfer events could be predicted depending on phylogenetic
incongruencies or parametric methods. Phylogenetic approaches are mainly based on
the accepted species taxonomy. First, align a set of genes of the candidate that have
the same function but in different species. Second, use the alignment to construct a
phylogenetic tree. Then, compare the phylogenetic tree with the species taxonomy or
the accepted phylogeny of the gene. The incongruencies between the two phylogenies
may indicate HGT events[13]. Parametric methods involve GC content, codon usage,
di- and tetra-nucleotide frequencies of the sequences[14]. Because these genomic
characteristics are specific to particular species, the atypical regions compared to the
general pattern of the genome suggest the possibility of HGT [13].
Because of different composition of tRNA pool, there are different level and
special codon usage bias in different species genome. Most amino acids are encoded
by more than one codon. In different organisms, there exist different preferences for
synonymous codons of the same amino acid. Therefore, the particular codon
frequencies in coding sequences, called codon usage bias, can sometimes represent
for particular organism[15]. In this study, we used GC content and codon usage bias
to explore the origin of the genes or to inference the causation relation for the
5
Chapter 2 Materials and Methods
2.1 Genome and sequences
The genome of Aspergillus fumigatus AF293 and Stigmatella aurantiaca
DW4/3-1 collected from NCBI ftp[16]. The genome size of Aspergillus fumigatus
Af293 is about 30 MB and organized in 8 chromosomes.
The genome size of Stigmatella aurantiaca DW4/3-1 is about 10 MB. All the
sequences used are available in NCBI website (www.ncbi.nlm.nih.gov).
2.2 Codon usage database
The codon bias usage of each genome obtain from Codon usage database[17].
The nucleotide sequences used for calculating the codon usage were obtained from
Genbank Genetic Sequence Database.
2.3 HGT candidate screening
I used BLASTp with Aspergillus fumigatus Af293 genome protein sequences
against all the sequences in the nr database downloaded from NCBI ftp[18]. Then, the
6
distant species within the e-value cutoff e-5. Based on the BLASTp results of the
candidates, construct FASTA files for each candidate.
2.4 Phylogenetic analysis
Using ClustalW aligned the set of protein sequences with higher similarity
based the BLASTp results. According to the alignments, I used neighbor-joining
method with poisson model and all the gaps were completely deleted for constructing
phylogenetic tree by MEGA5. To test the support for the tree topology, I ran the
method for 500 bootstrap replicates. Finally, analyze all the phylogenetic trees
manually to find out the nonsense distribution of the set of genes.
2.5 GC content
I counted the GC ratio of the sequences by a PHP file to compare the HGT gene
with other reference genes or genomes. In addition, I scanned the GC content of HGT
gene region sequence by the window of 150 bases and shift 10 bases every counting.
Meanwhile, whenever the window contained the protein coding regions, I counted the
7
2.6 Codon usage
Because of the preference of codon bias usage for particular species, I compare
the codon bias usage of HGT gene and the close genes in the same phylogenetic clade
with each other or with the genome bias data of the same species. I collect genome
codon usage data from codon usage database [17]. The sequences for the analysis are
compiled from NCBI-GenBank Flat File Release 160.0 and included only complete
sequenced protein coding genes. Besides, the ambiguous bases in the sequences, N,
are excluded from analysis. On the other hand, I collect ribosomal proteins of each
close gene species from NCBI. Then, I analyze the codon bias usage for HGT gene
and close genes individually but concatenate all ribosomal proteins for the codon
bias usage by the website [19]. After calculating the bias of each data set I included, I
download all the results and made a file to list all the percentage for each amino acid
in the sequences. In order to visualized composition for every amino acid, I draw pie
charts or bar charts and compared with the bias of genome data or other genes of close
8
Chapter 3 Results
3.1 HGT scan
To find HGT genes in eukaryotes, I scan Aspergillus fumigatus whole genome
protein sequences by using BLASTp. I got a blast output file that 9630 Aspergillus
fumigatus protein sequences as queries against NCBI nr database individually, and the
more similar sequences are recorded in the file by the e-value. First, I ran a PHP
pipeline for automatic filtration to find out HGT candidate genes, the cutoff e-value
were less than 10-5 and the best hit were distant species except the genus Aspergillus. I
got 57 candidate genes of Aspergillus fumigatus and the candidate genes were listed in
Table 1. Second, I constructed phylogenetic trees for each candidate gene and
analyzed the incongruence of the taxon distribution. There are seven phylogenies that
looked like horizontal transfer events patterns. Not all genomes are sequenced and
annotated, so losing some gene data may result in false horizontal transfer
phylogenetic patterns. To exclude the influences of sequencing data incompletion, I
do double check by running BLASTn against genome sequence data. I use HGT
nucleotide sequences as queries against to sequenced genome and all nucleotide
sequences to check if there are other closer sequences hit to genome sequences than
9
AFUA_5G10930, transferred from fungi to Stigmatella aurantiaca (Figure 1). In the
phylogenetic tree of gene AFUA_5G10930, the orthologous gene of Stigmatella
aurantiaca, STAUR_2131, group with the orthologs of Aspergillus clavatus, Aspergillus fumigatus, and Neosartorya fischeri in the same phylogenetic clade (Node
A), that distributed in a fungi clade. From this phylogeny, we couldn’t precisely infer
which species the HGT gene transferred from. It is possible that the HGT donor gene
hadn’t been sequenced yet.
3.2 GC content
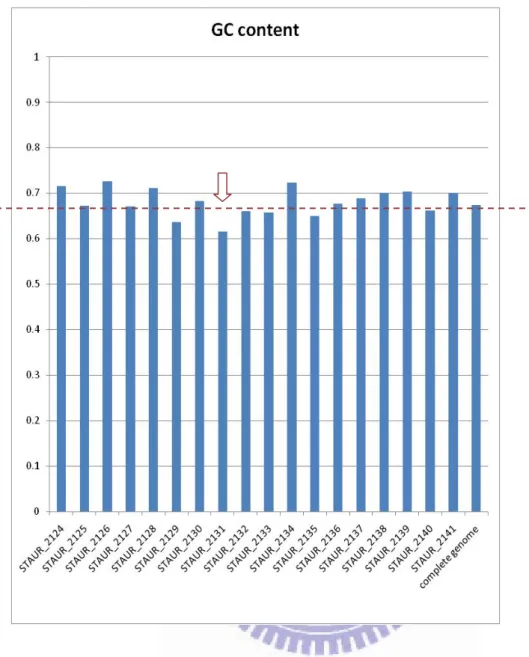
Stigmatella aurantiaca genome sequence contained 67% G+C nucleotides. I
compare the genes that next to the transferred gene, STAUR_2131, in Stigmatella
aurantiaca genome. The GC content of genes from STAUR_2124 to STAUR_2141
and Stigmatella aurantiaca complete genome contain more than 62% except the HGT
gene STAUR_2131. The GC content of the transferred gene is lower than other
Stigmatella aurantiaca genes but close to the orthologous gene ACLA_013950.
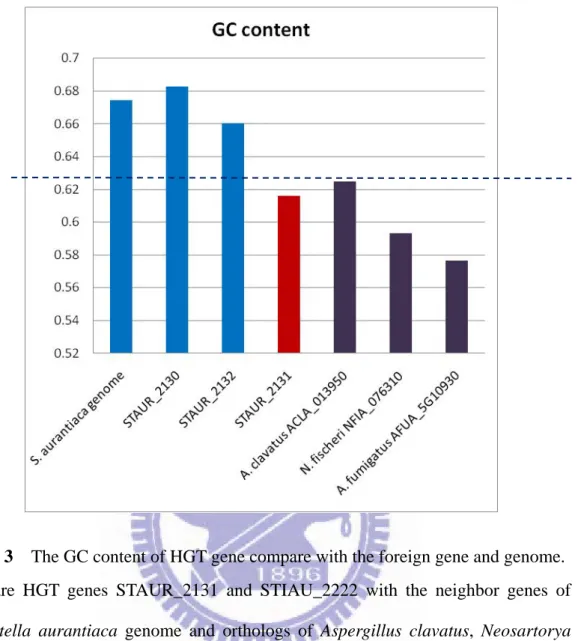
Moreover, I compare transferred gene STAUR_2131 and the foreign genes
STAUR_2130, STAUR_2132 and STAUR_2133 with the closest fungi genome,
Aspergillus clavatus, Aspergillus fumigatus, and Neosartorya fischeri (Figure 3). In
10
content of fungi are lower than 62%. In Figure 3, the GC content values of HGT
genes are between the values of Stigmatella aurantiaca and fungi. According to the
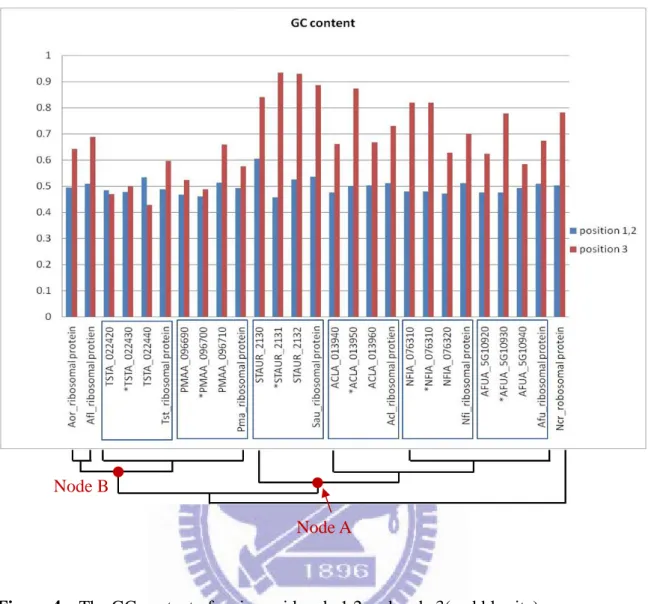
phylogeny of HGT orthologous gene, I compared the GC content for the coding
positions 1 + 2 and position 3 (wobble site), including the GC content of the orthologs
of HGT gene, neighbor genes and ribosomal protein genes. There are no obvious
differences of GC frequencies for positions 1 + 2 among the species in the phylogeny,
but the GC frequencies of position 3 are much higher than positions 1 + 2 within the
HGT gene clade (Node A). To summarize the data of GC content, the GC content of
positions 1 + 2 of STAUR_2131 is less than the neighbor genes, but the GC content of
the position 3 is very high. Mutations happen in the wobble sites of amino acid codes
that often contribute to synonymous mutations are more common than positions 1 + 2.
When one gene is transferred to another genome, the codon usage and the GC content
would become to the recipient genome gradually. The lower GC content of positions 1
+ 2 makes the GC content of STAUR_2131 lower than other neighbor genes in
Stigmatella aurantiaca, so the GC content of STAUR_2131 is between the GC
content of fungi and Stigmatella aurantiaca. Figure 4 displays that the GC content
level of positions 1 + 2 and wobble sites of Stigmatella aurantiaca are similar to the
level of species in the clade of Aspergillus clavatus. The nucleotide sequence
11
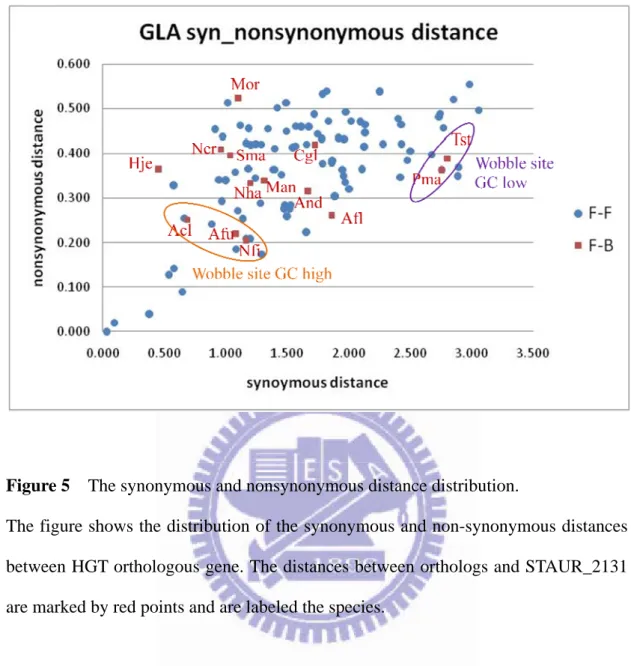
of the Aspergillus clavatus clade are smaller than the orthologs of the Penicillium
marneiffei clade (Figure 5). The group of orthologs that contain the same pattern of
the GC content level have smaller synonymous distances between the transferred gene
of Stigmatella aurantiaca; the orthologs of Penicillium marneffei and Talaromyces
stipitatus contain lower GC content of wobble sites have bigger distances between the
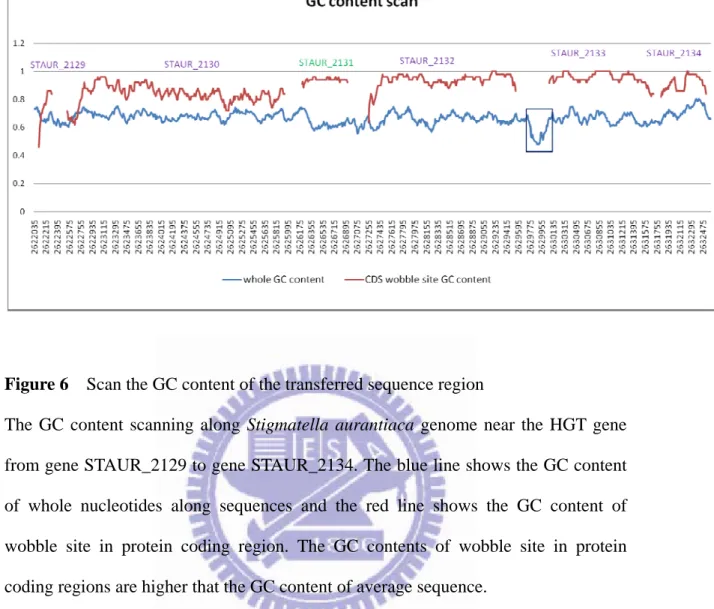
transferred gene. Then, I scanned the enlarged sequence fragment of the transferred
gene by 150 nucleotides window (Figure 6). The GC contents of wobble sites in
protein coding region are larger than the GC contents along the genome sequence.
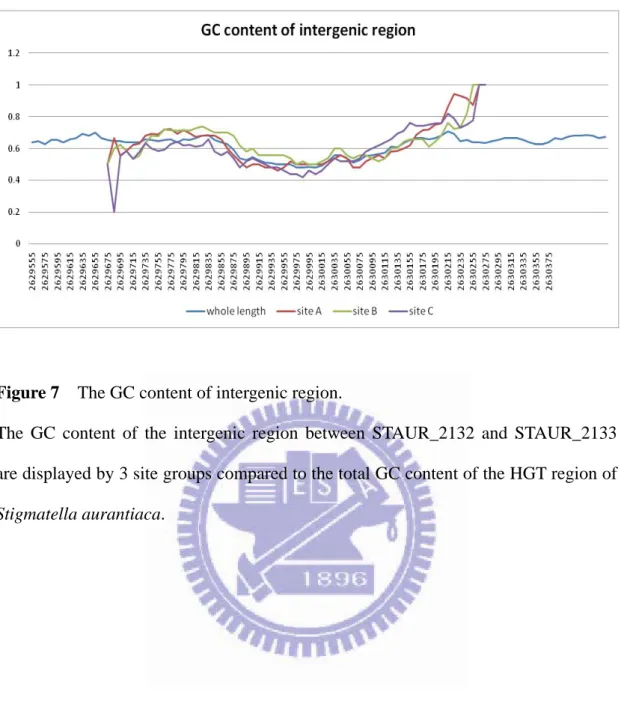
Besides, the GC contents in the intergenic region are less than the average GC
contents of protein coding region. To exam whether the sequence under positive
selection make the GC contents of protein coding region higher than intergenic region
to fit Stigmatella aurantiaca genome, I scan the GC content of intergenic regions by
dividing the sequences into three group in order, site A, site B, and site C. The GC
content of the three site groups are similar with the total GC content of the intergenic
region between gene STAUR_2132 and STAUR_2133. The coherence of GC content
of the three sites suggest that the protein coding regions are under positive selection,
so the wobble sites in protein coding region tend to mutate to G or C to fit Stigmatella
12
3.3 Codon usage bias
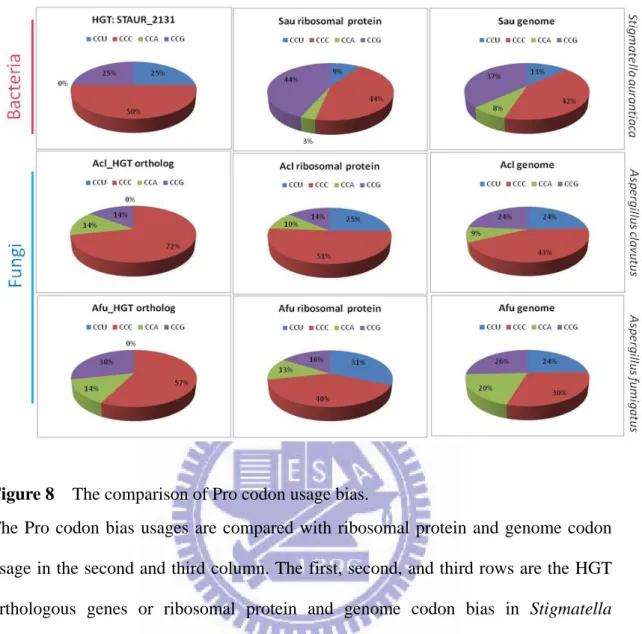
To infer the origin of the HGT gene, I compare the codon usage of HGT gene
with the orthologs and the neighbor genes to the HGT gene. In figure 8 first row, the
leucine usage bias of HGT gene is different from the neighbor genes, ribosomal
protein, and genome of Stigmatella aurantiaca. The Pro and Phe codon usage of
STAUR_2131 is different from the codon usage of Stigmatella aurantiaca genome
and the fungal orthologs and genomes (Figure 8-9). The Leu, Val, Ile, and Ser codon
usage bias of HGT gene is the same with the orthologous gene of Aspergillus clavutus
and similar with Aspergillus fumigatus but not consistent with the usage of both the
two fungal genome (Figure 10-13). The differences of the codon usage bias with the
Aspergillus clavatus genome indicate the transferred gene is not belonging to Aspergillus clavatus originally.
13
Chapter 4 Conclusion
The phylogenetic data suggests the gene STAUR_2131 is transferred from fungi
to Stigmatella aurantiaca. Inferred from the result of BLASTp, the GC content and
the codon usage, the most closed sequence is the gene ACLA_013950 of Aspergillus
clavatus. Based on the phylogeny and the codon usage bias, the HGT gene of Stigmatella aurantiaca is most closed to the orthologous gene of Aspergillus clavatus,
14
Chapter 5 Discussion
Stigmatella aurantiaca belongs to Myxobacterium. Myxobacterium existing
biphasic cell cycle, the cells migrate to aggregate center and form fruiting body when
nutrients depletion [20]. On the other hand, the transferred gene is most similar to
glucuronan lyase A, which is responsible for catalyzation of eliminative cleavage of
(1->4)-beta-D-glucuronans, which is from extracellular matrix. When cell migration,
the extracellular matrix have to degrade and reconstruct [21]. It is possible the gene
transferred between Stigmatella aurantiaca and fungi because Stigmatella aurantiaca
and fungi both contain fruiting body phase and become active when cell migration.
Horizontal gene transfer events can be explored by different methods, but HGT
can’t be sure by one method. Amounts of genome BLAST data through automatic
filtration, there are still many genes to be examined. Combining sequences similarity
and phylogenetic incongruence, I get seven candidate genes of HGT. Although the
phylogenies of the candidates gene seems like HGT events, there are some problems
would affect the accuracy of HGT. Not like bacterial genome, fewer fungal genomes
are sequenced. Besides, the phylogenetic trees of HGT are hard to verify. For example,
a phylogenetic tree that shows a bacteria gene or a clade out of a fungal clade looks
like a HGT event. From the phylogenetic tree, the closest gene to the bacterial gene is
15
genome data is not complete, so there are many leakages in the true phylogeny.
Sometimes, losing part of the true phylogeny lead to a HGT-like phylogenetic tree;
even though, I exam the protein and nucleotide sequences’ similarities and distance to
16
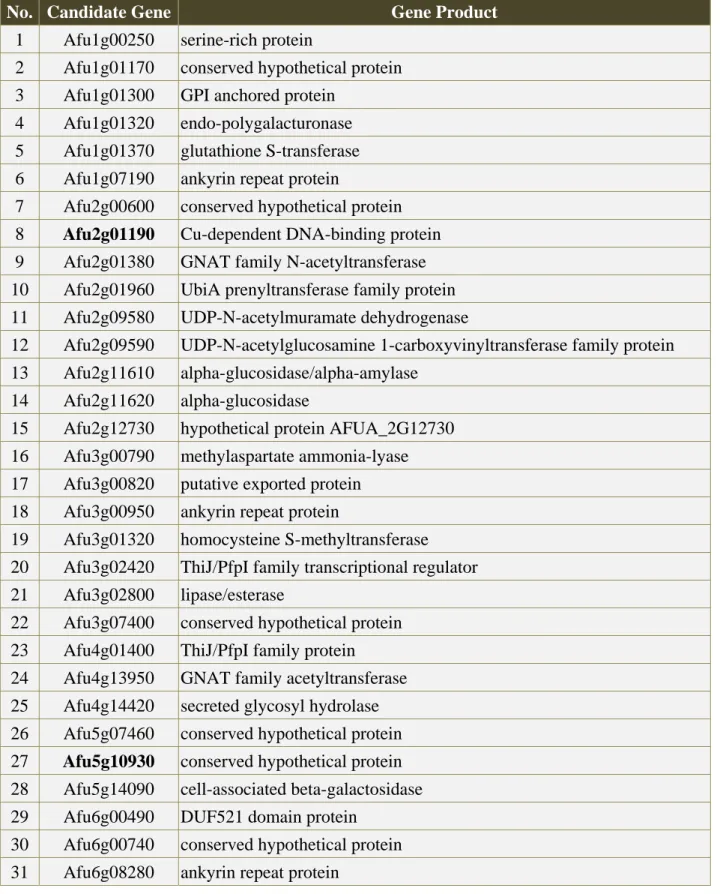
Table 1 The candidate genes through automatic filtration.
The bold labeled genes are the candidate genes after first step of phylogenetic analysis.
No. Candidate Gene Gene Product
1 Afu1g00250 serine-rich protein
2 Afu1g01170 conserved hypothetical protein 3 Afu1g01300 GPI anchored protein
4 Afu1g01320 endo-polygalacturonase 5 Afu1g01370 glutathione S-transferase 6 Afu1g07190 ankyrin repeat protein
7 Afu2g00600 conserved hypothetical protein 8 Afu2g01190 Cu-dependent DNA-binding protein 9 Afu2g01380 GNAT family N-acetyltransferase 10 Afu2g01960 UbiA prenyltransferase family protein 11 Afu2g09580 UDP-N-acetylmuramate dehydrogenase
12 Afu2g09590 UDP-N-acetylglucosamine 1-carboxyvinyltransferase family protein 13 Afu2g11610 alpha-glucosidase/alpha-amylase
14 Afu2g11620 alpha-glucosidase
15 Afu2g12730 hypothetical protein AFUA_2G12730 16 Afu3g00790 methylaspartate ammonia-lyase 17 Afu3g00820 putative exported protein 18 Afu3g00950 ankyrin repeat protein
19 Afu3g01320 homocysteine S-methyltransferase 20 Afu3g02420 ThiJ/PfpI family transcriptional regulator 21 Afu3g02800 lipase/esterase
22 Afu3g07400 conserved hypothetical protein 23 Afu4g01400 ThiJ/PfpI family protein
24 Afu4g13950 GNAT family acetyltransferase 25 Afu4g14420 secreted glycosyl hydrolase 26 Afu5g07460 conserved hypothetical protein 27 Afu5g10930 conserved hypothetical protein 28 Afu5g14090 cell-associated beta-galactosidase 29 Afu6g00490 DUF521 domain protein
30 Afu6g00740 conserved hypothetical protein 31 Afu6g08280 ankyrin repeat protein
17 32 Afu6g08770 ankyrin repeat protein
33 Afu6g09340 hypothetical protein AFUA_6G09340 34 Afu6g09360 proline-glycine rich protein
35 Afu6g10170 spherulin 4 family protein 36 Afu6g10750 conserved hypothetical protein 37 Afu6g12030 rhamnosidase
38 Afu6g12170 FKBP-type peptidyl-prolyl isomerase 39 Afu7g00690 aminotransferase
40 Afu7g00830 alpha/beta hydrolase
41 Afu7g05060 MgtC/SapB family membrane protein 42 Afu7g06990 GDP-D-mannose dehydratase
43 Afu8g00630 conserved hypothetical protein 44 Afu8g01050 lipase/esterase
45 Afu8g01700 haloalkane dehalogenase family protein 46 Afu8g05880 chaperonin (GroES/Cpn10)
47 Afu8g06480 rhamnogalacturonan acetylesterase superfamily protein 48 Afu8g06820 dihydrofolate reductase family protein
49 Afu8g07170 conserved hypothetical protein 50 Afu1g13790 Histone H3
51 Afu2g17750 ankyrin 2,3/unc44
52 Afu5g01180 RAN small monomeric GTPase (Ran) 53 Afu5g01300 integral membrane protein
54 Afu5g09030 similar to ankyrin 2,3/unc44
55 Afu8g00260 f-box domain and ankyrin repeat protein 56 Afu8g02140 ankyrin repeat protein
18
Figure 1 The phylogenetic tree of AFUA_5G10930.
The gene STIAU_2222 and STAUR_2131 are genes of Bacteria but they distribute in a fungi phylogeny. This phylogeny shows that the genes of Stigmatella aurantiaca transferred from fungi. The two genes are the same gene but different annotation by different sequencing release data. The expression of the node is “taxonomy|species|accession no.|protein product.” FA is fungi and ascomycete; B is Bacteria.
FA|A. oryzae|BAE66289.1|unnamed protein product FA|A. flavus|XP 002384622.1|conserved hypothetical protein FA|A. oryzae|XP 001827422.2|glucuronan lyase A FA|A. flavus|XP 002378726.1|conserved hypothetical protein
FA|A. oryzae|XP 001823345.1|glucuronan lyase A
FA|T. stipitatus|XP 002484423.1|integral membrane protein FA|P. marneffei|XP 002149242.1|conserved hypothetical protein B|S. aurantiaca|ZP 01465287.1|hypothetical protein STIAU 2222
B|S. aurantiaca|YP 003951762.1|hypothetical protein STAUR 2131 FA|A. clavatus|XP 001274384.1|conserved hypothetical protein FA|N. fischeri|XP 001259598.1|hypothetical protein NFIA 076310 FA|A. fumigatus|XP 753554.1|conserved hypothetical protein FA|A. fumigatus|EDP51830.1|conserved hypothetical protein
FA|S. macrospora|CBI55925.1|unnamed protein product FA|N. crassa|XP 959843.1|hypothetical protein NCU05852 FA|A. nidulans|XP 657616.1|hypothetical protein AN0012.2
FA|N. haematococca|XP 003041896.1|hypothetical protein NECHADRAFT 52969 FA|M. anisopliae|EFY98252.1|glucuronan lyase A
FA|H. jecorina|BAG80639.1|glucuronan lyase A
FA|C. globosum|XP 001226268.1|hypothetical protein CHGG 08341 FA|M. oryzae|XP 363838.1|hypothetical protein MGG 01764
100 100 100 100 100 82 100 100 100 96 67 42 55 63 54 56 36 0.05
Fungi
Fungi
Node A Bacteria19
Figure 2 The GC content of the neighbor genes of the HGT gene and Stigmatella
aurantiaca genome.
Almost all of the GC content of neighbor genes are higher than Stigmatella
aurantiaca genome or close to 67% except STAUR_2129 and STAUR_2131.
20
Figure 3 The GC content of HGT gene compare with the foreign gene and genome.
Compare HGT genes STAUR_2131 and STIAU_2222 with the neighbor genes of
Stigmatella aurantiaca genome and orthologs of Aspergillus clavatus, Neosartorya fischeri, and Aspergillus fumigatus. The GC level of HGT genes are more close to the
21
Figure 4 The GC content of amino acid code 1,2 and code 3(wobble site)
There show the ribosomal proteins’ GC content of position 1, 2 and position 3 of amino acid code. The genes with star marks represent HGT orthologs. The left and right genes to the star marked gene are the genes before and behind the gene of HGT orthologs. Aor stands for Aspergillus oryzae, Afl stands for Aspergillus flavus, Tst stands for Talaromyces stipitatus, Pma stands for Penicillium marneffei, Sau stands for Stigmatella aurantiaca, Acl stands for Aspergillus clavatus, Nfi stands for
Neosartorya fischeri, Afu stands for Aspergillus fumigatus, and Ncr stands for Neurospora crassa.
Node A Node B
22
Figure 5 The synonymous and nonsynonymous distance distribution.
The figure shows the distribution of the synonymous and non-synonymous distances between HGT orthologous gene. The distances between orthologs and STAUR_2131 are marked by red points and are labeled the species.
23
Figure 6 Scan the GC content of the transferred sequence region
The GC content scanning along Stigmatella aurantiaca genome near the HGT gene from gene STAUR_2129 to gene STAUR_2134. The blue line shows the GC content of whole nucleotides along sequences and the red line shows the GC content of wobble site in protein coding region. The GC contents of wobble site in protein coding regions are higher that the GC content of average sequence.
24
Figure 7 The GC content of intergenic region.
The GC content of the intergenic region between STAUR_2132 and STAUR_2133 are displayed by 3 site groups compared to the total GC content of the HGT region of
25
Figure 8 The comparison of Pro codon usage bias.
The Pro codon bias usages are compared with ribosomal protein and genome codon usage in the second and third column. The first, second, and third rows are the HGT orthologous genes or ribosomal protein and genome codon bias in Stigmatella
26
Figure 9 The comparison of Phe codon usage bias.
The Phe codon bias usages are compared with ribosomal protein and genome codon usage in the second and third column. The first, second, and third rows are the HGT orthologous genes or ribosomal protein and genome codon bias in Stigmatella
27
Figure 10 The comparison of Leu codon usage bias.
The Leu codon bias usages are compared with ribosomal protein and genome codon usage in the second and third column. The first, second, and third rows are the HGT orthologous genes or ribosomal protein and genome codon bias in Stigmatella
28
Figure 11 The comparison of Ile codon usage bias.
The Ile codon bias usages are compared with ribosomal protein and genome codon usage in the second and third column. The first, second, and third rows are the HGT orthologous genes or ribosomal protein and genome codon bias in Stigmatella
29
Figure 12 The comparison of Ser codon usage bias.
The Ser codon bias usages are compared with ribosomal protein and genome codon usage in the second and third column. The first, second, and third rows are the HGT orthologous genes or ribosomal protein and genome codon bias in Stigmatella
30
Figure 13 The comparison of Val codon usage bias.
The Val codon bias usages are compared with ribosomal protein and genome codon usage in the second and third column. The first, second, and third rows are the HGT orthologous genes or ribosomal protein and genome codon bias in Stigmatella
31
Reference
1. Brown, J.R., Ancient horizontal gene transfer. Nat Rev Genet, 2003. 4(2): p. 121‐32. 2. Gogarten, C.P.A.J.P., Biased gene transfer in microbial evolution. Nature Reviews Microbiology, 2011. 9: p. 543‐555. 3. Rivera, M.C. and J.A. Lake, The ring of life provides evidence for a genome fusion origin of eukaryotes. Nature, 2004. 431(7005): p. 152‐5. 4. C. G. Kurland, B.C., and Otto G. Berg*, Horizontal gene transfer: A critical view. PNAS, 2003. 100(17): p. 9658‐9662. 5. Jain, R., M.C. Rivera, and J.A. Lake, Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci U S A, 1999. 96(7): p. 3801‐6. 6. Shoemaker, N.B., et al., Evidence for extensive resistance gene transfer among Bacteroides spp. and among Bacteroides and other genera in the human colon. Appl Environ Microbiol, 2001. 67(2): p. 561‐8. 7. Becq, J., C. Churlaud, and P. Deschavanne, A benchmark of parametric methods for horizontal transfers detection. PLoS One, 2010. 5(4): p. e9989. 8. Regeard, C., et al., Indications for acquisition of reductive dehalogenase genes through horizontal gene transfer by Dehalococcoides ethenogenes strain 195. Appl Environ Microbiol, 2005. 71(6): p. 2955‐61. 9. Mehrabi, R., et al., Horizontal gene and chromosome transfer in plant pathogenic fungi affecting host range. FEMS Microbiol Rev, 2011. 35(3): p. 542‐54. 10. Ciuffetti, L.M., R.P. Tuori, and J.M. Gaventa, A single gene encodes a selective toxin causal to the development of tan spot of wheat. Plant Cell, 1997. 9(2): p. 135‐44. 11. Faris, J.D., et al., A unique wheat disease resistance‐like gene governs effector‐triggered susceptibility to necrotrophic pathogens. Proc Natl Acad Sci U S A, 2010. 107(30): p. 13544‐9. 12. Liu, Z., et al., The Tsn1‐ToxA interaction in the wheat‐Stagonospora nodorum pathosystem parallels that of the wheat‐tan spot system. Genome, 2006. 49(10): p. 1265‐73. 13. Whitaker, J.W., G.A. McConkey, and D.R. Westhead, Prediction of horizontal gene transfers in eukaryotes: approaches and challenges. Biochem Soc Trans, 2009. 37(Pt 4): p. 792‐5. 14. KO, R.H.B.a.H., Detecting Horizontally Transferred and Essential Genes Based on Dinucleotide Relative Abundance. DNA Research, 2008. 15: p. 267‐276.32 15. R.Grantham, C.G., M.Gouy, R.Mercier and A.Pave', Codon catalog usage and the genome hypothesis. Nucleic Acids Research, 1980. 8: p. r49‐r62. 16. NCBI. Available from: ftp.ncbi.nih.gov. 17. Nakamura, Y., Gojobori, T. and Ikemura, T., Codon usage tabulated from the international DNA sequence databases: status for the year 2000. Nucl. Acids Res., 2000. 28: p. 292. 18. Altschul, S., W Gish, W Miller, EW Myers, DJ Lipman, Basic local alignment search tool. J Mol Biol, 1990. 215: p. 403‐410. 19. Mobyle Codonw 1.4.4. Available from: http://mobyle.pasteur.fr/cgi‐bin/portal.py?#forms::codonw. 20. Heidelbach, M., H. Skladny, and H.U. Schairer, Heat shock and development induce synthesis of a low‐molecular‐weight stress‐responsive protein in the myxobacterium Stigmatella aurantiaca. J Bacteriol, 1993. 175(22): p. 7479‐82. 21. Reing, J.E., et al., Degradation products of extracellular matrix affect cell migration and proliferation. Tissue Eng Part A, 2009. 15(3): p. 605‐14.