Dissociation Rate Constant of the Hydrogen Fluoride Dimer by the ab Initio Anharmonic
RRKM Theory
†L. Yao*
Department of Physics, Dalian Maritime UniVersity, Dalian, China 116026
A. M. Mebel
Department of Chemistry and Biochemistry, Florida International UniVersity, Miami, Florida 33199
S. H. Lin
Institute of Atomic and Molecular Sciences, Academia Sinica, P.O. Box 23-166, Taipei, Taiwan 10764 and Department of Applied Chemistry, National Chiao-Tung UniVersity, Hsin-chu, Taiwan
ReceiVed: May 13, 2009; ReVised Manuscript ReceiVed: September 18, 2009
The dissociation rate constants for hydrogen fluoride dimers are calculated using the YL method, proposed by Yao and Lin (Yao, L.; Mebel, A. M.; Lu, H. F.; Neusser, H. J.; Lin, S. H. J. Phys. Chem. A 2007, 111, 6722). The dividing surface method and RRKM theory are also used to obtain pseudo-transition states and rate constants, respectively. For the dissociation of HF dimers, the anharmonic rate constants are around 3.02 × 1010to 3.46× 1012s-1, while the harmonic rate constants are in the range of 2.93× 1010to 1.66× 1013 s-1, at a temperature range of 243-1000 K, for the canonical case. For the microcanonical case, the anharmonic rate constants are in the range of 1.91× 1012to 1.32× 1013s-1and the harmonic rate constants are in the range of 1.89× 1012to 4.93× 1013s-1, with a total energy range of 1338-4500 cm-1. Both harmonic and anharmonic rate constants are only comparable to the experimental results 0.5× 1010s-1 and 1× 1011s-1 in an inert gas bath at room temperature. In this case the harmonic and anharmonic rate constants show similar results in this calculation. The results of this paper indicate that the YL method is suitable for calculating dissociation rate constants of small flexible HF dimers and the anharmonic effect should be included.
1. Introduction
Hydrogen bonded materials are important in many industrial and environmental processes, such as atmospheric aerosol formation, steam erosion of turbines, and atmospheric absorption of solar radiation.1-5 Experimental data on the IR, far-IR, vibration-rotation-tunneling and microwave spectra of HF clusters in the gas phase are available.6The primary step toward condensation and molecular dimer formation has been studied in detail via high-resolution laser7and Fourier transform infrared (FTIR) spectroscopy,8,9achieving instrumental bandwidths of about 1 and 100 MHz, respectively. IR spectroscopy of HF in supersonic jets reveals size-separated cluster absorptions in the fundamental H-F stretching region10-15consistent with simple ring structures.11Due to a vibrational Franck-Condon effect, combination bands with an F-F stretching mode gain intensity relative to the dimer.12-15For instance, Lee and co-workers16 showed that water and HF cluster formation in a molecular beam can be predicated by tunable, pulsed, infrared radiation in the frequency range 2900-3750 cm-1. Available experimental data for the dissociation of HF dimer, such as property deconvolu-tions and matrix spectra are very limited beyond the vapor phase for the HF and water dimer species. For instance, an upper limit to the excited vibrational state lifetime, 1 µs, was determined from the results reported. In the experiment, molecular beams containing small clusters of HF were prepared by supersonic expansion of 0.5-3.0% HF in a carrier of He, Ne, or Ar.17aAs
the laser was tuned through the frequency region of interest, the absorption of an infrared photon by (HF)n, which then
predissociates, was detected by the depletion of the (HnFn-1)*
mass spectrometer signal after the laser pulse. Vibrational predissociation lifetimes between 1 µs and 1 ms can be determined directly by the apparatus.17aNext the dimer bands of IR spectra can be assigned as follows: the 3720 cm-1band to the H-F stretch of the hydrogen-bonded proton, the 3878 cm-1band to the H-F stretch of the free proton, and the 3970 cm-1band to a combination band involving intra- and inter-molecular modes. The observed rotational structure of the 3970 cm-1 band with line widths of approximately 0.15 cm-1, combined with Lee’s results enables the predissociation lifetime τ to be limited to 30 ps< τ < 1 µs.17aThe predicted lifetime of (HF)2based on an empirical treatment of vibration to translation energy transfer is 200 ps, which falls within the above range, which corresponds to a rate constant of around k ) 0.5× 1010 s-1, for the dissociation of HF dimers was based on a room temperature gas phase.17b
Theoretical results can be used to simulate the corresponding experimental results. The surface of the electrostatic potential of optimized hydrogen-bonded clusters of HF dimers has been studied using the HF/6-31G level of theory.18Ovchinnikov et al. used the perturbative extension of diatomics-in-ionic-systems (DIIS) formulation as a practical method to study HF dimers.19 The dissociation energies reported in refs 19 and 20 are De) 1595 cm-1(4.56 kcal mol-1) and 1590 cm-1(4.55 kcal mol-1), respectively. Structure, binding energies, and vibrational spectra obtained by the DFT method are all reported in ref 21 for the
†Part of the “Vincenzo Aquilanti Festschrift”.
* Corresponding author, [email protected].
10.1021/jp9044379 2009 American Chemical Society Published on Web 10/14/2009
HF dimer. The other ab initio calculations carried out using MP2 (the Møller-Plessett second order perturbation method) for the HF dimer, transition state (TS) energies, and tunneling probability are reported in ref 22. As reported in ref 23, ab initio based six-dimensional semiempirical pair interaction potential for HF was used to give De) 19.1 kJ mol-1(3.06 kcal mol-1) and 4.2 kJ mol-1for the electronic hydrogen bond tunneling barrier. The bond lengths of H-F and F-F and the angles within the HF dimer are all reported in refs 18 and 24. Dissociation energy of the dimer into two monomers is 4.6 kcal mol-1.24VTST (variational transition state theory)25has been widely used for the modeling of barrierless radical-radical recombination, dissociation processes and has been applied successfully to a variety of reactions, in both the gaseous and condensed phases. In reactions without saddle points the variational criteria becomes essential for the application of TST (transition state theory).26 On the other hand, in the past few years, many chemists have observed significant anharmonic effects in the dissociation of molecular systems, especially of clusters and molecules with highly flexible TSs.26-32
The purpose of the present study is to compute accurate anharmonic rate constants for dissociation (evaporation) of the HF dimer in the temperature range of 243-1000 K for the canonical case and in the energy range of 1338-4500 cm-1 for the microcanonical case using the VTST method. Rate constants have been calculated using the systematic method developed recently by Yao and Lin (YL method),33 and the anharmonic effect on molecular dissociation has been examined.
2. Computational Methods
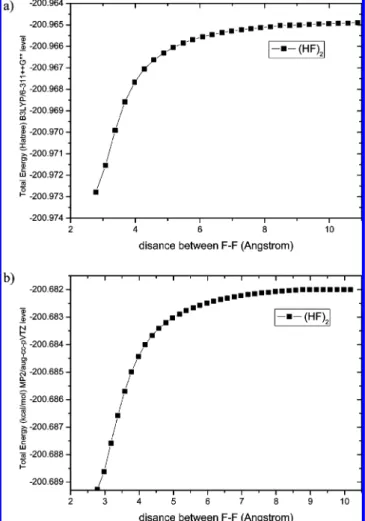
a. Ab Initio Calculations. Geometries of reactants and TSs have been optimized using the B3LYP/6-311++G** and MP2/ aug-cc-pVTZ levels. Vibrational harmonic and anharmonic frequencies, calculated at the same level, are used for charac-terization of stationary points and ZPE (zero-point energy) corrections. The optimized structure of the reactant HF dimer, at the MP2/aug-cc-pVTZ level, is given in Figure 1, where the numbers show optimized bond lengths, bond angles, and atomic labels. By use of the dividing surface method, at the MP2/aug-cc-pVTZ level, VTST is used to obtain the pseudo-transition state in the reaction, as a distinct TS for the dissociation of the HF dimer does not exist. To locate variational TSs, the potential energy surface (PES) was scanned along the reaction coordinate corresponding to the F-F distance between two HF monomers, shown in Figure 2. It is critical to compute the energy barrier as accurately as possible, because evaporation of the HF dimer is an activated process and such activated chemical reactions have rates that depend exponentially upon the height of an energy barrier. To achieve higher accuracy and reliability, single-point energies were recalculated by employing the coupled cluster CCSD(T) method with the aug-cc-pVTZ basis set. Grigorenko et al. used the method of extending the distance of an extended F-F bond from 2.7 to 6.0 Å to study the hydrogen bonding character of HF dimers.34,35They reproduced the HF dimer surface and concluded that the saddle points are far from
the minimum positions, which agrees with the results in this work. All the frequencies, energies of reactant, and TS structures in this work are calculated at 1 atm and 298.15 K (room temperature) using the GAUSSIAN 03 program.36The dissocia-tion rate constants of the HF dimer are calculated using these parameters.
(b) Rice-Ramsperger-Kassel-Marcus (RRKM)Theory and VTST Calculations. The YL method33in conjunction with a fitted Morse potential is used to calculate the rate constants of the HF dimer in the canonical and microcanonical cases. The RRKM theory is used to compute the dissociation rate constant of the HF dimer. Equation 1 gives the rate constant, k(E), for a unimolecular reaction A* f Aq
f P (where reaction A* and P are the reactant and products, respectively; Aq denotes the activated complex).27
where σ (set as 1) is the symmetry factor; h is Planck’s constant; F(E) represents the density of states of the energized reactant molecules; Wq(E - E
aq) is the total number of states for the TS (activated complex); E represents the total internal energy available to the system. (Aqcorresponds to an energy barrier of Eaq).
According to the TST theory, for a canonical system, the unimolecular rate constant is given by eq 2
Figure 1. The optimized structure of reactant HF dimer at the MP2/
aug-cc-pVTZ levels (bond lengths in angstroms and angles in degrees).
Figure 2. Changes of the relative B3LYP/6-311++G** (a) and MP2/
aug-cc-pVTZ (b) energies as function of coordinate distance between F1-F3 in angstroms for the HF dimer.
k(E) )σ h Wq (E - Ea q ) F(E) (1)
where Q(T) represents the partition function for the reactant and Q(T)qis the partition function for the activated complex. It is often more reasonable to include the ZPE in the energy barrier Eaq. For the case of the Morse oscillator, the partition function can be calculated by eq 3
where β ) 1/kT and N is the number of vibrating modes of reactant; the expression for Qq(β) is similar to eq 3.
In the calculations of the anharmonic effect in this paper, the Morse oscillator was chosen to fit the PES and to simulate the bonding. The potential energy curve for the HF dimer dissociation has also been calculated (Figure 2), and the VTST has been used to obtain the pseudo-transition states and the dissociation rate constants. Because no distinct TS exists on the PES for dissociation of the HF dimer, (HF · · · HF as for the case of a simple bond-cleavage process), one can consider different positions for the TS along the reaction path and calculate rate constants corresponding to each of them. VTST considers a one-way reactive flux through a dividing surface separating reactants and products and provides an upper boundary to the local-equilibrium reaction rate.25In the micro-canonical VTST, the minimum in the micromicro-canonical rate constant is found along the reaction path according to eq 4
where qqis the reaction coordinate, such that a different TS is found for each different energy. The individual microcanonical rate constants are minimized at the point along the reaction path where the total number of states Wq(E - Eq) has a minimum value. Thus, the reaction bottleneck is located where the minimal Wq(E - E
aq) values are found. That is, the sums of states for each TS candidate must be calculated along the reaction path. Each of these calculations requires the optimized structure, single-point energy, zero-point energy, and vibrational frequen-cies as functions of the reaction coordinate.37-39Using the above methods, rate constants and branch ratio provide a basis for future studies of a variety of collective phenomena such as dissociation and mass spectra of clusters and their IR fragmenta-tion spectra.
3. Results and Discussion
The dissociation (evaporation) process is defined as the removal of a hydrogen fluoride monomer from a cluster. The reactant is an HF dimer molecule, while the product is two HF monomers at infinite separation. Thus, the dissociation rate constant, k, for the HF dimer dissociating into two monomers corresponds to the following equation
Determination of the energetic quantities begins with the PES or interaction potential that is a function of all internal coordinates of the HF dimer. A series of energies at different distances between two dissociating fragments, corresponding to the length of a bond to be broken during dissociation, is calculated and is considered to be the reaction coordinates. To obtain these energies, partial geometry optimization was per-formed, with fixed values of the reaction coordinate and all other geometric parameters being optimized. The PES scan along the F-F distance (Figure 2) was carried out at the B3LYP/6-311++G** and MP2/aug-cc-pVTZ levels of the theory. As expected, Figure 2 indicates that no intrinsic barrier exists for the dissociation of the HF dimer, as the reverse process is barrierless. This is similar for the evaporation of a water dimer in ref 33. As no distinct barrier exists on the PES, the variational TS is represented by a dividing surface corresponding to a minimum in the reactive flux. The following procedure has been used for the VTST calculations. First, we scanned the PES, considering the dimer separating the two HF monomers with varying the F-F bond distance. Forty optimized geometries were calculated corresponding to the F-F distances from 2.78 to 10.28 Å with a step of 0.2 Å. For obtaining more accurate positions of the variational TSs, an additional 40 optimized geometries were calculated with F-F distances from 4.53 to 4.93 Å with a step of 0.01 Å. The relative energies at each point along the reaction path in parts a and b of Figure 2 were scaled by factors 0.958 and 0.942, respectively, which were obtained by comparing the HF dimer dissociation energies at the CCSD(T)/aug-cc-pVTZ and B3LYP/6-311++G**//MP2/ aug-cc-pVTZ levels, shown at the bottom of Table 1, and the results were used to evaluate the Morse potential energy function. The reaction energy calculated at the CCSD(T)/aug-cc-pVTZ level is 3.037 kcal mol-1with ZPEs in close agreement with other experimental values of 3.03538and 3.036 kcal mol-1 4 for dissociation of the HF dimer. The dissociation energy 4.83 kcal mol-1, without ZPE, in this work is also in close agreement with theoretical values of 4.5019and 4.60 kcal mol-1 24without ZPE, for the evaporation of the HF dimer. Minimal values for the total number of states at different energies were found using each structure along the internal reaction coordinate optimized at the B3LYP/6-311++G** level and used to fit the Morse potential energy function. Employing the harmonic model and using 3N - 7 harmonic vibrational frequencies (one imaginary vibrational frequency was excluded), the partition functions were obtained. Using the dividing surface method, the TS corresponds to an ensemble of configurations where the structure has the minimal total number of states. The TS, or pseudo-TS, for dissociation of the HF dimer is located at the F-F distance of 4.72 Å at 298.15 K and 1 atm, while for the water dimer, it was found at the O-O distance of 7.365 Å at 273 K.33 The bond energy of H · · · F is larger than the bond energy of H · · · O. As shown in Table 1, the reaction activation energy calculated at the B3LYP/6-311++G** and MP2/aug-cc-pVTZ levels is 3.64 and 2.60 kcal mol-1, respectively, with ZPE corrected, which are close to the dissociation energy of 3.03 kcal mol-1 in CCSD(T)/aug-cc-pVTZ level and ZPE at MP2/aug-cc-pVTZ level of this work. The geometric and energetic parameters of the reactant and pseudo-TS are collected in Table 1, including apparent activation energies, i.e., relative energies of the TS with respect to the reactant. We recomputed the single point energies at the CCSD(T)/aug-cc-pVTZ levels to obtain their more accurate estimates. Finally, the energy and number of states of pseudo-TS were employed to calculate the canonical and k(T) ) kT h Q(T)q Q(T)e -Eaq/kT (2) Q(β) )
∏
i)1 N∑
ni)0 ni(m) e-βEni (3) dk(E) dqq ) 0 (4) (HF)298 k 2HF (5)microcanonical rate constants of the direct dissociation processes by using the YL method within RRKM formalism.
The anharmonic and harmonic dissociation rate constants calculated using VTST are presented in Table 2, in a temperature range of 243-1000 K, for the canonical system. Corresponding to data in Table 2, the evaporation rate constants for the HF dimer are plotted in Figure 3. In earlier works, the experimental rate constants were predicted to be around 0.5× 1010s-1by Lee et al.,17aand to be around 1× 1011s-1at a room temperature by Klemperer et al.,17bwhich is close to the anharmonic results of 3.02× 1010s-1at the lower temperature of 243 K and 1.22 × 1011s-1at 303 K, for canonical systems. In comparison with the harmonic results, the canonical evaporation rate constant, for which the anharmonic effect is included, appears very different at high temperatures. Additional simulations using higher temperatures and different dividing surfaces may provide an insight as to the nature of the stability displayed by particular clusters.
The relationship between the total energy of a microcanonical system and the temperature of a canonical system is shown by eq 633
The total energies corresponding to the canonical temperatures of 243, 273, 303, 333, 500, 600, 700, 800, 900, 1000 K are 0. 668 (234 cm-1), 0.838 (293 cm-1), 1.017 (356 cm-1), 1.205 (421 cm-1), 2.347 (821 cm-1), 3.076 (1076 cm-1), 3.827 (1338 cm-1), 4.595 (1607 cm-1), 5.381 (1882 cm-1), 6.185 kcal mol-1 (2163 cm-1), respectively, which are also given in Tables 2 and 3. The first five energies are all lower than the calculated activation energy of 2.60 kcal mol-1. Table 2 shows the anharmonic and harmonic rate constants of the HF dimer dissociations in the temperature range of 243-1000 K, for the canonical case. From Table 2 and Figure 3, we can see that the anharmonic rate constants change from 3.02× 1010s-1at 243 K to 3.46 × 1012 s-1 at 1000 K, while the harmonic rate constants change from 2.93× 1010s-1at 243 K to 1.66× 1013 s-1at 1000 K. The magnitude of the harmonic dissociation rate constant is much higher than the anharmonic dissociation rate constant at high temperature and energies for the microcanonical and canonical cases. Hence, the microcanonical rate constant must be calculated at higher total energies of 1338-4500 cm-1, which corresponds to the vibrational frequencies of the HF dimer and high canonical temperatures above 500 K. Table 3 shows the anharmonic and harmonic rate constants of evaporation of the HF dimer at the given energy range. Data for the microca-nonical case are illustrated in Figure 4. The results show that the rate constant increases with the increasing total energy and the anharmonic result is in the range of 1.91× 1012to 1.32× 1013s-1, which is close to the predicted values within the literature.17The harmonic rate constants increase from 1.89× 1012s-1at 1338 cm-1to 4.93× 1013s-1at 4500 cm-1. As seen
TABLE 1: The Properties of HF Dimer and TS in B3LYP/6-311++G** Level and MP2/aug-cc-pVTZ Levela
HF dimer minimum B3LYP/6-311++G** TS HF dimer minimum MP2/aug-cc-pVTZ TS
zero-point energy (hartree) 0.0211868 0.0191855 0.0216429 0.0194269
imaginary frequencies cm-1 29.3742i 29.7284i
F1-F3 distance, Å 2.71889 4.72 2.75 4.72 F1-H2 distance, Å 0.934 0.929 0.928 0.923 F3-H4 distance, Å 0.930 0.928 0.925 0.922 angle (H2-F3-H4) 123.1 165.8 114.4 165.8 F1-F3-H2, deg 4.53 0.58 3.23 0.52 F3-F1-H2, deg 8.76 2.38 6.38 2.15
total energies (hartree) -200.9516787 -200.9438923 -200.6892854 -200.6832631
barrier (kcal/mol) 3.63b 2.60c
aThe dissociation energy De ) 3.03 kcal/mol CCSD(T)/aug-cc-pVTZ level with ZPE at MP2/aug-cc-pVTZ level in our work, which is close
to accurate spectroscopic measurements of the hydrogen bond dissociation energy De ) 3.035 kcal/mol38 and 3.036 kcal/mol.4 The energy
barriers calculated are 2.60 and 3.63 kcal/mol, which are close to the dissociation energy De ) 3.03 kcal/mol in our work and is close to the
bonding energies 4.56 kcal/mol in ref 25 and 3.035 kcal/mol in refs 4 and 38. b
The scaling factor is 0.958 of B3LYP/6-311++G** and
CCSD(T)/aug-cc-pVTZ (activation energy is 4.83 kcal/mol without ZPE).cThe scaling factor is 0.942 of MP2/aug-cc-pVTZ and CCSD(T)/
aug-cc-pVTZ (activation energy is 4.83 kcal/mol without ZPE).
TABLE 2: Evaporation Rate Constants for HF Dimer at a Temperature Range of 243-1000 K for the Canonical System, with Energy Barrier 2.60 kcal mol-1
temperature (K) 243.0 273.0 303.0 333.0 500.0 600.0 700.0 800.0 900.0 1000.0
corresponding energy (kcal/mol) 0. 668 0.838 1.017 1.205 2.347 3.076 3.827 4.595 5.381 6.185
correspond (cm-1) 234 293 356 421 821 1076 1338 1607 1882 2163
anharmonic rate constant of canonical case 1/s
3.02× 1010 6.59× 1010 1.22× 1011 1.99× 1011 9.47× 1011 1.50× 1012 2.05× 1012 2.56× 1012 3.03× 1012 3.46× 1012
harmonic rate constant of canonical case 1/s
2.93× 1010 7.03× 1010 1.43× 1011 2.58× 1011 1.99× 1012 4.00× 1012 6.62× 1012 9.70× 1012 1.31× 1013 1.66× 1013
Figure 3. Graph corresponding to canonical rate constant log(k) versus
temperature (K), for evaporation of HF dimer. The unit of rate constant
k is s-1. Data correspond to Table 2.
E )
∑
i
(
pωi
in Tables 2 and 3, the anharmonic VTST evaporation rate constant is nearly insensitive to the temperatures and total energies available, within the given range. However, the harmonic rate constant is highly sensitive to the temperatures and total energies available. The total number of states of the Morse potential is insensitive to energy changes when the energy is high. Generally, the anharmonic rate constants (Tables 2 and 3) are lower than the harmonic rate constants at higher temperatures and total energies, while the harmonic rates are slightly higher at lower temperatures and total energies. The difference is caused by the use of different models and harmonic and anharmonic potentials, which are utilized to simulate the vibrational bonds. For the different models and different vibrational states, the total number of states and density of states are counted, which affects the dissociation rate constant. It should be noted that the differences in the dependence of harmonic and anharmonic rate constants upon the temperature are due to the use of the harmonic surfaces and anharmonic surfaces in the RRKM calculations of the rate constants and the anharmonic oscillator (obtained from the corresponding anharmonic surfaces) will have nonequal energy spacings, decreasing with the increasing quantum numbers and its vibrational quantum numbers have a maximum limit. Figure 3 shows the differences when comparing the harmonic and anharmonic rate constants. The rate constant for the harmonic results increases sharply at temperatures ranging from 243 to 1000 K. The results in Table 3 show that at higher energies the anharmonic rate constants are much lower than the harmonic rate constants. The increasing rate of the anharmonic case decreases at the energy range that we calculated. The bonds may initially be broken very quickly at high energies, allowing the rate constant to initially increase sharply and then the increasing rate decrease at high energy. For instance, the calculated value is in reasonable agreement with experimental predictions17a for the hydrogen bond lifetime τ ) 200 ps (corresponds to a rate constant of around k ) 0.5× 1010s-1)
in HF dimers, based on an empirical treatment of the vibration to translation energy transfer. This should be compared to the anharmonic rate constant results of 3.02× 1010 s-1at lower temperature 243 K in this work. The lifetime estimate of τ∼ 10-11 s (corresponds to a rate constant of around k ) 1.0 × 1011s-1) by Klemperer et al. for the dissociation of HF dimers based on a room temperature gas phase, which is also very close to our anharmonic rate constant k ) 1.22 × 1011 s-1 at temperature 303 K.17bNormally, the microcanonical and canoni-cal variational criteria give similar results for the rate constants in the present paper, as well as before in ref 33.
The Gaussion 03 program was used in the calculation of anharmonic frequencies, and all the states which are lower than the dissociation energy of the oscillator are counted in calculat-ing the vibrational partition functions. We know that for the dissociation reaction one vibrating mode changed to a transla-tional mode, so the vibrating bond is significant. In this work only vibrational degrees of freedom are included, while the effect of rotational degree of freedom can be found in Tables 8 and 9 in ref 40. It should be noted that the contribution of rotational degrees of freedom can easily be taken into account in our method for the RRKM calculation through the calculation of rotational partition function. We can even take into account the effect of vibration-rotation interaction. This will be carried out in future work. Kinetic results, both predicted and calculated, have been interpreted and analyzed from the viewpoint of first-principle calculations. The major anharmonic effect of the dissociation rate constant is reported in this paper, which is very similar to that of the dissociation of the water dimer in ref 33. Anharmonic results of dissociation of HF dimer reported in our present work are in reasonable agreement with the experimental predictions.33This is the first anharmonic study of the temper-ature dependence upon the evaporation rate constant for an HF dimer.
4. Conclusions
The dividing surface VTST method and RRKM theory within the YL method are effectively used in the studies of HF clusters in this paper. In this work, the total number of states and density of states have been obtained, in order to calculate the rate constant. The results show that the behavior of the anharmonic VTST rate constants is quite different from that of the harmonic rate constants in the high energy (or temperature) range. Within both harmonic and anharmonic approaches, the canonical and microcanonical calculations give very close results in the low energy (or temperature) range. The method used in this paper can be extended to treat larger clusters of water or other small molecules. The calculations would then become more complex than for the HF and water dimers, not only due to increasing computation costs but also because there are more contributions to the anharmonic effect from numerous vibrational degrees of freedom. Another complication for larger clusters is the pos-sibility of a large variety of different isomers and conformations. Nevertheless, the present study clearly demonstrates that the role of anharmonic effects in the kinetics of cluster decomposi-tion is very important and should be taken into account. Similar behavior has been found when calculating the water dimers.
TABLE 3: Rate Constant of Evaporation of HF Dimer for Different Total Energies for Microcanonical System at Energy Barrier 2.60 kcal/mol
total energy (cm1) 1338 1607 1882 2163 3500 4000 4500
corresponding total energy (kcal/mol) 3.827 4.595 5.381 6.185 10.007 11.437 12.866
anharmonic rate constant (1/s) 1.91× 1012 7.97× 1012 1.29× 1013 1.23× 1013 1.14× 1013 1.23× 1013 1.32× 1013
harmonic rate constant (1/s) 1.89× 1012 5.10× 1012 9.42× 1012 1.44× 1013 3.72× 1013 4.37× 1013 4.93× 1013
Figure 4. Graph corresponding to microcanonical rate constant (log(k)
versus total energy (cm-1) for evaporation of HF dimer. The unit of rate constant k is s-1. Data correspond to Table 3.
Normally, the microcanonical and canonical variational criteria will give similar results for the rate constants, as shown in this paper and our previous paper. Actually, both of harmonic and anharmonic rate constants are only comparable to the experi-mental results 0.5× 1010s-1reported in ref 17b. In this case the harmonic and anharmonic show similar results in this calculation. Comparing the behavior of the water dimer and the anharmonic effect of dissociation of the HF dimer, we can conclude that the anharmonic effect of the HF dimer is as pronounced as that for the water dimer. Finally, the YL method has been found to be suitable for studying these kinds of small cluster systems.
Acknowledgment. This work was supported by the National Natural Science Foundation of China (Grant No. 10704012), NSC (Taiwan), Academia Sinica and the Natural Science Foundation of Liaoning Province (Grant Nos. 2006Z064, 20082140), and DLMU-ZL-200811.
References and Notes
(1) Maerker, C.; Schleyer, P. V. R.; Liedl, K. R.; Ha, T. K.; Quack, M.; Suhm, M. A. J. Comput. Chem. 1997, 18, 1695.
(2) Suhm, M. A. Bunsenges, Ber. J. Phys. Chem. 1995, 99, 1159. (3) Klopper, W.; Quack, M.; Suhm, M. A. Chem. Phys. Lett. 1996,
261, 35.
(4) Quack, M.; Suhm, M. A. J. Phys. Chem. 1991, 95, 28. (5) (a) Huong, P. V.; Couzi, M. J. Chim. Phys. 1969, 66, 1309. (b) Janzen, J.; Bartell, L. S. J. Chem. Phys. 1969, 50, 3611.
(6) (a) Desbat, B.; Huong, P. V. J. Chem. Phys. 1983, 78, 6377. (b) Cotton, F. A.; Wilkinson, G. AdVanced Inorganic Chemistry, 5th ed.; Wiley: New York, 1988; Chapter 3.
(7) (a) Chang, H. C.; Klemperer, W. J. Chem. Phys. 1994, 100, 1. (b) Signorell, R.; He, Y.; Muller, H. B.; Quack, M.; Suhm, M. A. Proc. 10th Int. Symp. on Atomic, Molecular, Cluster, Ion, and Surface Physics, VDF, Zurich 1996; pp 256-259. (c) Anderson, D. T.; Davis, S.; Nesbitt, D. J.
J. Chem. Phys. 1996, 104, 6225. (d) Bali, Z.; Miller, R. E. J. Phys. Chem.
1996, 100, 12945.
(8) Puttkamer, K. V.; Quack, M. Chem. Phys. 1989, 139, 31. (9) Quack, M.; Schmitt, U.; Suhm, M. A. Chem. Phys. Lett. 1997, 269, 29.
(10) Franck, E. U.; Meyer, F. Z. Elektrochem. 1959, 63, 571. (11) Suhm, M. A. Ber. Bunsen-Ges. Phys. Chem. 1995, 99, 1159. (12) Klopper, W.; Quack, M.; Suhm, M. A. Chem. Phys. Lett. 1996,
261, 35.
(13) (a) Quack, M.; Suhm, M. A. J. Chem. Phys. 1991, 95, 28. (b) Quack, M.; Suhm, M. A. Theor. Chim. Acta 1996, 93, 61. (c) Klopper, W.; Quack, M.; Suhm, M. A. Chem. Phys. Lett. 1996, 261, 35.
(14) Suhm, M. A. Ber. Bunsen-Ges. Phys. Chem. 1995, 99, 1159. (15) Huisken, F.; Kaloudis, M.; Kulcke, A.; Laush, C.; Lisy, J. M.
J. Chem. Phys. 1995, 103, 5366.
(16) Vernon, M. F.; Krajnovich, D. J.; Kwok, H. S.; Lisy, J. M.; Shen, Y. R.; Lee, Y. T. J. Chem. Phys. 1982, 77, 47.
(17) (a) Lisy, J. M.; Tramer, A.; Vernon, M. F.; Lee, Y. T. J. Chem.
Phys. 1981, 75, 4733. (b) Klemperer, W. Ber. Bunsen-Ges. Phys. Chem.
1974, 78, 1281.
(18) Abu-Awwad, F. M. Chem. Phys. Lett. 2002, 360, 340.
(19) Ovchinnikov, M.; Apkarian, V. A. J. Chem. Phys. 1999, 110, 9842. (20) Quark, M.; Suhm, M. A. Calais, J. L. K., Ed.; Kluwer Academic: Dordrecht, 1997; pp 415-463.
(21) Guedes, R. C.; do Couto, P. C.; Costa Cabral, B. J. J. Chem. Phys.
2003, 118, 1272.
(22) Liedl, K. R.; Kroemer, R. T.; Rode, B. M. Chem. Phys. Lett. 1995,
246, 455.
(23) Klopper, W.; Quack, M.; Suhm, M. A. Chem. Phys. Lett. 1996,
261, 35.
(24) (a) Liu, S. Y.; Michael, D. W.; Dykstra, C. E.; Lisy, J. M. J. Chem.
Phys. 1986, 84, 5032. (b) Zhang, C.; Freeman, D. L.; Doll, J. D. J. Chem. Phys. 1989, 91, 2489.
(25) (a) Kathmann, Shawn M.; Schenter, Gregory K.; Garrett, Bruce C.
J. Chem. Phys. 1999, 111, 4688. (b) Schenter, Gregory K.; Kathmann,
Shawn M.; Garrett, Bruce C. Phys. ReV. Lett. 1999, 82, 3484. (c) Schenter, Gregory K.; Kathmann, Shawn M.; Garrett, Bruce C. J. Chem. Phys. 2002,
116, 4275. (d) Garrett, B. C.; Truhlar, D. G. J. Am. Chem. Soc. 1979, 101,
4534. (e) Garrett, B. C.; Truhlar, D. G. J. Am. Chem. Soc. 1979, 101, 5207. (26) Kathmann, S. M.; Schenter, G. K.; Garrett, B. C. J. Chem. Phys.
1999, 111, 4688.
(27) (a) Krems, R.; Nordholm, S. Z. Phys. Chem. 2000, 214, 1467. (b) Shen, D.; Pritchard, H. O. J. Chem. Soc., Faraday Trans. 1996, 92, 1297. (c) Hobza, P.; Havlas, Z. Chem. ReV. 2000, 100, 4253.
(28) (a) Tou, J. C.; Lin, S. H. J. Chem. Phys. 1968, 49, 4187. (b) Lin, S. H.; Eyring, H. J. Chem. Phys. 1963, 39, 1577. (c) Lin, S. H.; Eyring, H.
J. Chem. Phys. 1964, 43, 2153.
(29) (a) McDowell, S. A. C. J. Mol. Struct. (THEOCHEM) 2006, 770, 119. (b) Bhuiyan, L. B.; Hase, W. L. J. Chem. Phys. 1983, 78, 5052.
(30) Peslherbe, G. H.; Hase, W. L. J. Chem. Phys. 1996, 105, 7432. (31) (a) Hase, W. L. Acc. Chem. Res. 1998, 31, 659. (b) Song, K.; Hase, W. J. Chem. Phys. 1999, 110, 6198.
(32) (a) Troe, J. Chem. Phys. 1995, 190, 381. (b) Troe, J. J. Phys. Chem.
1979, 83, 114. (c) Troe, J. J. Chem. Phys. 1983, 79, 6017. (d) Romanini,
D.; Lehmann, K. K. J. Chem. Phys. 1993, 98, 6437.
(33) (a) Yao, L.; Mebel, A. M.; Lu, H. F.; Neusser, H. J.; Lin, S. H. J.
Phys. Chem. A 2007, 111, 6722. (b) Yao, L.; Lin, S. H. Mod. Phys. Lett. B 2008, 22, 3043. (c) Yao, L.; Lin, S. H. Sci. China, Ser. B 2008, 51,
1146. (d) Yao, L.; He, R. X.; Mebel, A. M.; Lin, S. H. Chem. Phys. Lett.
2009, 470, 210.
(34) Dayton, D. C.; Jucks, K. W.; Miller, R. E. J. Chem. Phys. 1989,
90, 2631.
(35) Grigorenko, B. L.; Nemukhin, A. V.; Apkarian, V. A. J. Chem.
Phys. 1998, 108, 4413.
(36) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian03
(E.01); Gaussian, Inc.: Pittsburgh, PA, 2004.
(37) Chang, H. C.; Jiang, J. C.; Tsai, W. C.; Chen, G. C.; Lin, S. H. J.
Phys. Chem. B 2006, 110, 3302.
(38) (a) Tsoo, C.; Brooks, C. L. J. Chem. Phys. 1994, 101, 6405. (b) Pine, A. S.; Howard, B. J. J. Chem. Phys. 1986, 84, 590.
(39) Ovchinnikov, M.; Apkarian, V. A. J. Chem. Phys. 1999, 110, 9842. (40) Claes, L.; Francu¨ois, J. P.; Deleuze, M. S. J. Am. Chem. Soc. 2003,
125, 7129.