Articles
Hyperbranched Poly(aryl ether oxazole)s: Synthesis, Characterization,
and Modification
Zhi-Hao Gong, Chi-Meng Leu, Fang-Iy Wu, and Ching-Fong Shu* Department of Applied Chemistry, National Chiao Tung University, Hsin-Chu, Taiwan, 30035, R. O. C.
Received May 8, 2000; Revised Manuscript Received September 12, 2000
ABSTRACT: In this paper the synthesis and characterization of a hyperbranched poly(aryl ether oxazole) with terminal phenolic groups are described. An ABB′monomer containing a pair of phenolic groups and an aryl fluoride which is activated toward displacement by the attached oxazole ring was prepared. The nucleophilic substitution of the fluoride with the phenolic group leads to the formation of ether linkage and subsequently the hyperbranched poly(aryl ether oxazole) P1. As determined by a combination of model compound studies and 1H NMR integration experiments, the degree of branching of P1 is approximately 50%. The polymer P1 is thermally stable and readily soluble in polar organic solvents. The terminal phenolic groups in P1 were easily functionalized, yielding hyperbranched polymers with a variety of functional chain ends. Physical properties, such as the glass transition temperature and the solubility of the hyperbranched poly(aryl ether oxazole)s, depended significantly on the nature of the chain ends.
Introduction
Recently, hyperbranched macromolecules have re-ceived considerable attention since their unique highly branched structure is expected to exhibit some unusual properties.1-4Such polymers are conveniently synthe-sized in a single step via random one-pot polymerization of ABn-type monomers, yet they maintain many of the
architectural features and properties found in their more perfectly defined dendrimer counterparts,5which are built up by step-by-step sequences requiring isola-tion and purificaisola-tion in each step.6,7
Poly(aryl ether)s are a class of thermoplastics pos-sessing high thermal stability and good mechanical properties.8 It has been shown that aromatic nucleo-philic substitution reactions between activated aryl halide monomers and bisphenonates can lead to the formation of linear poly(aryl ether)s.9 Electron-with-drawing groups like ketone, sulfone, and some hetero-cycles, which may stabilize the anionic intermediate, were utilized as activating groups to facilitate nucleo-philic aromatic substitution in the synthesis of poly(aryl ether)s.9-12These synthetic strategies have been em-ployed for the preparation of hyperbranched poly(aryl ether)s in one-step polymerization from AB2monomers containing a phenolic group and two aryl fluorides which were activated toward nucleophilic displacement by either a sulfone or carbonyl group.13Recently, Hedrick et al. have extended this synthetic strategy to heterocycle-activated systems in the synthesis of hyperbranched poly(aryl ether quinoxaline)s.14
This report concerns the synthesis of a hyperbranched poly(aryl ether oxazole) by the one-step polymerization of an ABB′monomer which contains an aryl fluoride and two phenolic groups attached at C-2, C-4, and C-5
positions of an oxazole ring, respectively. The nucleo-philic substitution of the fluoride with the pheonilic groups, activated by the oxazole moiety, leads to the formation of ether linkage and subsequently the poly-(aryl ether oxazole). Furthermore, the modification of phenolic chain ends of the resulting polymers by reac-tion with several end-capping agents is described. The effects of the nature of the chain-end groups on the solubility and glass transition temperature of the hy-perbranched polymers have also been investigated.
Experimental Section
General Directions. THF was distilled from a sodium diphenyl ketyl solution just prior to use. Benzene was distilled over sodium. Anhydrous K2CO3was ground into fine powder and dried at 120 °C under vacuum. Other starting materials and reagents were used as obtained from the suppliers. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Unity 300 MHz or a Bruker-DRX 300 MHz spec-trometer. Differential scanning calorimetry (DSC) was per-formed on a SEIKO SSC 5200 DSC using a heating/cooling rate of 10 °C min-1. Samples were scanned from 30 to 300 °C and then cooled to 30 °C and scanned a second time from 30 to 300 °C. The glass transition temperature was determined from the second heating scan. Thermogravimetric analysis (TGA) was made on a SEIKO TG/DTA 200 instrument. The thermal stability of samples was determined in nitrogen by measuring weight loss while heating at a rate of 10 °C min-1. Size exclusion chromatography (SEC) was carried out on a Waters chromatography connected to a Waters 410 differential refractometer. Three 5 µm Waters styragel columns (300× 7.8 mm) connected in series in the decreasing order of pore size (105, 104, and 103Å) were used with DMF/0.05 M LiBr as eluent, and poly(methyl methacrylate) standard samples were used for calibration. Mass spectra were obtained on a JEOL JMS-SX/SX 102A mass spectrometer. Analytical TLC was performed on commercial Merck plated coated with silica gel 10.1021/ma000789y CCC: $19.00 © 2000 American Chemical Society
GF254. Silica gel for column chromatography was Merck Kieselgel 60 (70-230 mesh).
1,2-Di(4-methoxyphenyl)-2-oxyethyl-4-fluorobenz-oate (2). A mixture containing 4,4′-dimethoxybenzil 1 (12.0 g, 44.0 mmol), 4-fluorobenzoic acid (6.17 g, 44.0 mmol), 4-(dimethylamino)pyridine (0.22 g), and dicyclohexylcarbodi-imide (10.0 g) in dichloromethane (160 mL) was stirred at 25 °C under nitrogen for 15 h. The reaction mixture was filtered. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by column chromatography (hexane/EtOAc 3:1) to give 2 (11.71 g, 67.4%). 1H NMR (acetone-d6): δ 3.80 (s, 3 H), 3.88 (s, 3 H), 7.00 (d, 2 H, J ) 9.0 Hz), 7.01 (d, 2 H, J ) 9.0 Hz), 7.17 (s, 1H), 7.30 (dd, 2 H, J ) 8.7, 8.7 Hz), 7.60 (d, 2 H, J ) 9.0 Hz), 8.08 (d, 2 H, J ) 9.0 Hz), 8.13 (dd, 2H, J ) 9.0, 5.7 Hz).13C NMR (acetone-d 6): δ 192.5, 166.6 (d, JC-F) 253 Hz), 165.3, 164.7, 161.3, 133.2 (d, JC-F) 9 Hz), 131.8, 131.1, 128.2, 127.2 (d, JC-F) 2 Hz), 126.9, 116.4 (d, JC-F) 22 Hz), 115.2, 114.7, 78.3, 55.9, 55.5. MS (m/ z): 394.1215. Calcd. 394.1216 for C23H19O5F. 2-(4-Fluorophenyl)-4,5-di(4-methoxyphenyl)-1,3-oxa-zole (3). A mixture of 2 (2.50 g, 6.34 mmol) and ammonium acetate (2.95 g, 38.3 mmol) in AcOH (27 mL) was heated at reflux under nitrogen for 8 h. The resulting mixture was poured into water (100 mL) and extracted with EtOAc (3× 50 mL). The combined extracts were dried over MgSO4, and the solvent was removed in vacuo. The crude product was purified by recrystallization from hexane/EtOAc to give 3 (1.44 g, 60.5%).1H NMR (acetone-d 6): δ 3.80 (s, 3 H), 3.88 (s, 3 H), 7.00 (d, 2 H, J ) 9.0 Hz), 7.01 (d, 2 H, J ) 9.0 Hz), 7.17 (s, 1H), 7.30 (dd, 2 H, J ) 8.7, 8.7 Hz), 7.60 (d, 2 H, J ) 9.0 Hz), 8.08 (d, 2 H, J ) 9.0 Hz), 8.13 (dd, 2H, J ) 9.0, 5.7 Hz).13C NMR (acetone-d6): δ 192.5, 166.6 (d, JC-F) 253 Hz), 165.3, 164.7, 161.3, 133.2(d, JC-F) 9 Hz), 131.8, 131.1, 128.2, 127.2 (d, JC-F) 2 Hz), 126.9, 116.4(d, JC-F) 22 Hz), 115.2, 114.7, 78.3, 55.9, 55.5. MS (m/z): 394.1215. Calcd. 394.1216 for C23H19O5F. 2-(4-Fluorophenyl)-4,5-di(4-hydroxyphenyl)-1,3-oxa-zole (4). A suspension of 3 (4.00 g, 10.7 mmol) in AcOH (40 mL) and 48% aqueous HBr (125 mL) was heated at reflux for 48 h. The reaction mixture was cooled, and the precipitate was collected by filtration. The solid was then dissolved in 1.0 N NaOH solution, and the pH was adjusted to neutral using concentrated HCl. The resulting precipitate was filtered, washed with water, and dried to give 4 (3.54 g, 95,6%).1H NMR (DMSO-d6): δ 6.80 (d, 2 H, J ) 8.7 Hz), 6.83 (d, 2 H, J ) 8.4 Hz), 7.38 (dd, 2 H, J ) 8.7, 8.7 Hz), 7.44 (d, 2 H, J ) 8.7 Hz), 7.46 (d, 2 H, J ) 8.4 Hz), 8.09 (dd, 2 H, J ) 8.7, 5.4 Hz), 9.67 (s, 1H), 9.88 (s, 1H).13C NMR (DMSO-d 6): δ 163.4 (d, JC-F) 248 Hz), 158.1, 157.7, 157.5, 144.8, 134.7, 129.0, 128.3 (d, JC-F) 9 Hz), 128.1, 123.7 (d, JC-F) 2 Hz), 123.0, 119.5, 116.2 (d, JC-F ) 22 Hz), 115.8, 115.6. MS (m/z): 347.0963. Calcd. 347.0957 for C21H14O3NF. 2-{4-[3,5-Di(tert-butyl)phenoxy]phenyl} -4,5-di(4-meth-oxyphenyl)-1,3-oxazole (5). A mixture of 3 (0.20 g, 0.53 mmol), 3.5-di-tert-butylphenol (216 mg, 1.06 mmol), anhydrous K2CO3(150 mg, 1.07 mmol), benzene (4 mL), and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU) (0.8 mL) was heated at 110 °C. The water formed during the reaction was removed through an azeotropic distillation and collected in a Dean-Stark trap. After 4 h, the benzene was collected and removed from the system. The reaction mixture was then heated at 180 °C for 3 h. The resulting mixture was cooled, poured into water (10 mL), and extracted with EtOAc (3× 20 mL). The combined extracts were dried over Na2SO4, and the solvent was removed in vacuo. The product was purified by column chromatography (hexane/EtOAc 3:2) to give 5 (0,28 g, 93,4%).1H NMR (CDCl 3): δ 1.29 (s, 18 H), 3.83 (s, 6 H), 6.85-6.97 (m, 6 H), 7.05 (d, 2 H, J ) 9 Hz), 7.21 (t, 1 H, J ) 1.5 Hz), 7.57 (d, 2 H, J ) 9.3 Hz), 7.62 (d, 2 H, J ) 8.7 Hz), 8.07 (d, 2 H, J ) 9 Hz).13C NMR (CDCl 3): δ 159.7, 159.6, 159.4, 155.5, 152.9, 144.6, 135.2, 129.2, 128.0, 127.9, 125.1, 121.9, 121.8, 118.0, 117.9, 114.1, 113.99, 113.96, 113.8, 55.22, 55.19, 35.0, 31.3. MS (m/z): 561.2885. Calcd. 561.2879 for C37H39O4N. 2-{4-[3,5-Di(tert-butyl)phenoxy]phenyl} -4,5-di(4-hydroxy-phenyl)-1,3-oxazole (6). A suspension of 5 (1.20 g) in AcOH
(8 mL) and 48% aqueous HBr (25 mL) was heated at reflux for 48 h. The reaction mixture was cooled, and the precipitate was collected by filtration. The solid was then dissolved in 1.0 N NaOH solution and filtered. The filtrate was acidified with 1 N HCl, and the pH was adjusted to 7-8 using NaHCO3. The resulting precipitate was filtered, washed with water, and dried to give 6 (0.95 g, 83%).1H NMR (DMSO-d
6): δ 1.27 (s, 18 H), 6.80 (d, 2 H, J ) 9.0 Hz), 6.83 (d, 2 H, J ) 9.0 Hz), 6.92 (s, 2 H), 7.09 (d, 2 H, J ) 9.0 Hz), 7.25 (s, 1 H), 7.44 (d, 2 H, J ) 9.0 Hz), 7.45 (d, 2 H, J ) 9.0 Hz), 8.03 (d, 2 H, J ) 9.0 Hz), 9.63 (s, 1 H), 9.83 (s, 1 H).13C NMR (DMSO-d 6): δ 159.1, 158.1, 157.9, 157.3, 155.0, 152.6, 144.3, 134.5, 128.8, 128.0, 127.8, 122.9, 121.5, 119.4, 117.9, 117.8, 115.7, 115.4, 113.7, 34.6, 31.0. MS (m/z): 533.2560. Calcd. 533.2569 for C35H35O4N. Preparation of Hyperbranched Poly(aryl ether oxa-zole) (P1). A mixture of 4 (0.7 g, 2.01 mmol), anhydrous K2 -CO3(1.12 g, 8 mmol), benzene (5 mL), and DMPU (2.8 mL) was heated at 120 °C for 5 h. The water formed during the reaction was removed through an azeotropic distillation and was collected in a Dean-Stark trap. The reaction mixture was then heated to 180 °C while the benzene was removed through the Dean-Stark trap. After 10 h, the resulting mixture was poured into methanol (50 mL) and neutralized with 1 N HCl. The polymer was collected by filtration and purified by precipitating from DMF into methanol to give P1 (0.56 g, 85.2%). Anal. Calcd for (C21H13NO3)n: C, 77.06; H, 4.00; N, 4.28. Found: C, 77.47; H, 4.29; N, 4.19.
Preparation of Hyperbranched Poly(aryl ether oxa-zole) (P2). To a solution of P1 (0.30 g) and 4-(dimethylamino)-pyridine (0.17 g) in THF (7.0 mL) was added dropwise acetyl chloride (91 µL) under nitrogen. The reaction mixture was stirred at 25 °C for 48 h, later heated at reflux for 2 h, and then added to methanol. The collected polymer was purified by precipitating from CHCl3into methanol to give P2 (0.28 g, 82%). Anal. Calcd for (C23H15NO4)n: C, 74.79; H, 4.09; N, 3.79. Found: C, 74.24; H, 4.19; N, 3.37.
Preparation of Hyperbranched Poly(aryl ether oxa-zole) (P3). To a solution of P1 (0.20 g) and 4-(dimethylamino)-pyridine (113 mg) in THF (3.0 mL) was added dropwise hexanoic anhydride (190 µL) under nitrogen. The reaction mixture was stirred at 25 °C for 24 h and then added to methanol. The collected polymer was purified by precipitating from CHCl3into methanol to give P3 (0.21 g, 81%). Anal. Calcd for (C27H23NO4)n: C, 76.22; H, 5.54; N, 3.29. Found: C, 76.51; H, 5.96; N, 3.51.
Preparation of Hyperbranched Poly(aryl ether oxa-zole) (P4). To a solution of P1 (0.20 g), PPh3(0.488 g), and ethanol (103 µL) in THF (15 mL) was added dropwise diiso-propyl azodicarboxylate (0.36 mL) under nitrogen. The reaction mixture was stirred at 25 °C for 24 h and then added to methanol. The collected polymer was purified by precipitating from CHCl3into methanol to give P4 (0.209 g, 96%). Anal. Calcd for (C23H17NO3)n: C, 77.73; H, 4.82; N, 3.94. Found: C, 77.15; H, 4.65; N, 4.40.
Preparation of Hyperbranched Poly(aryl ether oxa-zole) (P5). P5 was prepared from P1 and 1-hexanol using the same procedure as for P4 (0.215 g, 95%). Anal. Calcd for (C27H25NO3)n: C, 78.81; H, 6.12; N, 3.40. Found: C, 78.49; H, 5.90; N, 3.67.
Results and Discussion
Synthesis and Characterization of the ABB′ Monomer. The ABB′oxazole monomer 4 was synthe-sized by the cyclocondensation reaction, similar to the method described by Moylan and co-workers15(Scheme 1). The 4-(dimethylamino)pyridine (DMAP) catalyzed the dicyclohexylcarbodiimide (DCC) coupling of the substituted benzoin 1 with 4-fluorobenzoic acid and gave the benzoin benzoate 2. Ring closure of the benzoin benzoate 2 with ammonium acetate in AcOH yielded the 2,4,5-triphenyloxazole 3 which was subsequently demethylated with HBr to afford the ABB′monomer 4.
The monomer 4 contains an aryl fluoride and two phenolic groups.
All compounds were characterized by 1H NMR,13C NMR, and high-resolution mass spectroscopy. The 4-and 5-positions in the oxazole ring are not identical due to the nonsymmetrical nature of this heterocycle, and therefore the chemical shift of the two hydroxyphenyl groups in 4 are not equal. The 1H NMR (DMSO-d
6) resonances for the aromatic protons of the two hydrox-yphenyl groups overlap somewhat and appear at 7.46, 6.83 ppm and 7.44, 6.80 ppm, respectively, while resonances for the proton of the hydroxyl groups are well-resolved and observed at 9.88 and 9.67 ppm, respectively. Though the two phenolic groups are chemi-cally nonequivalent, their corresponding phenolate an-ions have similar reactivity undergoing nucleophilic attack under the reaction conditions.
NMR has been used as an indicator for the ability of potential monomers to undergo nucleophilic displace-ment of the fluorine atom.16 Both 13C and 19F NMR spectrocopy were used to probe the electron density at the actual site of nucleophilic reaction, i.e., the C-F bond of aryl fluorides.19F NMR chemical shifts were the most sensitive probe for the reactivity of nucleophilic substitution of aryl fluorides, with a span of 9 ppm between the most activated monomer, 4,4′ -difluorophen-yl sulfone (-104.28 ppm), and nonactivated fluoroben-zene (-112.77 ppm).16 The 19F NMR of the oxazole monomer 4 is shown in Figure 1 together with that of 4,4-difluorophenyl sulfone and fluorobenzene. The19F NMR chemical shift of 4 (-109.68 ppm) indicates a downfield shift similar to that of 4,4-difluorophenyl sulfone. The magnitude of the downfield shift is com-parable to other polymerizable fluoromonomers acti-vated by heterocyclic rings.16The19F NMR data suggest that the fluorophenyl group in monomer 4 is likely to undergo nucleophilic aromatic substitution.
Model Compound Synthesis. To demonstrate the feasibility of the oxazole activated aryl ether synthesis, the reaction of potassium 3,5-di-tert-butylphenoxide with 3 was investigated as the model reaction for the polymerization of monomer 4. As shown in Scheme 2, l equiv of 3 and l equiv of 3,5-di-tert-butylphenol were reacted in the presence of anhydrous K2CO3in a solvent mix of 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidi-none (DMPU) and benzene under the conditions used for polymerization of 4. DMPU was chosen as the solvent for polymerization because it has been shown to be an excellent solvent for polyether synthesis.17 Benzene was used to remove water generated by phe-noxide formation during the initial stage through the Dean-Stark trap. Upon completion of phenoxide forma-tion and dehydraforma-tion, benzene was removed from the system, and the reaction mixture was then heated to 180 °C to effect the displacement reaction. After 3 h, TLC analysis indicated the complete conversion of 3 to 5, which was in high yield (93%) after column chroma-tography. The model reaction reveals that the aryl fluoride at the 2-position of the oxazole ring is cleanly substituted by a phenoxide, and this transformation is suitable for a polymer formation reaction.
Synthesis and General Properties of Hyper-branched Poly(aryl ether oxazole) P1. The one-step polymerization of 4 was performed under similar condi-tions used in the model reaction to give the correspond-ing hyperbranched poly(aryl ether oxazole) P1. High molecular polymer was attained within 10 h as judged by a pronounced increase in viscosity. A schematic Scheme 1
Figure 1. 19F NMR spectra in DMSO-d
6of a mixture of (a)
4,4′-difluorophenyl sulfone, (b) monomer 4, and (c) fluoroben-zene.
Scheme 2
structure of P1 and the general reaction are shown in Scheme 3. The molecular weight of P1 was determined by SEC analysis in DMF solution calibrated against linear poly(methyl methacrylate) standards and used only for a rough estimate. Because of the highly branched nature of hyperbranched macromolecules, SEC measurements tend to underestimate the true molecular weight.18Figure 2 shows the progression of molecular weight with reaction time for P1. There is an increasing gap in the growth of Mnand Mwthat leads to broad molecular weight distributions at higher con-versions. This observation resembles previous reports of other hyperbranched polymers and is consistent with Flory’s predictions on molecular distribution behavior for highly branched system.19 The glass transition temperature (Tg) of the hyperbranched poly(aryl ether oxazole) was determined by DSC. Tgof P1 was observed to increase modestly with molecular weight, ranging from 213 to 247 °C with Mw changing from 5600 to 76 500. Thermogravimetric analysis (TGA) was used to measure the thermal stability of P1, and because of its high aromaticity, P1 had high thermal stability. For a sample with Mw) 76 500 and Mw/Mn) 3.1, P1 lost 5 wt % at 392 °C and an additional 5 wt % at 458 °C.
Degree of Branching. Hyperbranched polymer P1 was formed by a sequence of condensation of ABB′ monomer, resulting in an irregular dendritic structure. The degree of branching (DB) is an important charac-teristic often used to reveal the structure of hyper-branched polymers. A combination technique of model compound studies and NMR spectroscopy has been used to quantify the different subunits appearing in the hyperbranched polymer and subsequently determine its DB.20Scheme 4 shows the model reaction performed to determine the DB of P1. Samples of 1 equiv of com-pound 6 and 1 equiv of comcom-pound 3 were reacted under the same conditions used for polymerization of monomer 4. After the reaction, TLC analysis indicated that complete conversion of 3 occurred. The model com-pounds were separated from the reaction mixture by preparative TLC, eluting with CH2Cl2/ethyl acetate 20: 1. Figure 3 shows the 1H NMR spectra of 6, which resembles the terminal unit, monoarylated products 7-4 and 7-5, which resemble the linear units, and diarylated product 8, which resembles the dendritic unit. The peak assignments are based on the peak positions of com-pounds 3 and 6 as well as the auxiliary of 2D (H,H)-COSY spectra. Because of the nonsymmetrical nature
of the oxazole ring, there were two different monoary-lated products, 7-4 and 7-5. Individual resonances associated with the aromatic protons of 7-4and 7-5are indistinguishable, whereas resonances due to the pro-tons of the phenolic groups of 7-4 and 7-5 occur at significantly different positions, 9.92 and 9.72 ppm, respectively. It was reported that in the oxazole ring the charge density at C-5 is more negative than that at C-4.21On the basis of the inductive effect, we tentatively assume that the resonance for the proton of the hydroxyl group of 7-5might appear at upfield 9.72 ppm and that of 7-4might occur downfield 9.92 ppm. The resonances of two hydroxyl protons of compound 6 are well-resolved and appear at 9.84 and 9.63 ppm. Figure 3 also includes 1H NMR spectra of the reaction mixture obtained directly from the model reaction without further separa-tion and the hyperbranched poly(aryl ether oxazole) P1. For the reaction mixture, the relative integration of the peaks for the signal at 9.92, 9.84, 9.72, and 9.63 ppm are 102, 100, 95, and 102 respectively, which indicates the abundance of compounds 6, 7-4, and 7-5 in the reaction mixture. The fact that almost the same quan-tity of the two monoarylated products 7-4and 7-5were produced in the reaction mixture reveals that the two phenoxide groups attached to the C-5 and C-2 positions of the oxazole ring of compound 6 have almost the same reactivity for undergoing nucleophilic attack under the reaction conditions. Additional comparison of the inte-gration values for the peaks in the regions of 8.4-8.0 and 7.8-7.6 ppm allows the ratio of compounds 6 and 8 in the reaction mix to be obtained, which is 1:1. On the basis of these data, the percentage of compounds 6, 7-4, 7-5, and 8 in the reaction mixture was calculated as approximately 25% for each compound. Good cor-relation is observed in the comparison of the1H NMR spectra of these model compounds with that of the hyperbranched poly(aryl ether oxazole) P1. The reso-nances at 9.92 and 9.71 ppm are attributed to the phenol protons of the two different linear subunits, whereas the resonances at 9.84 and 9.64 ppm are due to the two phenol protons of the terminal subunit. Integration of these resonances allows the relative percentage of each subunit to be determined. The total percentage esti-mated for the two linear subunits is approximately twice the percentage of the terminal subunit. According to the theoretical prediction, the number of terminal units is equal to the number of dendritic units for an AB2-type hyperbranched polymer possessing high molecular Figure 2. Molecular weight as determined by SEC analysis
vs reaction time plot for the polymerization reaction of monomer 4 at 180 °C.
weight.19,22The DB is given by22
where D, L, and T represent the fractions of dendritic, linear, and terminal units, respectively. On the basis of this formula, the calculated DB of the hyperbranched poly(aryl ether oxazole) is approximately 50%, which is the statistical value expected for a random AB2 poly-condensation.22This value is also in agreement with the ratio of the abundance of the model compounds obtained from the model reaction, which indicates that the reactivity of the nucleophilic substitution for ether linkage formation in the polymerization reaction is independent of molecular size.
Chemical Modification of Hyperbranched Poly-(aryl ether oxazole) P1. Unlike linear polyPoly-(aryl ether oxazole)s,12a,dthe hyperbranched polymer P1 is char-acterized by a large number of chain end groups. The terminal phenolic groups in P1 could be easily func-tionalized to yield hyperbranched polymers with a variety of functional chain ends. The nature of the end groups influences the physical and chemical properties of the hyperbranched polymers. As shown in Scheme 5, different functional groups could be introduced into P1 by reactions of the phenolic end groups. For the sake of simplicity, Scheme 5 only shows the overall compo-sitional repeat units, even though each polymer contains a combination of linear units, which could be in the 4-or 5-positions of the oxazole ring, dendritic units, and
terminal units. The phenolic groups of P1 were acylated with acid chloride or acid anhydride to give correspond-ing ester derivatives P2 and P3, respectively. Via the Mitsunobu reactions,23the phenolic groups of P1 were converted to ether groups to yield the ether derivatives P4 and P5. Successful modification of end groups of P1 could be confirmed by the 1H NMR spectra of the derivatives, which contain alkyl chain ends that exhibit 1H NMR peaks well separated from the peaks associated with aromatic units. Figure 4 shows the 1H NMR Figure 3. 1H NMR spectra in DMSO-d
6of model compounds (a) 8, (b) 7-4, (c) 7-5, (d) 6, and (e) the reaction mixture obtained
directly from the model reaction without further separation, compared with (f) the hyperbranched poly(aryl ether oxazole) P1.
DB ) D + T
D + L + T=
2T 2T + L
spectra of P2 and P4. The conversion of end-capping reaction was calculated by comparing the integration ratio of the protons attributed to the alkyl end groups versus those from the aromatic units. For all the modification reactions above, the use of excess reagents resulted in almost complete (95-100%) functionaliza-tion, indicating that the hydroxyl groups at the chain ends are readily accessible to reagents in solution.
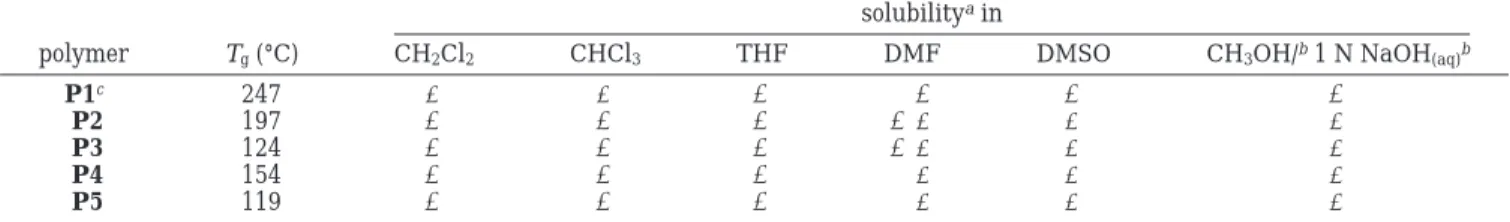
The glass transition temperature (Tg) and the solubil-ity of polymers P1-P5 are summarized in Table 1. The Tgvalues determined by differential scanning calorim-etry (DSC) of these polymers are very dependent on the nature of chain ends, which increases with increasing chain-end polarities. The Tg of P1, which has polar hydroxyl terminal groups, is 274 °C. The Tg values of P2 and P4, which have less polar terminal groups such as ester and ether groups, are 197 and 154 °C, respec-tively. A further decrease in Tg to 124 and 119 °C is observed for P3 and P5, respectively, due to increasing length of the alkoxyl chain of the terminal ester or ether groups.
The different chain ends also lead to differences in solubility. The phenolic-terminated polymer P1 is soluble in a solvent mixture consisting of NaOH(aq)/CH3OH and in polar solvents such as DMSO and DMF, whereas the ester-terminated polymers P2 and P3 are only partially soluble in DMF and insoluble in DMSO, and the ether-terminated polymers P4 and P5 are totally insoluble in both polar solvents. Conversely, in relatively nonpolar solvents such as CH2Cl2 and CHCl3 polymers P2-P5 are extremely soluble, whereas polymer P1 is insoluble.
Summary
A hyperbranched poly(aryl ether oxazole) with ter-minal phenolic groups was prepared by the one-step polymerization of an ABB′monomer containing an aryl fluoride and two phenolic groups. Though the two
phenolic groups are nonequivalent, the reactivity of their phenolates undergoing nucleophilic attack is simi-lar under the applied reaction conditions. The nucleo-philic substitution of the fluoride with the phenolate groups, which is activated by the oxazole moiety, resulted in the formation of ether linkage and subse-quently the hyperbranched poly(aryl ether oxazole) P1. The degree of branching of P1 as characterized by1H NMR spectroscopy is approximately 50%. The phenolic groups at the chain ends were readily accessible to reagents in solution and were converted to a variety of functional groups. The nature of the end groups could highly affect the physical properties of the hyper-branched macromolecule.
Acknowledgment. We thank the National Science Council (ROC) (NSC 89-2113-M009-001) for financial support.
References and Notes
(1) Literature for hyperbranched polymers published prior to 1998 see reviews: (a) Malmstro¨m, E.; Hult, A. J. Macromol.
Sci., Rev. Macromol. Chem. Phys. 1997, C37, 555. (b) Kim,
Y. H. J. Polym. Sci., Part A: Polym. Chem. 1998, 36, 1685 and references cited in the reviews.
(2) (a) Tao, X. T.; Zhang, Y.-D.; Wada, T.; Sasabe, H.; Susuki, H.; Watanabe, T.; Miyata, S. Adv. Mater. 1998, 10, 226. (b) Mueller, A.; Kowalewski, T.; Wooley, K. L. Macromolecules
1998, 31, 776. (c) Spetseris, N.; Ward, R. E.; Meye, T. Y. Macromolecules 1998, 31, 3158. (d) Miravet, J. F.; Fre´chet,
J. M. J. Macromolecules 1998, 31, 3461. (e) Trollsås, M.; Hedrick, J. L. Macromolecules 1998, 31, 4390.
(3) (a) Huber, T.; Voit, B.; Wolf, D. Macromol. Symp. 1999, 142, 133. (b) Yamakawa, Y.; Ueda, M.; Takeuchi, K.; Asai, M. J.
Polym. Sci., Part A: Polym. Chem. 1999, 37, 3638. (c) Shu,
C.-F.; Leu, C.-M. Macromolecules 1999, 32, 100. (d) Krichel-dorf, H. R.; Bolender, O.; Wollheim, T. Macromolecules 1999,
32, 3878. (e) Sunder, A.; Hanselmann, R.; Frey, H.; Mu¨ lhaupt, R. Macromolecules 1999, 32, 4240. (f) Thompson, D. S.; Markoski, L. J.; Moore, J. S. Macromolecules 1999, 32, 4764. (4) (a) Jayakannan, M.; Ramakrishnan, S. J. Polym. Sci., Part
A: Polym. Chem. 2000, 38, 261. (b) Yamanaka, K.; Jikei, M.;
Kakimoto, M. Macromolecules 2000, 33, 1111. (c) Paulasaari, J. K.; Weber, W. P. Macromolecules 2000, 33, 2005. (d) Magnusson, H.; Malmstro¨m, E.; Hult, A. Macromolecules
2000, 33, 3099. (e) Murtuza, S.; Harkins, S. B.; Long, G. S.;
Sen, A. J. Am. Chem. Soc. 2000, 122, 1867.
(5) (a) Fre´chet, J. M. J. Science 1994, 263, 1710. (b) Newkome, G. R., Ed. Advances in Dendritic Molecules; JAI Press: Greenwich, CT, 1994; Vol. 1. (c) Newkome, G. R.; Moorefield, C. N.; Vo¨gtle, F. Dendritic Molecules: Concepts, Syntheses,
Perspectives; VCH: Weinheim, FRG, 1996. (d) Zeng, F.;
Zimmerman, S. C. Chem. Rev. 1997, 97, 1681. (e) Frey, H.; Lach, C.; Lorenz, K. Adv. Mater. 1998, 10, 279. (f) Smith, D. K.; Diederich, F. Chem. Eur. J. 1998, 4, 1353.
(6) (a) Tomalia, D. A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. Polym. J. 1985,
17, 117. (b) Newkome, G. R.; Yao, Z.-Q.; Baker, G. R.; Gupta,
V. K. J. Org. Chem. 1985, 50, 2003.
(7) (a) Hawker, C. J.; Fre´chet, J. M. J. J. Am. Chem. Soc. 1990,
112, 7638. (b) Xu, Z. F.; Moore, J. S. Angew. Chem., Int. Ed. Engl. 1993, 32, 1354. (c) Miller, T. M.; Neenan, T. X. Chem. Mater. 1990, 2, 346.
Table 1. Effect of the Functionality of the Chain Ends on the Thermal and Solution Properties of the Hyperbranched Poly(aryl ether oxazole)s
solubilityain
polymer Tg(°C) CH2Cl2 CHCl3 THF DMF DMSO CH3OH/b1 N NaOH(aq)b
P1c 247 - - + + + +
P2 197 + + + + - -
-P3 124 + + + + - -
-P4 154 + + + - -
-P5 119 + + + - -
-aSolubility: +, soluble; + -, partially soluble; -, insoluble.bVolume ratio 1:1.cM
w) 76 500.
Figure 4. 1H NMR spectra in CDCl
3of the hyperbranched
poly(aryl ether oxazole)s P2 and P4. Asterisk: signal due to CDCl3.
(8) Rose, J. B. In High Performance Polymers: Their Origin and
Development; Seymour, R. B., Kirshenbaum, G. S., Eds.;
Elservier: New York, 1986; p 197.
(9) Labadie, J. W.; Hedrick, J. L.; Ueda, M. In Step-Growth
Polymers for High-Performance Materials; Hedrick, J. L.,
Labadie, J. W., Eds.; ACS Symposium Series 624; American Chemical Society: Washington, DC, 1996; Chapter 12. (10) (a) Attwood, Y. E.; Dawson, P. C.; Freeman, J. L.; Hoy, L. R.;
Rose, J. B.; Staniland, P. A. Polymer 1981, 22, 1096. (b) Singh, R.; Hay, A. S. Macromolecules 1992, 25, 1017. (c) Hay, A. S. Adv. Polym. Sci. 1967, 4, 496.
(11) (a) Hedrick, J. L.; Labadie, J. W. Macromolecules 1990, 23, 1561. (b) Hilborn, J. G.; Labadie, J. W.; Hedrick, J. L.
Macromolecules 1990, 23, 2854. (c) Hedrick, J. L.; Twieg, R. Macromolecules 1992, 25, 2021. (d) Carter, K. R.; Miller, R.
D.; Hedrick, J. L. Macromolecules 1993, 26, 2209. (e) Strukelj, M.; Hedrick, J. C. Macromolecules 1994, 27, 7511.
(12) (a) Maier, G.; Hecht, R.; Nuyken, O.; Burger, K.; Helmreich, B. Macromolecules 1993, 26, 2583. (b) Schneider, J. M.; Maier, G.; Nuyken, O. Macromol. Rep. 1994, A31, 179. (c) Maier, G.; Hecht, R. Macromolecules 1995, 28, 7558. (d) Maier, G.; Schneider, J. M. Macromolecules 1998, 31, 1798. (13) (a) Miller, T. M.; Neenan, T. X.; Kwock, E. W.; Stein, S. M.J.
Am. Chem. Soc. 1993, 115, 356. (b) Chu, F.; Hawker, C. J. Polym. Bull. 1993, 30, 265. (c) Hawker, C. J.; Chu, F. Macromolecules 1996, 29, 4370. (d) Morikawa, A. Macromol-ecules 1998, 31, 5999.
(14) (a) Srinivasan, S.; Twieg, R.; Hedrick, J. L.; Hawker, C. J.
Macromolecules 1996, 29, 8543. (b) Hedrick, J. L.; Hawker,
C. J.; Miller, R. D.; Twieg, R.; Srinivasan, S. A.; Trollsås, M.
Macromolecules 1997, 30, 7607.
(15) Moylan, C. R.; Miller, R. D.; Twieg, R. J.; Betterton, K. M.; Lee, V. Y.; Matray, T. J.; Nguyen, C. Chem. Mater. 1993, 5, 1499.
(16) (a) Carter, K. R. Macromolecules 1995, 28, 6462. (b) Carter, K. R. In Step-Growth Polymers for High-Performance
Materi-als; Hedrick, J. L., Labadie, J. W., Eds.; ACS Symposium
Series 624; American Chemical Society: Washington, DC, 1996; Chapter 17.
(17) Labadie, J. W.; Carter, K. R.; Hedrick, J. L.; Jonsson, H.; Kim, S. Y.; Twieg, R. J. Polym. Bull. 1993, 30, 25.
(18) Hawker, C. J.; Fre´chet, J. M. J. J. Chem. Soc., Chem.
Commun. 1990, 1010.
(19) Flory, P. J. J. Am. Chem. Soc. 1952, 74, 2718.
(20) Hawker, C. J.; Lee, R.; Fre´chet, J. M. J. J. Am. Chem. Soc.
1991, 113, 4583.
(21) van Es, T.; Backeberg, O. G. J. Chem. Soc. 1963, 1363. (22) Ho¨lter, D.; Burgath, A.; Frey, H. Acta Polym. 1997, 48, 30. (23) Mitsunobu, O. Synthesis 1981, 1.