細胞激素Interleukin-1受體抗頡質(1L-1 RA)基因多型與嚴重風濕性心臟病之關連; Association of lnterleukin-1 receptor antagonist (IL-1 RA)gene polymorphism with severe rheumatic heart disease
66
0
0
全文
(2) 結果: 在檢測的基因序列多形中, IL-1 RA intron 2 多形在病人與正常人的分布不 同達到統計上意義,其中正常人其因型 (genotype) 分布為 I/I: 96(93.2%),I/II: 6(5.8%), II/II: 1 (0.9%),而病人中 I/I: 68 (81.0%) ,I/II: 16 (19.0%),II/II: 0, p=0.014,對偶質(allele)頻率在正常人 I: 96.1%,II: 3.9%,而病人 I: 90.5%, II: 9.5%, p=0.023,故基因型 I/II 及對偶質 II 在病人中所佔比例顯著增加。 TGF-ß1 促進質(-509) 基因型 C/C 在病人中 (3.6%) 較常人 (23.3%)顯著減少 (p<0.001),但對偶質 T 及 C 在兩組人員分布並無差異 (p>0.1)。而 IL-1ß 促進 質 (-511),IL-1ß exon 5,IL-4 intron 3 及 ACE intron 16 之基因型及對偶質在兩 組人員分布均無差異 結論: 在一群選擇後之重度二尖瓣狹窄病患,IL-1RA 基因多型中對偶質 II (基因型 I/II) 比例較正常人有顯著升高,此一結果與其他文獻報告在發炎性 疾病中發現吻合,顯示 IL-1RA II 可能可以作為風濕性心臟病患嚴重二尖瓣狹 窄之標記基因,並待進一步更多病患的研究以證實,而 TGF-ß1 促進質(-509) 基因型 C/C 病人中減少,亦待證實及闡釋。. 2.
(3) ABSTRACT. Background: Rheumatic heart disease, as the major sequela of acute rheumatic fever, remains an significant problem in public health and clinical medicine in many developing countries. Balloon dilatation or surgical replacement of the diseased valves is the only effective treatment when the cardiac valves are severely scarred and deformed. Antigenic mimicry of the streptococcal and cardiac glycoproteins has been proposed to initiate an autoimmune reaction culminating in destruction the cardiac valves. However, little is known concerning the genetics in host susceptibility. A group of polymorphic genes, including the interleukin(IL)-1ß promoter(-511), IL-1 exon 5, IL-1 receptor antagonist(RA) intron 2, IL-4 intron 3, TGF-ß promoter(-509), and ACE intron 16, invovled in inflammatory reactions, fibrosis and cardiac diseases were selected and compared between patients with severe mitral stenosis (MS) and normal controls.. Methods: Six polymorphic genes, namely IL-1ß promoter -511 single neucleotide polymorphism (SNP), IL-1ß exon 5 SNP, IL-1 receptor antagonist (RA) intron 2 variable number of tandem repeats (VNTR), IL-4 intron 3 VNTR, TGF-ß1 promoter -509 SNP, and ACE intron 16 insertion/deletion were analyzed using polymerase chain reaction (PCR), restriction fragment analysis, and gel electrophoresis. The distribution of genotypes and alleles were determined and. 3.
(4) compared in a group of 84 patients with severe MS requiring percutaneous balloon mitral valvuloplasty and 103 normal controls.. Results: Among the 6 DNA sequences examined, the distribution of the polymorphism in IL-1RA intron 2 was significantly different between patients and controls. The genotypes in normal controls were I/I: 96(93.2%), I/II: 6 (5.8%), and II/II: 1(0.9%); whereas in patients the distribution was I/I: 68(81.0%), and I/II: 16 (19.0%), p=0.014. The allele frequencies of controls were I:96.1% and II:3.9% and those of patients were I:90.5% and II:9.5%, p=0.023. The genotype I/II and the allele IL-1RA II were significantly increased in patients. The incidence of TGF-ß1 promoter(-509) genotype C/C is significantly decreased in MS patients (3.6%)compared to controls(23.3%; p<0.001). But the frequencies of allele T and C showed a similar distribution in patients and controls.(p>0.1) The genotypes and alleles frequencies of IL-1ß promoter(-511), IL-1ß exon 5, IL-4 intron 3, and ACE intron 16 were not significantly different between patients and controls.. Conclusion: The finding of increased incidences of genotype I/II and allele frequency IL-1RA II in patients with severe MS was consistent with other reports on many diseases associated with chronic inflammation. IL-1RA II may be used as a genetic marker for patients of rheumatic heart disease with severe MS if the result can be confirmed by studies with large samples. The validity and. 4.
(5) significance of decreased incidence of TGF-ß1 promoter(-509) genotype C/C awaits further elucidation as well.. 5.
(6) INTRODUCTION. Rheumatic fever (RF) remains a very significant public health problem in many developing countries. It is characterized by damage to the collagen fibrils and the ground substance of connective tissue by an inflammatory reaction involving multiple organs, primarily the heart, the joints, and the central nervous system. The major importance of RF is its ability to cause scarring and dysfunction of the cardiac valves, leading to a crippling hemodynamics in the chronic stage. The symptoms of acute RF usually become manifest 3 weeks after a group A streptococcal (GAS) tonsillopharyngitis. However, there is no specific laboratory test that can establish a diagnosis of RF. To fulfill the Jones criteria,[1] either two major criteria, or one major criterion and two minor criteria, plus evidence of an antecedent streptococcal infection are required. Carditis is often regarded as the most specific manifestation, present in almost 80% of patients with acute RF.[2] It is almost always associated with a murmur of valvulitis, and mitral regurgitation is the hallmark. The Aschoff nodule in the proliferative stage is considered pathognomonic of rheumatic carditis.. The major factors that are related to the risk of RF are the magnitude of the immune response to the antecedent streptococcal pharyngitis, which is related to the virulence (“rheumatogenecity”) of the infecting strain, and persistence of the. 6.
(7) organism during convalescence. M proteins of rheumatogenic streptococci contain epitopes shared with human cardiac sarcolemmal membrane proteins and myosin.[3][4] Autoimmunity induced by antigenic mimicry between the streptococcal. glycoprotein and human cardiac myosin and laminin may be. responsible for the pathogenesis of rheumatic carditis[5]. The incidence of RF after streptococcal pharyngitis is significantly increased to 50% in patients with a previous history of RF.[6] The streptococcal M proteins may also act as superantigens to augment the immune response.[7] There has been also some evidence to suggest a genetic basis for susceptibility to RF. Familial predisposition to the disease has been indicated in numerous epidemiological studies. A specific B-cell alloantigen has been described in almost all patients with RF, [8] and susceptibility to RF has been liked with HLA-DR 1,2,3 and 4 haplotypes in various ethnic groups.[9] However, predisposing genetic factors of the hosts for its development remains incompletely defined.. The predominant cause of mitral stenosis (MS) is RF. Rheumatic involvement is present in 99% of stenotic mitral valves excised at the time of mitral valve replacement.[10] The valve leaflets are diffusely thickened by fibrous tissue and/or calcific deposits. The mitral commissures fuse, the chordae tendineae fuse and shorten, the valvular cusps become rigid, and these changes, in turn, lead to narrowing at the apex of the funnel-shaped (fish-mouth) valve. The anatomical. 7.
(8) changes in severe MS may result from the combined effect of a smoldering rheumatic process and a constant trauma to the mitral valve by the turbulent blood flow.[11] The principal symptom of MS is exertional dyspnea. Other clinical manifestations include pulmonary edema, hemoptysis, chest pain, systemic embolism and signs of right-sided heart failure. In temperate zones, patient who develop acute rheumatic fever have an asymptomatic period of approximately 15 to 20 years before symptoms of MS develop. It then takes approximately 5 to 10 years for most patients to progress from mild disability (i.e. early New York Heart Association [NYHA] Class II) to severe disability (i.e. NYHA Class III or IV). The progression can be much more rapid in patients in tropical and subtropical areas.[11]. Mitral valvotomy is indicated in patients with severe MS whose effective orifice (normal: 4-6 cm2) is reduced to less than approximately 1.0 cm2 /m2 body surface area (or < 1.6 cm2 in normal-sized adults), or in symptomatic patients with mild stenosis (orifice area 1.0-1.5 cm2/m2) who are symptomatic during ordinary activity and who develop pulmonary arterial systolic pressures exceeding 60 mmHg, or mean pulmonary capillary wedge pressures exceeding 25 mmHg during exercise.[12] Percutaneous mitral balloon valvotomy (PBMV) has been accepted as an alternative to surgical mitral commissurotomy in the treatment of patients with symptomatic rheumatic mitral stenosis. [13]. 8.
(9) Interleukin-1(IL-1), the prototypic “multifunctional” cytokine, affects nearly every cell type with potent inflammatory and stimulatory properties. It is one of the major cytokines produced at sites of inflammation and is involved in the initiation and progression of connective tissue destruction, and monocytes are the cell type responsible for the majority of IL-1 production.[14] The varied biological functions are due to its effects on the expression of over 80 other genes. The margin between clinical benefit and unacceptable toxicity in humans is exceedingly narrow. In addition to controlling gene expression, synthesis, and secretion, the regulation of its biological activities extends to surface receptors, soluble receptors, and a receptor antagonist. Among the members of the IL-1 gene family, IL-1ß is a systemic, hormone-like mediator intended to be released from cells, compared to IL-1a as primarily a regulator of intracellular events and mediator of local inflammation. Both molecules are translated in the cytosol associated with cytoskeletal rather than endoplasmic reticulum structure, and both precursors undergo myristoylation on lysine residues in their respective propieces. However, differences between these two cytokines are remarkable when examining regulation of gene expression, mRNA stability, translation, processing, and secretion. In addition, the affinities of pro and mature IL-lß binding to surface and soluble forms of the receptors are different from those of IL-la. Once released from cells, mature IL-lß encounters two antagonistic molecules: the soluble form of the type I1 receptor, which tightly binds IL-lß, and the soluble IL-1 receptor. 9.
(10) antagonist (IL-1RA), which competes with IL-1ß for cell surface receptor occupancy. The activity of IL-1ß is thus regulated by the large and complex nature of the IL-l ß promoter, the mRNA splicing processes, the phosphorylations of proteins required for translation, the stabilization of the 3’ untranslated region, the cleavage of proIL-1ß, and secretion, the effects of antagonistic molecules, and the presence of decoy receptors.. Unlike the promoter of IL-1a, the promoter region for IL-lß contains a clear TATA box, a typical motif of inducible genes. The half-life of IL-1ß mRNA depends on the cell type and the conditions of stimulation. Studied in cell lines derived from myelomonocytic. leukemias. established that endotoxin triggers transient. transcription and steady state levels of IL-lß mRNA, which accumulate for 4 hours followed by a rapid decrease due to synthesis of a transcriptional repressor.[15][16] Monocyte mRNA levels can be sustained for over 24 hours with cAMP-inducing agents or IL-1 itself.[17][18] Raising cAMP levels in human monocytes by histamine or prostaglandin (PG) E2 enhances IL-la-induced IL-lß gene expression and synthesis.[19] But cAMP reduces IL-lß synthesis by lipopolysaccharide (LPS) stimulation. Unlike most cytokine promoters, IL-lß regulatory regions can be found distributed over several thousand basepairs (bps) upstream and a few bps downstream from the transcriptional start site. IL-lß gene expression is regulated at different levels.[20] Sequences in the IL-lß promoter required for transcription. 10.
(11) has been confirmed. The enhancer region -2782 to -2729, strongly LPS-responsive, is required for transcription and contains cAMP response element(CRE) and binding sites for nuclear factor(NF)-?B and nuclear factor for IL-6 expression (NFIL-6). Another enhancer at -2896 to -2846, containing a cAMP response element, appears to act cooperatively. Activating protein-l (AP-l) sites also participate in endotoxin-induced IL-1ß gene expression. Proximal promoter elements between -131 and +14, required for maximal IL-1ß gene expression, contains binding sites for NF-ßA, which are identical to those of the hematopoietic transcription factor Spi-1/PU.1.[21] The requirement for Spi-1/PU.1 for IL-1ß gene expression imparts tissue specificity, because not all cells constitutively express this NF. Human blood monocytes, which constitutively express Spi-l/PU.1, are exquisitely sensitive to gene expression of IL-1ß by 1-10 pg/mL of LPS. The IL-1RA promoter also contains the proximal Spi-1PU.1 site, and is highly sensitive to LPS as well[20].. It is documented that there is a dissociation between transcription and translation of IL-1ß; for example, complement component C5a, hypoxia, adherence to surfaces or clotting of blood induces the synthesis of large amounts of IL-lß mRNA in monocytic cells without significant translation into the IL-1ß protein.[22][23] However, adding bacterial endotoxin or IL-1 itself to cells with high levels of steady state IL-1 ß mRNA results in augmented translation. Possible. 11.
(12) explanations include stabilization of the AU -rich 3’ untranslated region by adding LPS, or prevention of deadenylation of the poly (A) tail of IL-l mRNA by IL-1 itself. This stabilization of mRNA poly(A) tail may be seen in the stimulation of production of other cytokines, such as IL-8, by IL-1 as well.[24] by preventing deadenylation. An additional signal, transduced through mitogen-activated protein (MAP) kinase cascade, is required for the translation of IL-1 and can be inhibited by the dual cyclooxygenase/lipooxygnase inhibitors.[25]. After synthesis, proIL-1ß remains primarily cytosolic until it is cleaved by the interleukin-1ß converting enzyme (ICE) at the aspartic acid-alanine (116-117) peptideand transported out of the cell.[26] Although the proIL-1ß can be released from a cell independent of processing by ICE.[27] A Taq I polymorphism in the coding sequence of proIL-1ß has been correlated with IL-1ß secretion activity.[28] The 45-kD precursor of ICE requires two internal cleavages itself before becoming enzymatically active. Five isoforms of ICE, namely a, ß, ?, d, and e, can be produced from alternate RNA splicing.[29] Among them, ICEa cleaves the ICE precursor and proIL-lß, and ICEe inhibits the enzymatic activity by binding to the p20 chain of ICE to form an inactive complex, and suppresses apoptosis when overexpressed. ICE activity and thus the secretion of IL-1ß are regulated by inflammatory reaction as evidenced by the effects of interferon(INF)? and prostaglandin(PG)E2.[17][30] Although IL-1 can induce apoptosis in the. 12.
(13) pancreatic ß cell, mediated by increased inducible nitric oxide synthase(iNOS),[31] it can also be a growth factor for many other cells. The substrate for ICE overexpression to induce apoptosis remains unidentified.. The naturally occurring IL-1 receptor antagonist (IL-1RA) appears to be a unique situation in cytokine biology. It binds to the same IL-1 receptor but does not initiate signal transduction and acts as a competitive inhibitor of IL-1 without agonist activity.[32] [33] ProIL-1RA is processed in the Golgi apparatus and secreted. The expression of IL-1RA can be also induced by LPS stimulation of monocytes, but there is a delay of 1 to 2 hours, compared to the expression of IL-1ß.[34] The IL-1 signal can only be transduced when a heterodimer of the type I IL-1 receptor (IL-1RI) and the IL-1R accessory protein(IL-1R-AP) is formed and the GTPase domains in the cytoplasmic portions of IL-RI and IL-1R-AP are activated. While IL-1ß possesses 2 binding sites for each of the components, the IL-1RA has only one bidning site. Thus it binds the IL-1RI but cannot dock the IL-1R-AP to complete dimerization, and signaling cannot be initiated. IL-1RA has a nearly equal affinity for IL-1RI as IL-1ß[35] and occupancy of the IL-1RI by IL-1RA is an effective prevention of IL-1 signaling. But a 100 to 1000 fold excess of IL-1RA over IL-1 is necessary to block its action because of the spare receptor effect.[36] The observation of low and high binding affinities of IL-1 to various cells may be explained as that IL-l binds first to the IL-1RI with a low affinity,. 13.
(14) then a structural change may take place in IL-l allowing for docking of IL-1R-AP to the IL-1RI/IL-1 complex. The promoter region of the IL-1RI gene lacks a TATA or CAAT box characteristic of inducible genes, but the significance of its constitutive expression is unknown. IL-1RI expression is downregulated in T cells with a decreased mRNA half life and surface expression.[37] In cells that synthesize PGE2, IL-1 upregulates its own receptor via PGE2; however, when PGE2 synthesis is inhibited, IL-1 downregulates IL-1RI in the same cell.[38][39] The type II IL-1R(IL-1RII), lacking a cytosloic domain, binds IL-1ß tightly and acts as a decoy receptor.[40] Upregulation of IL-1RII is expected to reduce the activities of IL-1. IL-1 itself has differential effects on IL-1RI and IL-1RII gene expression: downregulates IL-1R1, but upregulates IL-lRII.[41] The transcription factor PU.l, present in cells of hematopoietic origin, is required for the expression of IL-1RII.[42] Dexamethasone and IL-4 has been shown to upregulates IL-1RII expression and thus counteract the activity of IL-1.[40] Of the three members of the IL-1 family, IL- 1ß has the lowest affinity for the cell bound form of IL-1RI, but it is the most avid binding member for the non-signal transducing type II receptor. In addition, the binding between IL-1ß and soluble IL-1RII is nearly irreversible because of a slow dissociation rate.[43] Because the type II receptor is more likely to bind to IL-1ß than IL-la, this can result in a diminished response to IL-1ß. The double antagonist mechanism of receptor antagonist and decoy receptor probably reflects the high functional level of only a few type I receptors. 14.
(15) and amplification of post-receptor events.. Interleukin-4(IL-4) is the primary cytokine involved in the humoral immune response leading to class switching to and production of immunoglobulin E (IgE) by B cells.[50] Naïve T cells, when presented with antigen in the presence of an early IL-4 burst, are driven to differentiate into T helper type 2 (TH 2) cells. TH 2 cells then provides the essential signals, IL-4 being one of them, to induce IgE production by B cells.[51] The IL-4 receptor(IL-4R) on B cells transduces the signal through the Janus kinase-signal transducer and activation of transcription (JAK-STAT) pathways. Mast cells, basophils and eosinophils also secret IL-4 to amplify the IgE response.[52][53] In addition to deviate the immune reaction to favor a T H 2 response, IL-4 is also a down-regulator of macrophages and depresses cell-mediated immunity.[54][55] The IL-4 genes are mapped to chromosome 5q, along with a cluster of tightly linked genes that includes those for IL-3, IL-5, IL-9, IL-12, IL-13, and granulocyte-macrophage colony-stimulating factor (GM-CSF). Various polymorphisms in the IL-4 family genes including IL-4 promoter -590T/C, IL-4 intron 3 VNTR, and IL-4Ra I50V and Q551R have been described. These polymorphisms. have. been. correlated. with. atopy,[56-58]. rheumatoid. arthritis,[59][60] inflammatory bowel disease,[61][62] periodontitis,[63] and IgA nephropathy.[64]. 15.
(16) Transforming growth factor (TGF-ß), a member of a family of dimeric polypeptide growth factors that includes bone morphogenic proteins and activins, regulates the proliferation and differentiation of cells, embryonic development, wound healing, and angiogenesis. Every cell in the body, including epithelial, endothelial, hematopoietic, neuronal, and connective-tissue cells, produces TGF-ß and has receptors for it.[65] Each of the three isoforms, namely TGF-ß1, TGF-ß2, and TGF-ß3, is encoded by a distinct gene and is expressed in both a tissuespecific and a developmentally regulated fashion, but their biologic properties are nearly identical. TGF-ß1 mRNA is expressed in endothelial, hematopoietic, and connective-tissue cells; TGF-ß2 mRNA in epithelial and neuronal cells; and TGF-ß3 mRNA primarily in mesenchymal cells.[66] During development, TGF-ß1 and TGF-ß3 are expressed early in structures undergoing morphogenesis, and TGF-ß2 is expressed later in mature and differentiating epithelium. It arrests the cell cycle in the G1 phase by stimulating production of the cyclin-dependent protein kinase inhibitor p15 and by inhibiting the function or production of essential cellcycle regulators, especially the cyclin-dependent protein kinases 2 and 4 and cyclins A and E.[67] The immunosuppressive effects of TGF-ß include inhibition of leukocyte proliferation and promotion of their differentiation, and suppression of inflammatory TH 1 cells and macrophages. It also provides chemotactic. stimuli. for. leukocyte. migration. and. regulates. adhesion. molecule –mediated localization of these cells.[68-71] Another important feature. 16.
(17) of TGF-ß that may be relevant to the chronic scarring of valves in patients with rheumatic heart disease is that it induces extracellular matrix (ECM) and mediates pathologic fibrosis. Fibrogenesis is not a unique pathologic process but is due to excesses in the same biologic events involved in normal tissue repair. TGF-ß is a key cytokine that initiates and terminates tissue repair and whose sustained production underlies the development of tissue fibrosis.[72] The TGF-ß1 gene is up-regulated in response to tissue injury, and TGF-ß1 is the isoform most implicated in fibrosis. TGF-ß is first synthesized as part of a large precursor molecule containing also a propeptide region, then cleaved from, but still attached to this propeptide through noncovalent bonds before secretion. Most TGF-ß is then stored in the ECM as an inactive, high-molecular-weight complex of TGF-ß, the propeptide, and the latent TGF-ß–binding protein. Active TGF-ß can be released. from. the. complex. by. plamin-mediated. cleavage. or. thrombospondin-1induced conformational change of the latent TGF-ß– binding protein.[73] There are 3 types of TGF-ß receptors on the cell surface. The most abundant type III receptor functions by binding TGF-ß and transferring it to the signaling receptors type I and II. These type I and II receptors contain serine–threonine protein kinases in the intracellular domains, which initiate intracellular signaling by phosphorylating the transcription factors Smads. The resulting Smad complex then moves into the nucleus, where it interacts in a cell-specific manner with various transcription factors to regulate the transcription. 17.
(18) of many genes.[74][75] TGF-ß not only stimulates fibroblasts and other cells to produce extracellular-matrix proteins and cell-adhesion proteins, including collagen, fibronectin, and integrins, but also decreases the production of enzymes that degrade the extracellular matrix, including collagenase, heparinase, and stromelysin, and increases the production of proteins that inhibit enzymes that degrade the extracellular matrix, including plasminogen-activator inhibitor type 1 and tissue inhibitor of metalloprotease.[76] Thus, the production of TGF-ß has been reported to be increased in patients with various diseases associated with fibrosis of the kidney, liver, lung or systemically.[65][77] In addition, polymorphisms in the TGF-ß1 gene regulate its level of expression in humans.[78][79] The SNP -509 T/C, which is in linkage disequilibrium with the SNP -800 G/A, has been associated with the plasma concentration of latent TGF-ß1.[80] creates two different alleles located in a region that is thought to be a negative regulatory area of the TGF-ß1 gene.[100] It is associated with risk for asthma and allergies,[101] promoting an increase in the levels of serum IgE, and susceptibility to osteoporosis.[102] However, the precise effect of T or C alleles on the transcriptional activity of the TGF-ß1 gene is not known.. The renin–angiotensin system (RAS), a two-enzyme cascade, plays an important role in the regulation of blood pressure, fluid balance, and electrolyte homeostasis and in the pathogenesis of cardiovascular disease. The initial enzyme, renin,. 18.
(19) cleaves its substrate, angiotensinogen, to angiotensin I, a decapeptide. Angiotensin I undergoes a second cleavage, mainly by tissue-bound angiotensin-converting enzyme (ACE) and serine proteinase to generate angiotensin II, an octapeptide , which via the angiotensin II type 1 receptor acts as a potent vasoconstrictor and aldosterone stimulating peptide. The ACE also inactivates the nonapeptide bradykinin and blocks the tissue kallikrein system.[84] Interests in the possible role of RAS as a risk factor for cardiovascular disease have been generated as large clinical trials demonstrated the effects of the ACE inhibitors (ACEI) in decreasing morbidity and mortality in patients with heart failure.[81-83] Inhibition of the ACE and antagonism of the angiotensin II type 1 receptor decrease blood pressure in hypertensive patients, and more importantly prevent mortality and morbidity in patients with symptomatic or asymptomatic congestive heart failure, acute myocardial infarction, or diabetic nephropathy. A tissue-based RAS can elevate blood pressure quite independently of the plasma system. Although renin appears to be a rate-limiting enzyme in the endocrine RAS, tissue ACE may be a more important independent factor in influencing plasma and tissue angiotensin II levels in the local autocrine or paracrine RAS.[85-88] Thus, the RAS acts systemically and locally to influence vascular tone, blood volume, myocardial contractility, thromboresistance, and tissue responses to injury. The most prominent polymorphism in the angiotensin-converting enzyme gene is defined by the insertion(I) or deletion(D) of a 287 base pair insert in intron 16 of. 19.
(20) the gene. This insert harbors a sequence very similar to a silencer element,[89] which may explain why subjects with one or two D alleles have approximately 25% and 50% higher angiotensin-converting enzyme levels than subjects with the other genotype both in plasma and at tissue sites.[90-93] Membrane-bound ACE, rather than soluble ACE, is responsible for the regional conversion of angiotensin I into angiotensin II. Since the plasma or tissue angiotensin I levels are six orders of magnitude below the Michaelis constant (Km) for angiotensin I, and thus this process follows first-order kinetics, angiotensin I–II conversion was found to be similar over a wide range of arterial angiotensin I levels.[94-96] Increased ACE gene expression and ACE activity in the vessel wall stimulate the local rate of angiotensin II production. This increased ACE activity in DD homozygotes has been correlated with higher risk of hypertension in men, excess risk of left ventricular hypertrophy in untreated hypertensive patients, and an increased risk of nephropathy in the presence of diabetes or other concomitant diseases.[97] There is also an significant inter-racial variation of the allele frequencies, so that the prevalence of the D allele was 56.2% in Caucasians, higher in blacks (60.3%), but substantially lower in Asians (39.1%). [98]. We hypothesized that polymorphisms of the IL-1, IL-4, TGF-ß, and ACE families affects the inflammatory destruction of cardiac valves in patients with rheumatic heart disease and the development of heart failure. Polymorphisms in IL-1ß. 20.
(21) promoter, IL-1ß exon 5, IL-1RA intron 2, IL-4 intron 3, TGF-ß promoter, and ACE intron 16 were compared between a group of MS patients receiving PBMV and normal controls.. 21.
(22) PATIENTS AND METHODS. A group of 84 consecutive patients (mean age 43.6 ± 5.8 years; 12 males and 72 females) with severe MS were collected. All were symptomatic during ordinary activity and underwent PBMV after a detailed clinical and echocardiographic evaluation to assess the valve morphology and cardiac function. Indications for PBMV were effective mitral orifice less than approximately 1.0 cm2/m2 body surface area, and New York Heart Association (NYHA) class III or IV symptoms. Patients with more than grade 2 mitral regurgitation (MR) on left ventriculogram, persistent left atrial thrombus on transesophgeal echocardiography (TEE), extensive commissural calcification, or concomitant cardiac disease requiring surgical intervention were excluded.. There were 103 age- and sex- matched ( mean age 48.1 ± 8.1 years; 17 males and 86 females) normal controls. All people enrolled were ethnic Chinese living in Taiwan and have signed the informed consent. The study was reviewed and approved by our research ethics committee.. Polymerase chain reaction and typing for IL-1 related allelic polymorphisms. The genomic DNA was prepared from peripheral blood using a DNA Extractor. 22.
(23) WB kit (Wako, Japan). Polymerase chain reactions (PCRs) were carried out in a total volume of 50 ml, containing genomic DNA, 2-/6 pmole of each primer, 1 x Taqpolymerase buffer (1.5 mM MgCl2), and 0.25 units of AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA). PCR amplification was performed in a programmable thermal cycler GeneAmp PCR System 2400 (Applied Biosystems). The primers for the IL-1ß promoter region, exon 5 and IL-1RA intron 2 gene polymorphisms and the cycling conditions are given in Table 1.. The IL-1ß promoter polymorphism at position -511 was analyzed by PCR amplification followed by restriction analysis using Ava I (New England Biolabs, Beverly, USA) digestion. The allele with cytosine (‘C’) at position -511 showed up as 190-bp and 114-bp on agarose ele ctrophoresis, while the thymine (‘T’ ) allele was 304-bp. (Figure 1). The region containing the polymorphic site at position +3953 within exon 5 of the IL-1ß gene was amplified and then digested by Taq I (New England Biolabs). Class ‘E1’ was 135+114 bp and ‘E2’ was 249 bp as revealed by electrophoresis. (Figure 2). For IL-1RA intron 2, 10 ml of the products were loaded into 3% agarose gel. 23.
(24) containing ethidium bromide for electrophoresis and each allele was identified according to its size. The 86-bp variable number tandem repeat (VNTR) of IL-1RA gene was classified as ‘I’for 410-bp, ‘II’ for 240-bp, ‘III’for 500-bp, ‘IV’ for 325-bp and ‘V’ allele for 595-bp. (Figure 3). The IL-4 intron 3 contains a 70-bp length of VNTR. The upstream primer sequences for fragment amplification and PCR was 5’-AGGCTAAAGGGGGAAAGC-3’ and the downstream primer 5’-CTGTTCACCTCAACTGCTCC-3’. Denaturation, annealing and extension were performed at 95°C for 30 seconds, 60°C for 42 seconds, and 72° for 42 seconds, respectively, for 30 cycles.[99] PCR products were analyzed by electrophoresis on agarose gel without digestion. Each allele was recognized according to its size. The 183 bp fragment was designated RP1 and the 253 bp fragment RP2.. In the analysis of TGF-ß1 -509 T/C, the sequences of PCR primers were 5’-TTTTGCCATGTGCCCAGTAG -3’ (upstream) and 5’-CACCAGAGAAAGAGGACCAG- 3’ (downstream). PCR was carried out for 35 cycles of 1 min at 95°C, 1 min at 57°C and 1 min at 72°C, with a final extension of 721C for 7 min. The product then underwent restriction endonuclease digestion. A 15-ml aliquot of PCR products was mixed with a 5 ml solution containing 2 ml 10_NE Buffer (50mM NaCl, 10mM Tris-HCl, 10mM MgCl2, 1mM dithiothreitol, pH 7.9), 0.1 ml Eco. 24.
(25) 81I(20 U/ml) (New England Biolabs, Inc., Beverly, MA, USA) and 2.5 ml sterile deionized H2O incubated at 37°C. The total amount aliquot of the digest was mixed with 3 ml of loading buffer and electrophoresed on a 10% vertical non-denaturing polyacrylamide gel at 20 mA. The gel was silver stained by DNA Silver Staining Kit (Amersham Pharmacia Biotech AB, Uppsala, Sweden)[100]. The primer pair used to amplify of the ACE gene consisted of 5’-CTGGAGACCACTCCCATCCTTTCT-3’ and 5’-GATGTGGCCATCACATTCGTCAGAT-3’. Amplification was performed for 35 cycles with steps of denaturation at 94°C for 2 minutes, annealing at 58°C for 15 seconds, and extension at 72°C for 30 seconds. PCR products were subjected to electrophoresis in agarose gels and stained by ethidium bromide for visualization. To avoid mistyping of the ID genotype as DD, dimethyl sulfoxide was added to the PCR reaction mix to enhance amplification of the I allele, and repeated the procedure with samples with the DD genotype. [103-105]. Statistical analysis. Continuous variables are expressed as the mean ± SD. The genotype distribution and allele frequency of the gene polymorphisms among patients and controls were compared by use of the Chi-square test and the Fisher’s exact test. A P value <. 25.
(26) 0.05 was considered to be significant. The statistical analysis was done by use of the software SPSS for windows (SPSS Inc, Chicago, Ill). 26.
(27) RESULTS. There were 84 patients who have received PBMV for severe MS. Their ages ranged from 32 to 68 years (mean 43.6 ± 5.8 years). Seventy (83.3%) of them were female and 14 (16.7%) were male. Three patients received mitral valve replacement surgery with a mechanical valve because of recurrent, calcific MS from 8 to 13 years after PBMV. All patients were in NYHA functional class I or II currently. The mean area of the post-dilated mitral valve estimated by echocardiographic planimetry was 1.62 ± 1.8 cm2 ( range 1.28 – 1.83 cm2 ) as of the last follow-up, excluding the 3 patients who had a mechanical valve implanted.. Genotypes and allele frequencies of the three genomic polymorphisms of the IL-1 gene family were analyzed in MS patients and in healthy controls by PCR, restriction digestion, and gel electrophoresis. The genotype distribution and allele frequency of polymorphisms in IL-1ß promoter were shown in Table 2 and 3 respectively. There were no significant difference between patients and controls.. As shown in Table 4 and 5, there were no significant difference in the genotype distribution and allele frequency of polymorphisms in IL-1ß exon 5 between patients and controls.. 27.
(28) In the analysis of the IL-1RA gene polymorphism, neither of the rare alleles III, IV, V, or the recently reported single copy VNTR was identified in the samples studied. Genotype II/II was found only once in the control group, and none in the patients. Genotype I/II was significantly increased in the patient group compared to controls ( p<0.05; Table 6 ). The odds ratio for people with genotype IL-1RA I/II to develop severe MS was 3.80 compared to people with non- IL-1RA I/II genotypes.. Allele II of the IL-1RA gene was significantly increased in the patient group compared to controls ( p<0.05; table 7 ). The odds ratio for IL-1RA II carriers to develop severe MS was 2.61 compared to noncarriers. Analysis of linkage between alleles IL-1RA II and alleles of the IL-1ß exon 5 or IL-1ß promoter restriction polymorphisms showed an equal distribution of IL-1Ra alleles in carriers of the different IL-1ß exon 5 or IL-1ß promoter 5alleles (Table 8), which suggested that the results of our study were not affected by unsuspected linkage between the polymorphisms studied.. For the genotypes and allele frequencies the IL-4 intron 3 VNTR polymorphism, there is no significant difference between controls and MS patients (Table 9 & 10).. The genotype C/C in the polymorphism of TGF-ß promoter(-509) appeared in only 3.6% of the MS patients, compared with 23.3% in the control group.( p <. 28.
(29) 0.001, Table 11), and genotypes T/C and T/T were 71.4% vs. 45.6% and 25.0% vs. 31.1%, respectively, in patient vs. control group. The odds ratio for IL-1RA II carriers to develop severe MS was 2.61 compared to noncarriers. But further analysis of the individual T and C allele frequencies revealed a similar distribution in patient and control groups.(p > 0.1, Table 12). Both the genotype and allele frequency of the ACE intron 16 I/D polymorphism showed similar distribution in patient and control groups. (Table 13 & 14). 29.
(30) DISCUSSION. Carditis is the most severe complication of post-streptococcal rheumatic fever, which remains a significant problem worldwide, especially in developing countries because of the persistently high incidence. When congestive heart failure is caused by deformed and dysfunctional valves, mechanical dilation and surgical replacement of the diseased valves are the only effective treatment, with the inherent costs and risks. Very little is know concerning host susceptibility. Certain HLA class II alleles/haplotypes were also associated with risk or protection from rheumatic heart disease.[106][107] As the basic rheumatic process is inflammation and destruction of connective tissue, and IL-1, IL-4 TGF-ß are cytokines involved in the immuno-inflammatory response and tissue repair, and ACE may affect the cardiac adaptation to hemodynamic changes, we examined the effects that polymorphisms of the these genes might have on the development of rheumatic heart disease.. For the sake of completeness in data collection, only patients with MS severe enough to receive PBMV are included in this study. Our results revealed significantly increased incidences of IL-1RA I/II genotype and IL-1RA II allele frequency in the patient group compared to controls, and this difference in distribution of the IL-1RA polymorphism is not linked to IL-1ß promoter or IL-1ß. 30.
(31) exon 5 polymorphisms. As these are a homogenous group of patients with severe disease, the association of IL-RA polymorphism with severe MS may reflect its effect on either the pathogenesis of rheumatic carditis, or the progression and severity of inflammatory reaction in general.. The loci for the human IL-1a, IL-1ß and IL-1RA are all linked within the proximal region of the long arm of chromosome 2.[44] Different polymorphisms have been described in the IL-1ß genes, and at least two of them could influence protein production: one is located within the promoter region, [45] the other in exon 5. [46] The polymorphism in intron 2 of IL-1RA is a variable number of tandem repeats (VNTR) of an 86-bp sequence. So far 6 alleles of one to six copies of the 86-bp sequence respectively have been reported.[47][48] Because three potential protein binding sites are located in this 86-bp sequence, the number of repeats may influence gene transcription and protein production. The IL-1RA gene polymorphism affects both IL-1Ra and IL-1 production. Possession of allele II of the IL-1RA gene has been associated with variable productions of IL-1RA and IL-1RA/ IL-1ß ratios among conditions associated with chronic inflammation. Persons with IL-1RA II homozygosity have a more prolonged and more severe proinflammatory. immune. response. than. genotypes.[49]. 31. persons. with. other. IL-1RA.
(32) IL-1RA inhibits activities of IL-1 by competitive binding to the same functional receptor without initiating signal transduction, thus the balance between IL-1RA and IL-1 levels influences the biological effects of IL-1. Many inflammatory diseases such as ulcerative colitis[108], multiple sclerosis[109], psoriasis[110], silicosis[111], periodontitis[112], etc. have been associated with IL-1RA II. However, IL-1RA II has been associated with variable production of IL-RA in different disease states[113][114], and. increased IL-1ß production was. observed.[115][116] Conversely, the participation of the IL-1ß gene in the regulation of IL-1RA production has also been implicated.[117] This reflects the complex interactions of cytokines involved in inflammation. In addition, it was suggested that the regulation of IL-1RA and IL-1 production were different in men and women[114]. Rheumatic heart disease is also characterized by chronic inflammation and female preponderance. Unfortunately, patients with milder forms of MS were not included in this study. With the biological actions of IL-1 and the reports of increased incidences of the allele IL-1RA II in other inflammatory diseases taken into account, our findings provide some evidence that this allele is associated with severe MS in patients with rheumatic heart disease. Functional correlation of the polymorphisms with IL-1 and IL-1RA tissue levels in the mitral valve was not performed in this study as most patients received PBMV only and tissue sample was not obtained. Although the IL-1RA allele was reported to be in linkage disequilibrium with IL-1ß polymorphism[118], our findings. 32.
(33) revealed no linkage between the three polymorphisms of the IL-1 family. The possible association of IL-1RA polymorphisms with rheumatic heart disease warrants further study. If the findings can be confirmed in large samples, IL-1RA II may be used as a marker for selection in patients with MS for novel therapeutic modalities such as the use of specific IL-1 receptor antagonist before the disease progresses to a late stage.. TGF-ß mediates a complex mix of pro- and anti-inflammatory activities. In immunity, however, its predominant functions are anti-inflammatory.[69][70] It also induces formation of the extracellular matrix and fibrosis.[72] The base exchange at -509 in the TGF-ß promoter was linked to elevated total IgE in asthmatic patients.[101] This polymorphism represented a C-to-T base exchange which induced a YY1 consensus sequence and is present in a region of the promoter associated with negative transcription regulation.[119][120] The C allele was associated with lower levels of acid-activatable latent TGF, with a dose-response effect of the T allele.[80] The genotype T/T was found to be increased in patients with severe periodontitis.[121] Our result revealed a significantly lower incidence of the C/C genotype in patients with MS.(Table11) However, this possible protective effect, if any, of the C allele cannot be demonstrated in the comparison of allele frequencies. (Table 12) A sampling error due to the small size of our cohort shoulde also be considered.. 33.
(34) CONCLUSION. The finding of increased incidence of IL-1RA II in patients with severe MS suggests this allele may be used as a marker for disease severity and progression in patients with rheumatic heart disease. The incidence of the TGF-ß1(-509) C/C genotype was found to significantly decreased in our small group of patients with MS. Confirmation awaits further studies with large samples.. 34.
(35) REFERENCES. [1] Special Writing Group of the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease of the Council on Cardiovascular Disease in the Young of the American Heart Association: Guidelines for the diagnosis of rheumatic fever. Jones criteria, 1992 Update. JAMA 1992; 268:2069-73.. [2] Stollerman GH. Rheumatic fever. Lancet 1997; 349:935 42.. [3] Krisher K, Cunningham MW. Myosin: A link between streptococci and heart. Science 1985; 227:413-5.. [4] Dale JB, Chiang EC. Intranasal immunization with recombinant group A streptococcal M fragment fused to the B subunit of Escherichia coli labile toxin protects mice against systemic challenge infections. J Infect Dis 1995; 171: 1038–41.. [5] Cunningham MW. Autoimmunity and molecular mimicry in the pathogenesis of post-streptococcal heart disease.. Front Biosci 2003;8:S533-43.. [6] Dajani AS. Current status of nonsuppurative complications of group A. 35.
(36) streptococci. Pediatr Infect Dis J 1991;10:S25-7.. [7] Watanabe-Ohnishi R, Aelion J, LeGros L, et al. Characterization of unique human TCR V-beta specificities for a family if streptococcal superantigens represented by rheumatogenic serotypes of M protein. J Immunol 1994;152: 2066-73.. [8] Khanna, A. K., Buskirk, D. R., Williams, R. C., et al. Presence of non-HLA B cell antigen in rheumatic fever patients and their families as defined by a monoclonal antibody. J Clin Invest 1989;83:1710-6.. [9] Ayoub, E. M., Barrett, D. J., Maclaren, N. K., et al. Association of class II human histocompatibility leukocyte antigens with rheumatic fever. J Clin Invest 1986;77:2019-26.. [10] Olson LJ, Subramanian R, Ackerman DM, et al. Surgical pathology of the mitral valve: a study of 712 cases spanning 21 years. Mayo Clin Proc 1987;62:22-34.. [11] Dalen JE and Alpert JS (eds). Valvular heart disease. 2nd ed. Boston, Little, Brown and Co, 1987, 600 pp.. 36.
(37) [12] Bonow RO, Carabello B, de Leon AC Jr. ACC/AHA Guidelines for the management of patients with valvular heart diseases. J Am Coll Cardiol 1998; 32:1486-582.. [13] Igor F. Palacios. Farewell to surgical mitral commissurotomy for many patients. Circulation 1998;97:223-6.. [14] Dinarello CA. Biologic basis for interleukin-1 in disease.. Blood. 1996;87:2095-147.. [15] Fenton MJ, Clark BD, Collins KL, Webb AC, Rich A, Auron PE. Transcriptional regulation of the human prointerleukin 1 beta gene. J Immunol 1987;138:3972-9.. [16] Fenton MJ, Vermeulen MW, Clark BD, Webb AC, Auron PE. Human pro-IL-l beta gene expression in monocytic cells is regulated by two distinct pathways. J Immunol 1988;140:2267-73.. [17] Schindler R, Ghezzi P, Dinarello CA. IL-l induces IL-I .IV. IFN-? suppresses IL-1 but not lipopolysaccharide-induced transcription of IL-I. J Immunol 1990; 144:2216-22.. 37.
(38) [18] Serkkola E, Hurme M. Synergism between protein-kinase C and cAMPdependent pathways in the expression of the interleukin-1ß gene is mediated via the activator-protein-l (AP-l) enhancer activity. Eur J Biochem 1993; 213:243-9.. [19] Vannier E, Dinarello CA. Histamine enhances interleukin (1L)-l-induced IL-l gene expression and protein synthesis H2 receptors in peripheral blood mononuclear cells: Comparison with IL-I receptor antagonist. J Clin Invest 1993; 92:281-7.. [20] Auron PE, Webb AC. Interleukin-l. A gene expression system regulated at multiple levels. Eur Cytokine Netw 1994;5:573-92.. [21] Kominato Y, Galson DL, Waterman WR, Webb AC, Auron PE. Monocyte expression of the prointerleukin-lß gene is dependent on promoter sequences which bind the hematopoietic transcription factor Spi-1/PU.1. Mol Cell Biol 1995;15:59-68.. [22] Schindler R, Clark BD, Dinarello CA. Dissociation between interleukin-l beta mRNA and protein synthesis in human peripheral blood mononuclear cells. J Biol Chem 1990;265:10232-7.. 38.
(39) [23] Kaspar RL, Gehrke L. Peripheral blood mononuclear cells stimulated with C5a or lipopolysaccharide to synthesize equivalent levels of IL-1ß mRNA show unequal IL-1ß protein accumulation but similar polyribosome profiles. J Immunol 1994;153:277-86.. [24] Stoeckle MY, Guan L. High-resolution analysis of gro a mRNA poly (A) shortening: Regulation by interleukin-l ß. Nucleic Acids Res 1993;21:1613-7.. [25] Han 3, Richter B, Li 2, Kravchenko VV, Ulevitch RJ. Molecular cloning of human p38 MAP kinase. Biochim Biophys Acta 1995;1265:224-7.. [26] Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA,Livingston DJ. Structure and mechanism of interleukin-1ß converting enzyme. Nature 1994;370:270-5.. [27] Thornbeny NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Schmidt JA, Tocci M: A novel heterodimeric cysteine protease is required for interleukin-l beta processing in monocytes. Nature 1992;356:768-74.. [28] Pociot F, Molvig J, Wogensen L, Worsaae H, Nerup J. A TaqI polymorphism. 39.
(40) in the human interleukin-l beta (IL-I beta) gene correlates with IL-I beta secretion in vitro. Eur J Clin Invest 1992;22:396-402.. [29] Alnemri ES, Femandes-Alnemri T, Litwack G: Cloning and expression of four novel isoforms of human interleukin-l beta converting enzyme with different apoptotic activities. J Biol Chem 1995;270:4312-7.. [30] Lonnemann G, Barndt I, Kaever V, Haubitz M, Schindler R, Shaldon S, Koch KM: Impaired endotoxin-induced interleukin-1 beta secretion, not total production, of mononuclear cells from ESRD patients. Kidney Int 1995;47:1158-67.. [31] Mandrup-Poulsen T, Zumsteg U, Reimers J. Pociot F, MØ rch L, Helkvist S, Dinarello CA, Nerup J. Involvement of interleukin-1 and interleukin-1 antagonist in pancreatic ß-cell destruction in insulin-dependent diabetes mellitus. Cytokine 1993;5:185-91.. [32]. Boch. JA,. Wara-aswapati. N,. Auron. PE.. Interleukin. 1. signal. transduction--current concepts and relevance to periodontitis. J Dent Res 2001;80:400-7.. [33] McIntyre KW, Stepan GJ, Kolinsky KD, et al. Inhibition of interleukin 1. 40.
(41) binding and bioactivity in vitro and modulation of acute inflammation in vivo by IL-1 receptor antagonist and anti IL-1 receptor monoclonal antibody. J Exp Med 1991; 173:931-9.. [34] Arend WP. Interleukin-l receptor antagonist. Adv lmmunol 1993;54:167-227.. [35] Hannum CH, Wilcox CJ, Arend WP. Joslin FG, Dripps DJ, Heimdal PL, Armes LG, Sommer A, Eisenberg SP, Thompson RC. Interleukin-l receptor antagonist activity of a human interleukin-l inhibitor. Nature 1990;343:336-40.. [36] Ohlsson K, Bjork P, Bergenfeldt M, Hageman R, Thompson RC. Interleukin-1 receptor antagonist reduces mortality from endotoxin shock. Nature 1990;348:550-2.. [37] Ye K, Koch K-C, Clark BD, Dinarello CA: Interleukin-l down regulates gene and surface expression of interleukin-1 receptor type I by destabilizing its mRNA whereas interleukin-2 increases its expression. Immunology 1992;75:427-34.. [38] Takii T, Akahoshi T, Kat0 K, Hayashi H, Marunouchi T, Onozaki K. Interleukin-1 upregulates transcription of its own receptor in a human fibroblast cell line TIG-l . Role of endogenous PGE 2 and cAMP. Eur J Immunol. 41.
(42) 1992;22:122-7.. [39] Takii T, Hayashi H, Marunouchi T, Onozaki K: Interleukin-1 down-regulates type I interleukin-l receptor mRNA expression in a human fibroblast cell line TIG-l in the absence of prostaglandin E2 synthesis. Lymphokine Cytokine Res 1994;3:213-9.. [40] Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, Gin J, Dower SK, Sims JE, Mantovani A. Interleukin-l type II receptor: A decoy target for IL-1 that is regulated by IL-4. Science 1993;261:472-5.. [41] Bristulf J, Gatti S, Malinowsky D, Bjork L, Sundgren AK, Bartfai T. Interleukin-l stimulates the expression of type I and type I1 interleukin-l receptors in the rat insulinoma cell line Rinm5F; sequencing a rat type I1 interleukin-l receptor cDNA. Eur Cyokine Netw 1994;5:319-30.. [42] Vannier E, Ye K, Fenton MJ, Sims JE, Dinarello CA. IL-1 receptor type II gene expression: A role for PU.1. Cytokine 1995;7:596.. [43]Arend WP, Malyak M, Smith MF, Whisenand TD, Slack JL, Sims JE, Giri JG, Dower SK: Binding of IL-1a, IL-1ß, and IL-1 receptor antagonist by soluble IL-1. 42.
(43) receptors and levels of soluble IL-I receptors in synovial fluids. J Immunol 1994; 153:4766-74.. [44] Steinkasserer A, Spurr NK, Cox S, et al. The human IL-1 receptor antagonist gene (IL1RN) maps to chromosome 2q14-q21, in the region of the IL-1a and IL-1b loci. Genomics 1992;13:654-7.. [45] Di Giovine FS, Takhsh E, Blakemore AIF, et al. Single base polymorphism at -/511 in the human interleukin-1 beta gene (IL 1 beta). Hum Mol Genet 1992;1:450.. [46] Pociot F, Molvig J, Wogensen L, et al. A TagI polymorphism in the human interleukin-1b (IL-1b) gene correlates with IL-1b secrection in vitro. Eur J Clin Invest 1992;22:396-402.. [47] Tarlow JK, Blakemore AIF, Lennard A, et al. Polymorphism in human IL-1 receptor antagonist gene intron 2 is caused by variable numbers of an 86 base pair tandem repeat. Hum Genet 1993;91:403-4.. [48] Vamvakopoulos JE, Taylor CJ, Morris-Stiff GJ, Green C, Metcalfe S. The interleukin-1 receptor antagonist gene: a single-copy variant of the intron 2. 43.
(44) variable number tandem repeat (VNTR) polymorphism. Eur J Immunogenet 2002;29(4):337-40.. [49] Witkin SS, Gerber S, Ledger WJ. Influence of interleukin-1 receptor antagonist gene polymorphism on disease. Clin Infect Dis 2002;34:204-9.. [50] Gessner A, Rollinghoff M. Biologic functions and signaling of the interleukin-4 receptor complexes. Immunobiology 2000;201:285-307.. [51] Haas H, Falcone FH, Holland MJ, Schramm G, Haisch K, Gibbs BF, Bufe A, Schlaak M. Early interleukin-4: its role in the switch towards a Th2 response and IgE-mediated allergy. Int Arch Allergy Immunol 1999;119:86-94.. [52] Nelms K, Huang H, Ryan J, Keegan A, Paul WE. Interleukin-4 receptor signalling mechanisms and their biological significance. Adv Exp Med Biol 1998;452:37-43.. [53] Izuhara K, Shirakawa T. Signal transduction via the interleukin-4 receptor and its correlation with atopy. Int J Mol Med 1999;3:3-10.. [54] Bonecchi R, Sozzani S, Stine JT, Luini W, D'Amico G, Allavena P, Chantry. 44.
(45) D, Mantovani A. Divergent effects of interleukin-4 and interferon-gamma on macrophage-derived chemokine production: an amplification circuit of polarized T helper 2 responses. Blood 1998;92:2668-71.. [55] Dello Sbarba P, Rovida E, Caciagli B, Nencioni L, Labardi D, Paccagnini A, Savini L, Cipolleschi MG. Interleukin-4 rapidly down-modulates the macrophage colony-stimulating factor receptor in murine macrophages. J Leukoc Biol 1996; 60:644-50.. [56] Novak N, Kruse S, Kraft S, et al. Dichotomic nature of atopic dermatitis reflected by combined analysis of monocyte immunophenotyping and single nucleotide polymorphisms of the interleukin-4/interleukin-13 receptor gene: the dichotomy of extrinsic and intrinsic atopic dermatitis. J Invest Dermatol 2002;119:870-5.. [57] Gao PS, Mao XQ, Hopkin JM, Adra CN, Yang X, Shirakawa T. Functional significance of polymorphisms of the interleukin-4 and interleukin-13 receptors in allergic disease. Clin Exp Allergy 2000;30:1672-5.. [58] Elliott K, Fitzpatrick E, Hill D, et al. The -590C/T and -34C/T interleukin-4 promoter polymorphisms are not associated with atopic eczema in childhood. J. 45.
(46) Allergy Clin Immunol 2001;108:285-7.. [59] Cantagrel A, Navaux F, Loubet-Lescoulie P, Nourhashemi F, Enault G, Abbal M, Constantin A, Laroche M, Mazieres B. Interleukin-1beta, interleukin-1 receptor antagonist, interleukin-4, and interleukin-10 gene polymorphisms: relationship to occurrence and severity of rheumatoid arthritis. Arthritis Rheum 1999;42:1093-100.. [60] Buchs N, Silvestri T, di Giovine FS, Chabaud M, Vannier E, Duff GW, Miossec P. IL-4 VNTR gene polymorphism in chronic polyarthritis. The rare allele is associated with protection against destruction. Rheumatology (Oxford) 2000;39:1126-31.. [61] Klein W, Tromm A, Griga T, Fricke H, Interleukin-4 and interleukin-4 receptor gene polymorphisms in inflammatory bowel diseases. Genes Immun 2001;2:287-9.. [62] Aithal GP, Day CP, Leathart J, Daly AK, Hudson M. Association of single nucleotide polymorphisms in the interleukin-4 gene and interleukin-4 receptor gene with Crohn's disease in a British population. Genes Immun 2001;2:44-7.. 46.
(47) [63] Michel J, Gonzales JR, Wunderlich D, Diete A, Herrmann JM, Meyle J. Interleukin-4 polymorphisms in early onset periodontitis. J Clin Periodontol 2001;28:483-8.. [64] Masutani K, Miyake K, Nakashima H, Hirano T, Kubo M, Hirakawa M, Tsuruya K, Fukuda K, Kanai H, Otsuka T, Hirakata H, Iida M. Impact of interferon-gamma and interleukin-4 gene polymorphisms on development and progression of IgA nephropathy in Japanese patients. Am J Kidney Dis 2003;41:371-9.. [65] Blobe GC , Shiemann WP , Lodish H : Role of TGF-ß in human disease . N Engl J Med 2000;342:1350-8.. [66] Taipale J, Saharinen J, Keski-Oja J. Extracellular matrix-associated transforming growth factor-ß: role in cancer cell growth and invasion. Adv Cancer Res 1998;75:87-134.. [67] Ravitz MJ, Wenner CE. Cyclin-dependent kinase regulation during G1 phase and cell cycle regulation by TGF-ß. Adv Cancer Res 1997;71:165-207.. [68] Letterio JJ, Roberts AB. Regulation of immune responses by TGF-ß. Annu. 47.
(48) Rev Immunol 1998;16:137-61.. [69] Wahl SM , Hunt DA , Wong HL , et al. TGF-ß is a potent immunosuppressive agent that inhibits IL-1 dependent lymphocyte proliferation . J Immunol 1988;140: 3026-32.. [70] Tsunawaki S , Sporn M , Nathan C. Deactivation of macrophages by tansforming growth factor-beta. Nature 1988;334:260-2.. [71] Rook AH , Kehrl JH , Wakefield LM , et al. Effects of transforming growth factor-ß on the functions of natural killer cells: Depressed cytolytic activity and blunting of interferon responsiveness. J Immunol 1986;136:3916-3920.. [72] Border WA, Noble NA. Transforming growth factor ß in tissue fibrosis. N Engl J Med 1994;331:1286-92.. [73] Crawford SE, Stellmach V, Murphy-Ullrich JE, et al. Thrombospondin-1 is a major activator of TGF-ß1 in vivo. Cell 1998;93:1159-70.. [74] Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-ß receptor. Nature 1994;370:341-7.. 48.
(49) [75] Nakao A, Imamura T, Souchelnytskyi S, et al. TGF-ß receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J 1997;16:5353-62.. [76] Border WA , Ruoslahti E. TGF-ß in disease: The dark side of tissue repair. J Clin Invest 1992;90:1-6.. [77] Awad MR, El-Gamel A, Hasleton P, Turner DM, Sinnott PJ, Hutchinson IV. Genotypic variation in the transforming growth factor-ß1 gene: association with transforming growth factor-ß1 production, fibrotic lung disease, and graft fibrosis after lung transplantation. Transplantation 1998;66:1014-20.. [78] Yamada Y, Miyauchi A, Goto J, et al. Association of a polymorphism of the transforming growth factor-ß1 gene with genetic susceptibility to osteoporosis in postmenopausal Japanese women. J Bone Miner Res 1998;13:1569-76.. [79] Li B, Khanna A, Sharma V, Singh T, Suthanthiran M, August P. TGF-ß1 DNA polymorphisms, protein levels, and blood pressure. Hypertension 1999;33:271-5.. [80] Grainger DJ, Heathcote K, Chiano M, et al. Genetic control of the circulating concentration of transforming growth factor type ß1. Hum Mol Genet 1999;8:93-7.. 49.
(50) [81] Pfeffer MA, Braunwald E, Moye LA, Basta L, Brown EJ Jr, Cuddy TE, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after acute myocardial infarction. Result of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med 1992;327:669-77.. [82] Yusuf S, Pepine CJ, Graces C, Pouleur H, Salem D, Kostis J, et al. Effects of enalapril on myocardial infarction and unstable angina in patients with low ejection fraction. Lancet 1992;340:1173-8.. [83] The SOLVD investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 1992;327:685-91.. [84] Urata H, Healy B, Stewart RW, Bumpus FM, Husain A. Angiotensin II-forming pathways in normal and failing human hearts. Circ Res 1990;66:883-90.. [85] Dzau VJ. Vascular renin-angiotensin system and vascular protection. J Cardiovasc Pharmacol 1993;22(suppl 5):S1-9.. [86] Cook JL, Chen L, Bhandaru S, Bakris GL, Re RN. The use of antisense. 50.
(51) oligonucleotides to establish autocrine angiotensin growth effects in human neuroblastoma and mesangial cells. Antisense Res Devel 1992;2:199-210.. [87] Oliver JA, Sciacca RR. Local generation of angiotensin II as a mechanism of regulation of peripheral vascular tone in the rat. J Clin Invest 1984;74:1247-51.. [88] Lavoie JL, Sigmund CD. Minireview: overview of the renin-angiotensin system--an endocrine and paracrine system. Endocrinology 2003;144:2179-83.. [89] Hunley TE, Julian BA, Phillips JA 3rd, Summar ML, Yoshida H, Horn RG, Brown NJ, Fogo A, Ichikawa I, Kon V. Angiotensin converting enzyme gene polymorphism: potential silencer motif and impact on progression in IgA nephropathy. Kidney Int 1996;49:571-7.. [90] Schunkert H. Polymorphism of the angiotensin-converting enzyme gene and cardiovascular disease. J Mol Med 1997;75:867-75.. [91] Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F. An insertion/deletion polymorphism of the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest 1990;86,1343-6.. 51.
(52) [92] Mizuiri S, Yoshikawa H, Tanegashima M, Miyagi M, Kobayashi M, Sakai K, Hayashi I, Ikawa A, Ohara T, Hasegawa A. Renal ACE immunohistochemical localization in NIDDM patients with nephropathy. Am J Kidney Dis 1998;31:301-7.. [93] Danser AHJ, Deinum J, Osterop APRM, Admiraal PJJ, Schalekamp MADH. Angiotensin I-to-II conversion in the human forearm and leg. Effect of the ACE gene I/D polymorphism. J Hypertens 1999;17: 1867-72.. [94] Danser AHJ, Koning MMG, Admiraal PJJ, Derkx FHM, Verdouw PD, Schalekamp MADH. Metabolism of angiotensin I by different tissues in the intact animal. Am J Physiol 1992; 263:H418-H428.. [95] Muller DN, Fischli W, Clozel J-P, Hilgers KF, Bohlender J, Menard J, Busjahn A, Ganten D, Luft FC. Local angiotensin II generation in the rat heart. Role of renin uptake. Circ Res 1998;82:13-20.. [96] Van Dijk MA, Kroon I, Kamper AM, Boomsma F, Danser AHJ, Chang PC. The angiotensin-converting enzyme gene polymorphism and responses to angiotensins and bradykinin in the human forearm. J Cardiovasc Pharmacol 2000;35:484-90.. 52.
(53) [97] Staessen JA, Wang JG. Genetic polymorphisms in the renin – angiotensin system: relevance for susceptibility to cardiovascular disease. Eur J Pharmacol 2000;410:289-302. [98] Staessen JA, Ginocchio G, Wang JG, Saavedra AP, Soubrier F, Vlietinck, R, Fagard R. Genetic variability in the renin–angiotensin system: prevalence of alleles and genotypes. J Cardiovasc Risk 1997;4:401-22.. [99] Cantagrel A, Navaux F, Loubet-Lescoulie P, et al. Interleukin-1beta, interleukin-1. receptor. antagonist,. interleukin-4, and interleukin-10. gene. polymorphisms: Relationship to occurrence and severity of rheumatoid arthritis. Arthritis Rheum 1999;42:1093-100.. [100] De Souza A, Trevilatto P, Scarel-Caminaga R, De Brito R, Line S. Analysis of the TGF-beta1 promoter polymorphism (C-509T) in patients with chronic periodontitis. J Clin Periodontol 2003;30:519-23.. [101] Hobbs K, Negri J, Klinnert M, Rosenwasser LJ. Borish L. Interleukin-10 and transforming growth factor-ß promoter polymorphisms in allergies and asthma. Am J Crit Care Med 1998;158:1958-62.. 53.
(54) [102] Yamada Y, Miyauchi A, Takagi Y, Tanaka M, Mizuno M, Harada A. Association of the C-509-T polymorphism, alone or in combination with the T-869-C polymorphism, of the transforming growth factor-beta1 gene with bone mineral density and genetic susceptibility to osteoporosis in Japanese women. J Mole Med 2001;79:149-56.. [103] Tiret L, Rigat B, Visvikis S, et al: Evidence, from combined segregation and linkage analysis, that a variant of the angiotensin I-converting enzyme (ACE) gene controls plasma ACE levels. Am J Hum Genet 1992;51:197-205.. [104] Shanmugan V, Sell KW, Saha BK: Mistyping ACE heterozygotes. PCR Methods Applic 1993;3:120-1.. [105] Lindpaintner K, Pfeffer M, Kreutz R, et al: A prospective evaluation of an angiotensin-converting-enzyme gene polymorphism and the risk of ischemic heart disease. N Engl J Med 1995;332:706-11.. [106] Carlquist JF, Ward RH, Meyer KJ, Husebye D, Feolo M, Anderson JL. Immune Response Factors in Rheumatic Heart Disease: Meta-Analysis of HLA-DR Associations and Evaluation of Additional Class II Alleles. J Am Coil Cardio11995;26:452-7. 54.
(55) [107] Guedez Y, Kotby A, El-Demellawy M, et al.: HLA class II associations with rheumatic heart disease are more evident and consistent among clinically homogeneous patients. Circulation 1999;99:2784-90.. [108] Mansfield JC, Holden H, Tarlow JK, et al. Novel genetic association between ulcerative colitis and the anti-inflammatory cytokine interleukin-1 receptor antagonist. Gastroenterology 1994;106:637-42.. [109] Schrijver HM, Crusius JBA, Uitdehagg BMJ, et al. Association of interleukin-1ß and interleukin-1 receptor antagonist genes with disease severity in MS. Neurology 1999;52:595-9.. [110] Tarlow JK, Cork MJ, Clay FE, et al. Association between interleukin-1 receptor antagonist (IL-1ra) gene polymorphism and early and late-onset psoriasis. Br J Dermatol 1997;136:132-48.. [111] Yucesoy B, Vallyathan V, Landsittel DP, et al. Polymorphisms of the IL-1 gene complex in coal miners with silicosis. Am J Ind Med 2001;39:286-91.. [112] Parkhill JM, Henning BJW, Chapple ILC, Heasman PA, Taylor JJ. Association of interleukin-1 gene polymorphisms with early-onset periodontitis. J. 55.
(56) Clin Periodontol 2000;27:682-9.. [113] Danis VA, Millington M, Hyland VJ, Grennan D. Cytokine production by normal human monocytes: inter-subject variation and relationship to an IL-1 receptor. antagonist. (IL-1Ra). gene. polymorphism.. Clin. Exp. Immunol. 1995;99:303-10.. [114]Arend WP, Malyak M, Guthridge CJ, Gabay C. Interleukin-1 receptor antagonist: role in biology. Annu Rev Immunol.1998; 16:27-55.. [115] Santtila S, Savinainen K, Hurme M. Presence of the IL-1RA allele 2 (IL1RN*2) is associated with enhanced IL-1ß production in vitro. Scand J Immunol 1998;47:195-8.. [116] Vamvakopoulos J, Green C, Metcalfe S. Genetic control of IL-1 I bioactivity through differential regulation of the IL-1 receptor antagonist. Eur J Immunol 2002;32: 2988-96.. [117] Hurme M, Santtila S. IL-1 receptor antagonist (IL-1Ra) plasma levels are co-ordinately regulated by both IL-1Ra and IL-1 genes. Eur J Immunol 1998;28:2598-602.. 56.
(57) [118] El-Omar EM, Carrington M, Chow W-H. et al.: Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000;404:398-402.. [119] Kim SJ, Glick A, Sporn MB, Roberts AB. Characterization of the promoter region of the human transforming growth factor ß1 gene. J Biol Chem 1989;264:402-408.. [120] Shrivastava A, Calame K. An analysis of genes regulated by the multi-functional transcriptional regulatory Yin Yang-1. Nucleic Acids Res 1994;24:5151-5.. [121] De Souza A, Trevilatto P, Scarel-Caminaga R, De Brito R, Line S. Analysis of the TGF-beta1 promoter polymorphism (C-509T) in patients with chronic periodontitis. J Clin Periodontol 2003;30:519-23.. 57.
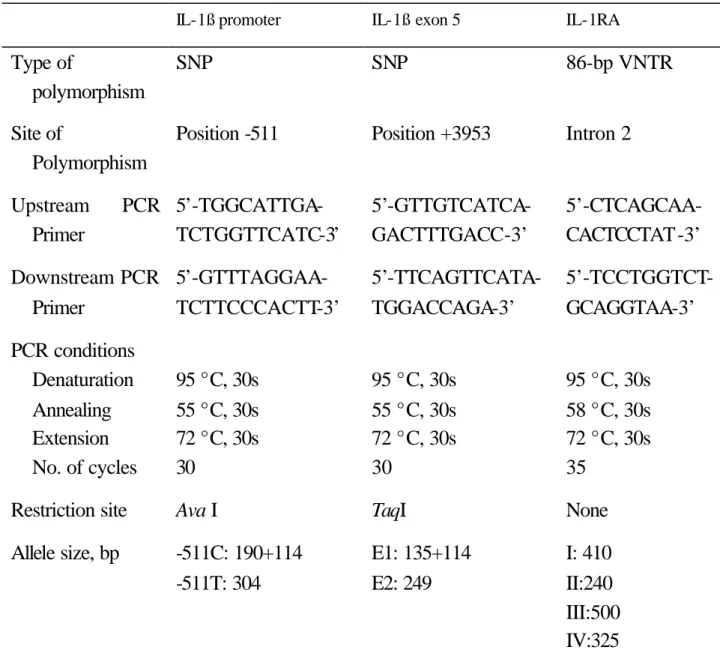
(58) Table 1. Main characteristics of IL-1ß and IL-1RA gene polymorphisms and techniques used for screening IL-1ß promoter. IL-1ß exon 5. IL-1RA. Type of polymorphism. SNP. SNP. 86-bp VNTR. Site of Polymorphism. Position -511. Position +3953. Intron 2. 5’-GTTGTCATCAGACTTTGACC-3’. 5’-CTCAGCAACACTCCTAT-3’. Downstream PCR 5’-GTTTAGGAAPrimer TCTTCCCACTT-3’. 5’-TTCAGTTCATATGGACCAGA-3’. 5’-TCCTGGTCTGCAGGTAA-3’. PCR conditions Denaturation Annealing Extension No. of cycles. 95 °C, 30s 55 °C, 30s 72 °C, 30s 30. 95 °C, 30s 55 °C, 30s 72 °C, 30s 30. 95 °C, 30s 58 °C, 30s 72 °C, 30s 35. Restriction site. Ava I. TaqI. None. Allele size, bp. -511C: 190+114 -511T: 304. E1: 135+114 E2: 249. I: 410 II:240 III:500 IV:325 V:595. Upstream Primer. PCR 5’-TGGCATTGATCTGGTTCATC-3’. IL-1ß: interleukin-1ß; IL-1RA: interleukin-1 receptor antagonist; SNP: single nucleotide polymorphism; VNTR: variable number of tandem repeats.. 58.
(59) Table 2. Genotype distribution of IL-1ß promoter in MS patients and normal controls Genotype IL-1ß promoter. CC. Control MS. CT. TT. Total. 26(25.2%) 53(51.5%). 24(23.3%). 103 (100%). 26(31.0%) 42(50.0%). 16(19.0%). 84 (100%). Chi-square test: x2 = 0.953, p = 0.621. Table 3. Allele frequency of IL-1ß promoter in MS patients and normal controls Allele IL-1ß promoter. C. T. Total. Control. 105(51.0%). 101(49.0%). 206 (100%). MS. 94 (56.0%). 74 (44.0%). 168 (100%). Chi-square test: x2 = 0.922, p = 0.196. Table 4. Genotype distribution of IL-1ß exon 5 in MS patients and normal controls Genotype IL-1ß exon 5. E1E1. E1E2. Total. Control. 98(95.1%). 5(4.9%). 103 (100%). MS. 80(95.2%). 4(4.8%). 84 (100%). Fisher exact test: x2 < 0.001, p = 0.626. 59.
(60) Table 5. Allele frequency of IL-1ß exon 5 in MS patients and normal controls Allele IL-1ß exon 5. E1. E2. Total. Control. 201(97.6%). 5(2.4%). 206 (100%). MS. 164 (97.6%). 4(2.4%). 168 (100%). Fisher exact test: x2< 0.001, p = 0.626. Table 6. Genotype distribution of IL-1RA in MS patients and normal controls Genotype IL-1RA. I/I. I/II. II/II. Total. Control. 96 (93.2%). 6 (5.8%). 1 (0.9%). 103 (100%). MS. 68 (81.0%). 16(19.0%). 0. 84 (100%). Fisher exact test: x2 = 8.483, p = 0.014; Odds ratio for genotype I/II:3.80. Table 7. Allele frequency of IL-1RA in MS patients and normal controls Allele IL-1RA. I. II. Total. Control. 198 (96.1%) 8 (3.9%). 206 (100%). MS. 152 (90.5%) 16(9.5%). 168 (100%). Chi-square test: x2 = 4.902, p = 0.023; Odds ratio for allele II:2.61. 60.
(61) Table 8.. Allele linkage of the 3 IL-1 gene polymorphisms in MS patients IL-1RA II (+). IL-1RA II (-). p. IL-1ß promoter C (+) IL-1ß promoter C (-). 13 3. 55 13. NS. IL-1ß exon 5 E2 (+) IL-1ß exon 5 E2 (-). 1 15. 3 65. NS. Table 9. Genotype distribution of IL-4 intron3 in MS patients and normal controls Genotype IL-4 intron 3. RP1/RP1. RP1/RP2. RP2/RP2. Total. Control. 72(69.9%). 29(28.2%). 2(1.9%). 103 (100%). MS. 57(67.9%). 25(29.8%). 2(2.4%). 84 (100%). Fisher exact test: x2< 0.001, p = 0.626. Table 10. Allele frequency of IL-4 intron3 in MS patients and normal controls Allele. RP1. RP2. Total. Control. 173(84.0%). 33(16.0%). 206 (100%). MS. 139(82.7%). 29(17.3%). 168 (100%). IL-1RA. Chi-square test: x2 = 0.46, p = 0.49. 61.
(62) Table 11. Genotype distribution of TGF-ß promoter(-509) in MS patients and normal controls Genotype. T/T. T/C. C/C. Total. Control. 32(31.1%). 47(45.6%). 24(23.3%) 103 (100%). MS. 21(25.0%) 60(71.4%) 3(3.6%). TGF-ß(-509). Chi-square test: x2= 17.92. 84 (100%). , p < 0.001. Table 12. Allele frequency of TGF-ß promoter(-509) in MS patients and normal controls Allele TGF-ß(-509). T. C. Total. Control. 111(53.9%). 95(46.1%). 206 (100%). MS. 102(60.7%). 66(39.3%). 168 (100%). Chi-square test: x2 = 1.73 , p > 0.1. 62.
(63) Table 13. Genotype distribution of ACE intron 16 in MS patients and normal controls Genotype ACE intron 16. I/I. D/D. I/D. Total. Control. 38(36.9%). 12(11.7%). 53(51.5%). 103 (100%). MS. 36(42.9%). 14(16.7%). 34(40.5%). 84 (100%). Chi-square test: x2 = 2.46. , p = 0.29. Table 14. Allele frequency of ACE intron 16 in MS patients and normal controls Allele ACE intron 16. I. D. Total. Control. 129(62.6%). 77(37.4%). 206 (100%). MS. 106(63.1%). 62(36.9%). 168 (100%). Chi-square test: x2 < 0.01. , p > 0.9. 63.
(64) Figure 1. Gel electrophoresis of IL-1ß promoter sequence after Ava I digestion. TT. CT. CC. TT. M. 304bp 190bp 114bp. Figure 2. Gel electrophoresis of IL-1ß exon 5 sequence after Taq I digestion. M. E1/E2. E1/E1 E2/E2. 249bp. 135bp 114bp. 64.
(65) Figure 3. Gel electrophoresis of IL-1RA intron 2 sequences M. M. 595bp 410bp 325bp 240bp. 65.
(66) 作者簡歷 姓名:李秉純 出生 : 民國 48 年 11 月 16 日 學歷:國立陽明醫學院醫學系畢業 經歷: 1983-1984. 台北榮民總醫院實習醫師. 1984-1990. 台北榮民總醫院外科部住院醫師 台北榮民總醫院心臟血管外科主治醫師. 1991 1990-1992. 宜蘭員山榮民醫院代外科主任. 1992-1997. 台中榮民總醫院心臟血管外科主治醫師. 1994-1995. 荷蘭鹿特丹大學醫院心臟外科研究員. 1997-1998. 中山醫學院附設醫院心臟血管外科主任. 1998-迄今. 中國醫藥學院附設醫院心臟血管外科主任. 現任:中國醫藥學院附設醫院心臟血管外科主任 資格: 1989. 中華民國外科醫學會專科醫師. 1990. 中華民國胸腔及心臟血管外科專科醫師. 1993. 中華民國心臟學會專科醫師. 66.
(67)
數據


相關文件
Laser Microdissection Microdissection for pure DNA, RNA, for pure DNA, RNA, Proteins and Living Cells Proteins and Living
the lymphocyte function-associated antigen 1, or LFA-1, was so named because antibodies recognizing this structure interfere with lymphocyte cell adhesion events and
1.4 Exponential and Logarithmic Functions 1.5 Finding Limits Graphically and Numerically 1.6 Evaluating Limits Analytically.. 1.7 Continuity and One-Sided Limits 1.8
mathematical statistics, statistical methods, regression, survival data analysis, categorical data analysis, multivariate statistical methods, experimental design.
Oral and maxillofacial metastasis of male breast cancer: Report of a rare case and literature review
Less than 1% of all breast cancers occur in male patients, and to date, only 8 cases of metastatic breast adeno- carcinoma to the oral and maxillofacial region in a male patient
Higher immunoexpression of HIF-1 a, NOTCH1, ADAM-12, and heparin-binding epidermal growth factor like growth factor (HB-EGF) in epidermoid cells in compari- son with mucous cells
Orthokeratinized odontogenic cyst with an associated keratocystic odontogenic tumor component and ghost cell Table 1 Previous case reports of multiple orthokeratinized
Unit 1 Unit 1 - How many people are in your familyA. Read
