ARTICLES
Excited-State Double-Proton Transfer on 3-Methyl-7-azaindole in a Single Crystal:
Deuterium Isotope/Tunneling Effect
Wei-Shan Yu,† Chung-Chih Cheng,‡ Chen-Pin Chang,‡ Guo-Ray Wu,† Chin-Hao Hsu,†and Pi-Tai Chou*,†,§
Department of Chemistry, National Chung-Cheng UniVersity, Chia-Yi, Taiwan, Republic of China, Department of Chemistry, Fu Jen Catholic UniVersity, Shin Chuang, Taiwan, Republic of China, and Department of Chemistry, National Taiwan UniVersity, Taipei, Taiwan, Republic of China
ReceiVed: February 12, 2002; In Final Form: June 8, 2002
Unlike 7-azaindole consisting of the tetrameric configuration, 3-methyl-7-azaindole (3MAI) exists solely as intact double hydrogen-bonded dimeric forms in a single crystal. Both steady-state and time-resolved measurements down to 8.0 K reveal remarkable deuterium isotope effects on the rate of excited-state double proton transfer (ESDPT) in the N(1)-deuterated 3MAI (3MAI-d) single crystal. The rates of ESDPT for the 3MAI-d dimer resolved at <150 K are mainly governed by the proton tunneling mechanism. At <12 K, the nearly temperature-independent ESDPT dynamics lead us to qualitatively deduce a barrier height of∼1.73 kcal/mol for the 3MAI-d dimer. The results provide an ideal model to investigate the intrinsic ESDPT dynamics for 7-azaindole analogues in which the structural information is well documented.
1. Introduction
The excited-state double proton transfer (ESDPT)1-3in the 7-azaindole (7AI) dimer (Figure 1) has long been recognized as a prototype to mimic the photoinduced mutation of the DNA base pair.4,5 Much research has focused on the dynamics of ESDPT, especially in solution phase.6-10 Early studies have revealed that the rate of proton transfer is within 0.3-1.0× 1012s-1 for the 7AI dimer in nonpolar solvents.6,7 Recently, 0.2-ps and 1.1-ps dynamics were resolved by Takeuchi and Tahara8 and first assigned to the rates of internal conversion and proton transfer, respectively, based on a concerted ESDPT model. Subsequently, Chachisvilis et al.9reported the 1-ps and 12-ps dynamics and interpreted on the basis of nonconcerted double proton-transfer reaction, but it was then claimed that the 12-ps component is due to the vibrational cooling.10Then, Fiebig et al. reassigned the 0.2-ps and 1-ps components to the two-steps double proton transfer11 and concluded that both trajectories of the symmetric and asymmetric vibrational motion coupled with the solvent dynamics must be considered for the ESDPT in the solution phase. Only when the internal energy is small, such as at sufficiently low temperatures, can one examine the process of tunneling and nonconcertedness. Despite different viewpoints on the mechanism, both claimed observation of the kinetic isotope effects on the rate of ESDPT at ambient temperature.
Temperature-dependent ESDPT dynamics on 7AI were once suggested to be inaccessible due to the formation of thermo-dynamically more favorable oligomers at low temperature.12,13
In a 77 K methylcyclohexane glass containing concentrated 7AI, the dominant steady-state emission maximum at∼460 nm was reported to originate from phosphorescence of the oligomers in which the cooperative double proton transfer is prohibited.12,13 Recently, via a clever approach of applying sufficiently low 7AI concentrations, Catala´n and Kasha14were able to resolve the dimeric form at low temperature and to study its
corre-* To whom correspondence should be addressed.
†Department of Chemistry, National Chung-Cheng University. ‡Department of Chemistry, Fu Jen Catholic University. §Department of Chemistry, National Taiwan University.
Figure 1. Differences in photophysical properties for the 7AI-associated structures: a. the dual hydrogen-bonded dimer (in solution) and b. tetrameter (in a single crystal).
10.1021/jp020411x CCC: $22.00 © 2002 American Chemical Society Published on Web 08/09/2002
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
sponding proton-transfer spectroscopy. Instead of the exclusive proton-transfer tautomer fluorescence in the 7AI dimer, the appearance of normal dimeric fluorescence at <200 K for the deuterated 7AI rendered a convincing deuterium isotope effect on the ESDPT dynamics14 (hereafter N(1)-protonated and deuterated-7AI analogues are denoted by the suffix -h and -d, respectively). However, from the thermodynamic point of view, the hydrogen-bonding equilibrium should be dominated by the enthalpic factor and hence favors the oligomer formation at sufficiently low temperatures (e.g., 77 K and lower). It is thus rather difficult to determine the lower-limit temperature at which the existence of dual hydrogen-bonded dimer is free from the interference of oligomers. This in combination with possible perturbations resulting from solvent dielectric relaxation and/ or viscosity (e.g., solvent cage) may further smear and com-plicate the ESDPT dynamics.
From yet another approach, the hydrogen bond in the solid state can be well-characterized.15It has been demonstrated that the hydrogen-bonding structure in the single crystal provides an ideal model for investigating proton-transfer dynamics free from the solvent perturbation.16,17 Unfortunately, the crystal structure of 7AI reveals tetrameric units of approximate S4 symmetry, in which molecules are associated by means of four complementary N-H‚ ‚ ‚N hydrogen bonds18(Figure 1b). As a result the cooperative ESDPT is prohibited. Recently, we have found that chemical modification at the C(3)-position of 7AI, forming 3-iodo-7-azaindole (3IAI), exhibited a dimeric structure in the single crystal. Steady-state approaches have shown the dominant ESDPT process even at 10 K for the 3IAI-h dimer, resulting in a unique tautomer emission.19Unfortunately, studies on ESDPT dynamics of 3IAI, particularly the deuterium isotope effect, were rather difficult due to its photochemical liability as well as fast S1-T1 intersystem crossing enhanced by the iodine heavy atom effect.19,20Thus, chemical modifications on 7AI suitable for investigating intrinsic ESDPT in the single-crystal became one of our focuses. In this study 3-methyl-7-azaindole (3MAI, Figure 2) was synthesized, of which the photostability is superior to that of 3IAI. Furthermore, unlike 3IAI where the association of iodine substituents induces strong perturbation due to the enhanced spin-orbit coupling and resonance effects, the much more inert methyl substitution in 3MAI provides an ideal model to mimic the intrinsic ESDPT dynamics of the 7AI dimer.
2. Experimental Section
2.1. Materials. Synthesis of 3-methyl-7-azaindole (3MAI). A precursor 7-azaindole-3-carboxaldehyde has to be prepared prior to the synthesis of 3MAI, which was synthesized according to the previous report.21Subsequently, a solution of semicarbazide
hydrochloride (1.35 g) and sodium acetate trihydrate (2.3 g) was added to a solution containing 7-azaindole-3-carboxalde-hyde (0.80 g) in 80 mL of boiling water. After the appearance of a transitory yellow coloration, a white solid gradually precipitated. The mixture was then heated on the steam-bath for ∼15 min, then cooled to yield semicarbazone (1.1 g). Semicarbazone (1.1 g) was added to a solution containing sodium (0.96 g) in dry diethylene glycol (40 mL). The solution was refluxed at 205°C under nitrogen atmosphere for 1h, and the mixture was cooled and poured into ∼230 mL of water containing 3.1 g of glacial acetic acid. The resulting solution, which contained solid in suspension, was extracted with ether. The extracts were then washed with water, dried, and evapo-rated. The crude product was chromatographed on silica gel (eluent: ethyl acetate) to obtain 0.41 g (55%) 3MAI.1H NMR (400 MHz), δ 9.66 (s, 1H), 8.28 (dd, J ) 1.6, 4.8 Hz, 1H), 7.89 (dd, J ) 1.4, 7.6 Hz, 1H), 7.08(s, 1H), 7.04 (dd, J ) 4.4, 8.0 Hz, 1H), 2.31(s, 3H). A single crystal of 3MAI-h with dimensions of∼0.50 × 0.45 × 0.45 mm3was obtained by slow evaporation in a CH3OH solution. To obtain the 3MAI-d single crystal, a similar recrystalization procedure was repeated three times in the CD3OD solution under the N2atmosphere. After monitoring the disappearance of the N(1)-H proton in1H NMR (in CDCl3), we concluded that∼95% of the N-H proton had been deuterated. The transfer of 3MAI-d to the cryostat was handled in a N2filled drybox to avoid the D/H exchange on the surface of the crystal.
The compound 1,3-dimethyl-7-azaindole (3MM(1)AI, Figure 2) was synthesized by adding sodium hydride (57%, 60 mg) to the THF solution containing 3MAI (0.15 g), followed by the addition of methyl iodide (30 mg).1H NMR (CDCl
3, 400 MHz), δ 2.32 (s, 3H), 4.10 (s, 3H), 7.0 (s, 1H), 7.17 (t, 1H), 8.05 (d, J ) 7.2 Hz, 1H), 8.33 (d, J ) 5.2 Hz, 1H). The compound 3,7-dimethyl-1H-pyrrolo[2,3-b]pyridine (3MM(7)AI, Figure 2) was synthesized by the reaction of 3MAI (0.12 g) and CH3I (0.58 g) in THF under a N2atmosphere. NaOH (2.5 N, 3 mL) was then added and the mixture was stirred for∼20 min to obtain 3MM(7)AI (60 mg).1H NMR (CDCl
3, 400 MHz),δ 2.37 (s, 3H), 4.74(s, 3H); 7.37(t, 1H); 7.55(s, 1H); 7.98(d, J ) 7.64 Hz, 1H); 8.34(d, J ) 6.8 Hz, 1H).
2.2. Measurements. A closed-helium-cycle cryostat (Oxford, Model CCC1104) was used for the temperature-dependent study in the range of 298-8 K. The solid sample (i.e., single crystal) was placed tightly between two thin quartz plates that came into close contact with the Indium part of the cooling compart-ment to ensure maximum thermal conduction. The solid sample was excited by the fourth harmonic (266 nm) of the Nd:YAG laser under a front-face configuration. The resulting fluorescence was detected by an intensified charge coupled detector (ICCD, Princeton Instrument, Model 576G/1) coupled with a polychro-mator in which the grating is blazed with a maximum of 500 nm. Occasionally, to obtain excitation spectra the output of an Nd:YAG (355 nm, 8 ns, Continuum Surlite II) pumped optical parametric oscillator was tuned between 650 and 720 nm and was then frequency doubled by a BBO crystal to obtain a tunable 325-360 nm excitation frequency. The intensity of the laser pulse was measured by a joule meter, which was then normal-ized to obtain the corrected excitation spectra. A combination of filters was used in front of the exit polychromator to isolate the emission wavelengths of interest. The crystal orientation was 45°with respect to the direction of the excitation light.
Detailed fluorescence lifetime measurement has been de-scribed in the previous report.22Briefly, a third harmonic of the Ti-Sapphire oscillator (100 fs, 82 MHz, Spectra Physics) Figure 2. Structures of 3MAI and their corresponding proton-transfer
isomer as well as methylated derivatives.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
coupled with a pulse picker (NEOS, model N17389) was used as an excitation source, giving a tunable wavelength in the range of 255-275 nm with a repetition rate of 8 MHz. An Edinburgh OB 900-L single-photon counter was used as a detecting system. The resolution of the time-correlated photon-counting system is limited by the detector response of∼30 ps. The fluorescence decays were analyzed by the sum of exponential functions with an iterative convolution method reported previously,23which allows partial removal of the instrument time broadening and consequently renders a temporal resolution of∼15 ps. 3. Results
3.1. Structure of 3MAI in a Single Crystal. As shown in Figure 3, the crystal structure of 3MAI-h is composed of a cyclic dimer via dual N-H‚‚‚N hydrogen bonds. Parallel cyclic dimers are stacked via the interaction between their ringπ-systems to form alternating slabs. The orientation of the parallel molecules in one slab is nearly perpendicular to those in adjacent slabs. The space group is p21/C, and Z ) 8. Thus, there are two kinds of crystallographically independent molecules. Each of them forms a hydrogen-bonding dimer by the operation of the inversion center, while there is no symmetry relation between the two forms of dimers. To examine whether these two kinds
of hydrogen-bonding dimers (specified as dimer 1 and dimer 2) are in the same circumstances in the single crystal, we have closely examined these two components in the X-ray data. The results of X-ray structural analyses are shown in Figure 3 and Table 1. The sameness in configuration of these two hydrogen-bonded dimers under different spatial arrangements can be supported by several critical bond lengths and angles shown in Table 1. For example, N(1A)‚‚‚N(2) and N(1A)‚‚‚H(2)N(2) were calculated to be 2.980 and 2.131 Å, respectively, for dimer 1. In addition, the angle of N(1A)‚‚‚H(2)N(2) was calculated to be 170.9°. In comparison, similar results of 2.979 Å (N(3A)‚‚ ‚N(4)) and 2.121 Å (N(3A)‚‚‚H(4)N(4)) with an N(3A)‚‚‚H(4)N-(4) angle of 171.4°were obtained for dimer 2. These, in combination with similar bond angles and bond distances among the rest of the atoms between dimer 1 and 2 (see Table 1) indicate that within the standard deviation these two hydrogen-bonding dimers are in the same configuration and hence should behave in practically the same way.
To ensure the same crystal structure in both fluorescence and X-ray measurements, we first determined the structure by X-ray spectroscopy, followed by performing temperature-dependent fluorescence spectroscopy and dynamics of the same single crystal. During the fluorescence measurement an ultrahigh vacuum was maintained in the cryostat so that the D/H isotope exchange was negligible. In the case of 3MAI-d, great care was taken to perform the X-ray measurement in dry air conditions, and the results are attached in the Supporting Information. Comparing X-ray data shown in Table 1 and Supporting Information, it is obvious that single crystals of 3MAI-h and 3MAI-d used in the fluorescence studies are isomorphous. The X-ray crystallography was also performed at -80°C. The results showed negligible differences in structure from that obtained at 295 K, indicating no temperature-dependent structural transformation for 3MAI in a single crystal.
Figure 3. a. The projection of 3MAI-h dimer in a unit cell of the single crystal. b. Two types of spatially arranged dimeric forms, specified as dimer 1 and 2. Bond distance and angle are in angstroms and degree (degrees), respectively.
TABLE 1: Bond Lengths [Ångstroms] and Angle [Degrees] for 3MAI-h in a Single Crystal
Dimer 1 N(1)-C(5) 1.332(2) N(1)-C(1) 1.335(2) N(2)-C(5) 1.365(2) N(2)-C(7) 1.368(2) C(1)-C(2) 1.387(2) C(2)-C(3) 1.377(2) C(3)-C(4) 1.394(2) C(4)-C(5) 1.416(2) C(4)-C(6) 1.425(2) C(6)-C(7) 1.367(2) C(6)-C(8) 1.502(2) N(2)-H(2) 0.856(2) N(1A)‚‚‚H(2) 2.131(2) N(1A)‚‚‚N(2) 2.980(2) C(5)-N(1)-C(1) 113.95(14) C(5)-N(2)-C(7) 108.30(14) N(1)-C(1)-C(2) 124.99(16) C(3)-C(2)-C(1) 119.98(16) C(2)-C(3)-C(4) 117.77(15) C(3)-C(4)-C(5) 116.74(14) C(3)-C(4)-C(6) 135.71(15) C(5)-C(4)-C(6) 107.54(14) N(1)-C(5)-N(2) 125.88(14) N(1)-C(5)-C(4) 126.57(14) N(2)-C(5)-C(4) 107.54(14) C(7)-C(6)-C(4) 105.41(14) C(7)-C(6)-C(8) 128.49(16) C(4)-C(6)-C(8) 126.10(15) C(6)-C(7)-N(2) 111.22(15) N(1A)‚‚‚H(2)-N(2) 170.90 Dimer 2 N(3)-C(13) 1.335(2) N(3)-C(9) 1.335(2) N(4)-C(13) 1.366(2) N(4)-N(15) 1.368(2) C(9)-C(10) 1.390(2) C(10)-C(11) 1.376(2) C(11)-C(12) 1.386(2) C(12)-C(13) 1.415(2) C(12)-C(14) 1.434(2) C(14)-C(15) 1.365(2) C(14)-C(16) 1.500(2) N(4)-H(4) 0.865(2) N(3A)‚‚‚H(4) 2.121(2) N(3A)‚‚‚N(4) 2.979(2) C(13)-N(3)-C(9) 113.75(14) C(13)-N(4)-C(15) 108.42(14) N(3)-C(9)-C(10) 125.03(16) C(11)-C(10)-C(9) 119.82(15) C(10)-C(11)-C(12) 117.89(15) C(11)-C(12)-C(13) 117.01(14) C(11)-C(12)-C(14) 135.84(14) C(13)-C(12)-C(14) 107.15(13) N(3)-C(13)-N(4) 125.77(14) N(3)-C(13)-C(12) 126.50(14) N(4)-C(13)-C(12) 107.71(14) C(15)-C(14)-C(12) 105.55(14) C(15)-C(14)-C(16) 128.67(17) C(12)-C(14)-C(16) 125.77(15) C(14)-C(15)-N(4) 111.16(15) N(3A)‚‚‚H(4)-N(4) 171.4
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
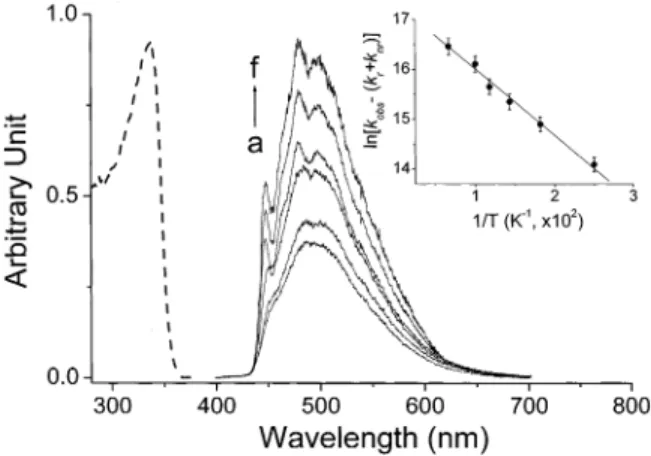
3.2. Spectroscopy and Dynamics in the Single Crystal. In contrast to a normal fluorescence (λmax∼ 370 nm) observed in 7AI,19 3MAI-h in a single crystal exhibited a unique, large Stokes-shifted fluorescence (λmax∼ 500 nm) throughout 298-8.0 K (Figure 4). In comparison, 3MM(1)AI (see Figure 2), which serves as a nonproton-transfer model for 3MAI, revealed a fluorescence with a peak maximum at 410 nm (τf∼ 0.9 ns) in the solid state. Conversely, 3MM(7)AI, a model compound of the proton-transfer tautomer, exhibited room-temperature fluorescence maximized at 510 nm (Figure 5,τf∼ 2.1 ns), of which the spectral and dynamical features resemble those observed in the 3MAI-h single crystal. These steady-state approaches unambiguously lead to two concluding remarks: 1. ESDPT is apparently operative in the 3MAI-h single crystal, resulting in a 500-nm proton-transfer tautomer emission. 2. Due to the lack of any observable normal emission, the rate of ESDPT must be fast even at a temperature of as low as 8.0 K. The latter viewpoint was supported by the subsequent time-resolved measurement. When monitored at the 500-nm emission band, the rise component of the tautomer fluorescence was too fast to be resolved (, 15 ps) throughout 298-8.0 K. Con-versely, the decay of the tautomer emission can be well fitted by single exponential kinetics. The lifetime-revealed slight temperature dependence, varying from 2.7 (298 K) to 7.4 ns (8.0 K). From the standpoint of a temperature-independent
fluorescence radiative decay rate, the result simply indicates the decrease in nonradiative deactivation rates (vide infra), which is consistent with the steady-state measurement, showing that the intensity of the tautomer emission increased as the temper-ature decreased (see Figure 4a-f).
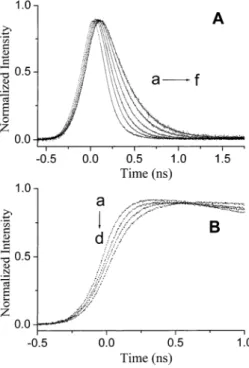
Remarkable temperature-dependent fluorescence spectra were observed for the 3MAI-d single crystal. Dual emission became obvious at <200 K, in which a normal emission band (specified as the F1band) maximized at 395 nm gradually appeared (Figure 6). Conversely, the 500-nm tautomer emission (specified as the F2band) intensity versus temperature was slightly irregular (see insert of Figure 6); it increased upon decreasing the temperature from 298 to 190 K, followed by a decrease in the intensity at 150-120 K. At < 12 K the intensity as well as the ratio of dual emission was nearly temperature independent. By tuning the excitation wavelength, the resulting excitation spectrum, within experimental error, was independent of the emission wavelength monitored at either the F1or F2band, which is also effectively identical with the absorption profile (see Figure 6). In accordance with the steady-state approach, a remarkable deuterium isotope effect on the ESDPT dynamics was also observed in the 3MAI-d single crystal. While the rise component of the F1band was beyond the instrument response of∼ 15 ps at 298-8.0 K, at <150 K the decay was resolvable and revealed drastic temperature dependence. As shown in Figure 7A and Table 2, the results clearly showed a decrease of the single-exponential decay rate constant, kobsN, upon decreasing the temperature. For instance, kobsNmeasured to be 3.8× 109s-1 at 8 K is smaller than that of 2.6× 1010s-1measured at 100 K by∼7-fold. Conversely, the fluorescence dynamics of the F2 band resolved at <150 K could only be fitted by dual exponential kinetics, which consist of a rise and a decay component as indicated by the fitted negative and positive preexponential factors, respectively. When monitored at >520 nm region free from the interference of the normal emission, the absolute values for both rise and decay preexponential factors, within experimental error, are similar (see Table 2). Slight differences are possibly attributed to the existence of∼5% of nondeuterated 3MAI-h in which fast ESDPT takes place. Therefore, the fitted preexponential value for the decay com-ponent is greater than that for the rise comcom-ponent. Table 2 also Figure 4. (s) The fluorescence of the 3MAI-h crystal as a function
of temperature at a. 298, b. 240, c. 195, d. 90, e. 50, and f. 8 K. (- -) The absorption spectra of 3MAI-h in a solid form at room temperature. Inset: The plot of ln[kobs - (kr+ knr)] versus the reciprocal of the temperature.
Figure 5. The fluorescence and fluorescence excitation spectra of 3MM(1)AI (s) and 3MM(7)AI (- -) in the crystal form at ambient temperature.
Figure 6. (s) The fluorescence of 3MAI-d crystal as a function of temperature at a. 298, b. 200, c. 165, d. 135, e. 110, f. 85, g. 35, h. 12, and i. 8 K. The excitation spectrum of the 3MAI-d crystal monitored at the F2(• •) and F1bands (o o). The excitation wavelength was tuned from 360 to 325 nm. (- -) The absorption spectra of 3MAI-d in a solid form at room temperature. Inset: The plot of tautomer emission intensity at 500 nm versus the temperature (in Kelvin).
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
shows an interesting correlation between decay and rise components for the F1and F2 bands, respectively, at various temperatures, in which the rise time of the F2 band, within experimental error, was the same as the decay time of the F1 band. In summary, the time-resolved measurement in combina-tion with steady-state approaches led us to conclude that both F1(normal emission) and F2(tautomer emission) bands originate from the same ground-state species, i.e., the normal dimeric form, and the F1band is apparently the precursor of the F2band. Thus, the existence of a nonnegligible ESDPT barrier for the 3MAI-d dimer is apparent, and the deuterium isotope effect becomes significant on the rate of ESDPT at sufficiently low temperatures. The irregular temperature-dependent tautomer emission intensity for 3-MAI-d can thus be rationalized by two competing relaxation processes: 1. As the temperature de-creases, the decrease of the ESDPT rate results in a smaller yield of population to the tautomer state, 2. The tautomer fluorescence quantum yield tends to increase at lower temper-ature due to the decrease of the rate of radiationless deactiva-tions. Details on the relaxation dynamics will be discussed in the following section.
4. Discussion
For the F2band, the observed temperature-dependent decay rate can be expressed as
where knr denotes the temperature independent radiationless decay rate constant, possibly involving internal conversion, intersystem crossing, etc. The temperature-dependent radiation-less decay rate constant knr(T) can be further expressed as an Arrhenius type of thermally deactivated pathway, namely, knr -(T) ) Ae-Ea/RT where Ea is the barrier for the thermally
deactivated process. As indicated by the time-resolved measure-ment, the lifetime of the tautomer emission (the F2band) was invariable at <12 K. Thus, it is reasonable to assume that knr -(T)is negligible, and the decay rate of 1.36× 108s-1measured at, e.g., 8 K is equivalent to kr+ knr. The plot of ln[kobs- (kr + knr)] versus the reciprocal of the temperature reveals straight-line behavior (see insert of Figure 4), supporting the validity of the thermal deactivation pathway for the tautomer emission. A best linear least-squares fit for the insert of Figure 4 gives Ea and the frequency factor to be 0.35 kcal/mol and 5.2 × 108 s-1, respectively.
For the F1band, the observed temperature-dependent decay rate can be expressed as
where knr(T)denotes the thermally deactivated pathways except for the rate of proton transfer kpt(T). It is not feasible to obtain kr+ knr+ knr(T) for the normal emission (i.e., the F1band) of 3MAI in a single crystal because the dynamics of relaxation of the excited normal species are still dominated by the rate of double proton-transfer reaction even at low temperatures. Alternatively, we have attempted to obtain kr + knr + knr(T) values by performing the temperature-dependent relaxation dynamics of the 3MM(1)AI crystal in which the excited-state proton-transfer reaction is prohibited due to the lack of pyrrolic hydrogen. The results showed a slightly temperature-dependent decay rate from 4× 108s-1 (150 K) to 1.1× 108s-1(8 K). Taking the kobsvalue of the 3MM(1)AI crystal to be kr+ knr+ knr(T) for the 3MAI-d dimer, a plot of ln[kobs- (kr + knr + knr(T))] versus 1/T is shown in Figure 8. Apparently, the contribution of kr+ knr+ knr(T) is rather small in this plot simply due to its much smaller value in comparison to the rate of double proton-transfer reaction. If the ESDPT dynamics, kpt(T), mainly Figure 7. A. The time-dependent fluorescence decay dynamics of the
3MAI-d crystal monitored at the F1 band (380 nm) at various temperatures of a. 150, b. 100, c. 70, d. 40, e. 20, and f. 10K. B. The rise dynamics of the F2band (520 nm) monitored at a. 190, b. 150, c. 70, and d. 10 K.
TABLE 2: Temperature-Dependent Fluorescence Relaxation Dynamics for 3MAI-d in a Single Crystal
fluorescence lifetime F2(λmax) 500 nm)a temperature (K) F1(λmax)395 nm) τ (ns) τ1(ns) a1 τ2(ns) a2 150 NA 6.66 1.000 100 0.038 0.040 -0.208 6.80 0.239 70 0.061 0.060 -0.205 7.15 0.235 40 0.127 0.130 -0.010 7.28 0.014 20 0.210 0.226 -0.011 7.33 0.017 10 0.260 0.260 -0.011 7.36 0.013 8 0.263 0.265 -0.011 7.38 0.015 aThe F
2band was fitted by dual exponential kinetics expressed as
F(t) ) a1e-τ1-1t+ a2e-τ2-1t.
Figure 8. The plot of ln[kobs- (kr+ knr+ knr(T))] versus the reciprocal of the temperature for a. the decay of the F1band (2) and b. rise kinetics of the F2band (O) in a 3MAI-d single crystal at e150 K. See text for the definition of kr+ knr+ knr(T).
kobs) kr+ knr+ knr(T) (1)
kobs) kr+ knr+ knr(T) + kpt(T) (2)
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
incorporate a barrier crossing, i.e., a classical thermally activated process, straight-line behavior is expected upon plotting ln[kobs - (kr + knr + knr(T))] as a function of the reciprocal of the temperature. In contrast, the plot depicted in Figure 8a revealed an asymptote-like curve, which approached a constant value at <12 K. A similar pattern was also obtained when the rise dynamics of the F2band (i.e., the tautomer emission) are plotted against the reciprocal of the temperature (see Figure 8b).
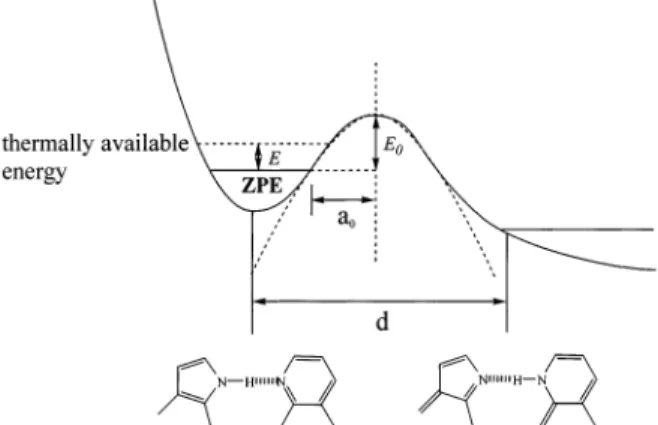
The concave asymptote for the kpt(T)-versus-1/T plot in combination with a nearly constant ESDPT rate at 8-12 K led us to conclude that the dynamics of ESDPT for the 3MAI-d crystal are not governed by a thermally activated process, but more plausibly proceed through a proton-tunneling mechanism. To apply such theories, simplified approximation about the ESDPT potential energy surface (PES) is necessary. Both steady-state and dynamic approaches indicate that the ESDPT reaction in the 3MAI dimer is exergonic and irreversible. Accordingly, a simplified one-dimensional PES regarding the ESDPT can be represented by a bound-free potential curve shown in Figure 9, in which the proton-tunneling rate is given by
where V(x) is the energy barrier along the reaction coordinate, ν is the bound vibrational frequency, i.e., the reaction-coordinate frequency of the N-H (or N-D) mode in the case of 3MAI, m is the effective mass of the proton or deuterium, and a0is the distance from the center of the barrier to the classical vibrational turning points. Equation 3 can easily be evaluated if the potential barrier is further represented by a semiempirical, one-dimen-sional approach based on a parabolic shape-like activation energy, i.e., V(x) ) E0(1 - x2/a02) where E0 denotes the maximum barrier height above the zero point energy (ZPE). The resolvable ESDPT time scales for 3MAI-d are in the range of few tens to hundreds picoseconds. Thus, it is reasonable to assume a fast thermal equilibrium prior to the ESDPT reaction. Further support of this viewpoint can be given by varying the excitation wavelength from 255 to 275 nm, giving the same results on the ESDPT dynamics (not shown here). Accordingly, the tunneling rate for a molecule possessing thermal energy E above ZPE can thus be expressed as
A rough estimate of a0can be obtained from the average N-H‚‚‚N hydrogen bond distances,24the estimated displacement of the proton transfer (denoted d in Figure 9) and classical turning points for the zero-point motion of the N-H (or N-D) harmonic vibration from the equilibrium position. For the case of 7AI, a0, has been estimated to be 0.27 and 0.36 Å for N(1)-H and N(1)-D substituents, respectively.3,11,25The same a
0values were used for 3MAI in the following calculation, assuming that methyl substitution at the C(3) position has negligible perturba-tion on the dimeric structure. Since the tunneling rate was measured to be independent of the temperature in the range of 8-12 K, the thermally available energy E can be neglected. Accordingly, the barrier height E0could be deduced by applying known reaction-coordinate frequencyν, effective mass m and the estimated a0value to eq 4. For the case of 3MAI-d, taking ν to be∼6.0 × 1013s-1 (∼2000 cm-1), an E
0value of 605 cm-1 (1.73 kcal/mol) was thus deduced. At a relatively high temperature such as >20 K (see Figure 8) the available thermal energy is nonnegligible. The actual tunneling rate has to be calculated by the sum of all possible thermally populated states and is thus complicated. Given the difference in ZPE of∼400 cm-1 between N-H and N-D stretching, a barrier height of ESDPT was further deduced to be ∼205 cm-1 (∼0.58 kcal/ mol) for the 3MAI-h dimer. Plugging E0of 205 cm-1, a0H) 0.27 Å, m ) 1, andν ) 2800 cm-1(8.4× 1013s-1) to eq 4, the proton tunneling rate, ktunnel, was calculated to be 4.3 × 1012s- 1for 3MAI-h at <12 K. This time scale of k
tunnel- 1) 233 fs is too fast to be resolved by our picosecond time-correlated single photon counting system. On the basis of the energy barrier of 0.58 and 1.73 kcal/mol, respectively, rates of ESDPT for 3MAI-h and 3MAI-d were calculated to be negligibly small via a thermally activated process at <12 K. Certainly, it should be noted that the proton tunneling could rarely be satisfactorily described as a one-dimensional process. The motion of atoms between which the proton is being exchanged may have a major effect on the tunneling rate.26-32 Although the above results conclude a finite barrier to ESDPT, it is rather small, and subtle changes in the proton-transfer distance or energetics could have large effects on the proton-transfer rates. This recognition has led to the introduction of two-dimensional approaches in which the second dimension represents the effective motion of the other atoms during the proton-transfer reaction. Theoretical approaches33-35have re-vealed that the KIE (kinetic isotope effect) temperature depen-dence may be caused by thermally excited reactants and/or products that modulate the barrier width and shape. Although two-dimensional models may produce a more qualitative insight into the kinetic isotope effect and the temperature dependence of the transfer process, they still require the introduction of empirical parameters and thus cannot render quantitative predic-tions. For more precise quantitative work it is necessary to consider all degrees of freedom of the global system for an overall proton-transfer reaction.
At this stage, we are unable to differentiate the one-step versus two-step, nonconcerted ESDPT mechanism. Although the experimental results indicate a single decay and rise component for normal and tautomer emissions, respectively, it is also possible that ESDPT takes place via a two-step mechanism, and the rate in one of the steps is too fast to be resolved by the picosecond resolution. Alternatively, fluorescence upconversion with femtosecond resolution may provide valuable information. Unfortunately, the temperature-dependent study in the 3MAI single crystal is intrinsically difficult due to the photolysis and/ Figure 9. A simplified one-dimensional PES regarding the ESDPT
based on a bound-free potential curve. See text for the detailed description of notations.
ktunnel) ν exp
[
-x2mp
∫
- a0a0
V(x)1/2dx
]
(3)ktunnel) ν exp
[
-πa0p
x
2m(E0- E)]
(4)Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
or thermal reaction upon high repetition rate and high power laser excitation. Focus on this subject is currently in progress. 5. Conclusion
In conclusion, both steady-state and time-resolved measure-ments down to 8.0 K have revealed a remarkable deuterium isotope effect on the rate of ESDPT in the 3MAI-d single crystal. A barrier height of ESDPT of 1.73 and 0.58 kcal/mol was estimated for the 3MAI-d and 3MAI-h dimers, respectively, in a single crystal, and the ESDPT rates are mainly governed by a proton/deuterium tunneling mechanism. The results provide a prototype to mimic the intrinsic ESDPT dynamics of the 7AI-like dual hydrogen-bonded dimer with complete structural information, which are believed to bring up a broad spectrum of interest in the field of proton-transfer studies.
Acknowledgment. This work was supported by the National Science Council NSC (90-2113-M-002-055).
Supporting Information Available: X-ray data for 3MAI-h and 3MAI-d. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) Taylor, C. A.; El-Bayoumi, M. A.; Kasha, M. Proc. Nat. Acad. Sci. U.S.A. 1969, 63, 253.
(2) Ingham, K. C.; Abu-Elgheit, M.; El-Bayoumi, M. A. J. Am. Chem. Soc. 1971, 93, 5023
(3) Ingham. K. C.; El-Bayoumi, M. A. J. Am. Chem. Soc. 1974, 96, 1674.
(4) Watson, J. D.; Crick, F. H. C. Nature (London) 1953, 171, 964. (5) (a) Lowdin, P.-O. AdV. Quantum Chem. 1965, 2, 213. (b) Lowdin, P.-O. ReV. Mod. Phys. 1963, 35, 724.
(6) Hetherington, W. M., III; Micheels, R. H.; Eisenthal, K. B. Chem. Phys. Lett. 1979, 66, 230.
(7) Share, P.; Sarisky, M.; Pereira, M.; Repinec, S.; Hochstrasser, R. M. J. Lumin. 1991, 48/49, 204.
(8) Takeuchi, S.; Tahara, T. Chem. Phys. Lett. 1997, 277, 340. (9) Chachisvilis, M.; Fiebig, T.; Douhal, A.; Zewail, A. H.J. Phys. Chem. A. 1998, 102, 669.
(10) Takeuchi, S.; Tahara, T. J. Phys. Chem. A. 1998, 102, 7740.
(11) Fiebig, T.; Chachisvilis, M.; Manger, M.; Zewail, A. H.; Douhal, A.; Garcia-Ochoa, I.; de La Hoz Ayuso, A. J. Phys. Chem. A. 1999, 103, 7419.
(12) Bulska, H.; Chodkowska, A. J. Am. Chem. Soc. 1980, 102, 3259. (13) Bulska, H.; Grabowska, A.; Pakula, B.; Sepiol, J.; Waluk, J.; Wild, U. P. J. Lumin. 1984, 29, 65.
(14) Catala´n, J.; Kasha, M. J. Phys. Chem. A 2000, 104, 10812. (15) Steiner, T. Angew. Chem., Int. Ed. 2002, 41, 48.
(16) Sekikawa, T.; Kobayashi, T.; Inabe, T. Phys. Chem. A 1997, 101, 644.
(17) Sekikawa, T.; Kobayashi, T.; Inabe, T. J. Phys. Chem. B 1997, 101, 10645.
(18) Dufour, P.; Dartiguenave, Y.; Dartiguenave, M.; Dufour, N. Lebuis, A. M.; Belanger-Gariepy, F.; Beauchamp, A. L. Can. J. Chem. 1990, 68, 193.
(19) Chou, P. T.; Liao, J. H.; Wei, C. Y.; Yang, C. Y.; Yu, W. S.; Chou, Y. H. J. Am. Chem. Soc. 2000, 122, 986.
(20) After prolong photolysis of 3IAI the crystal turned yellow, accompanied by a decrease of the proton-transfer tautomer emission. Under the same experimental conditions similar photolysis reaction did not occur in 3MAI. It is thus believed that iodine substituents play a role for the observed photochemistry in 3IAI.
(21) Robison, M. M.; Robison, B. L. J. Am. Chem. Soc. 1956, 78, 1247. (22) Chou, P. T.; Chen, Y. C.; Yu, W. S.; Chou, Y. H.; Wei, C. Y.; Cheng, Y. M. J. Phys. Chem. A 2001, 105, 1731.
(23) Demas, J. N.; Crosby, G. A. J. Phys. Chem. 1971, 75, 991-1024. (24) Vinogradov, S. N.; Linnell, R. H. Hydrogen Bonding; Van Nostrand-Reinhold: New York, 1971; Chapter 7.
(25) Kim, S. K.; Breen, J. J.; Willberg, D. M.; Peng, L. W.; Heikal, A.; Syage, J. A.; Zewail, A. H. J. Phys. Chem. 1995, 99, 7421.
(26) Fernandez-Ramos, A.; Smedarchina, Z.; Siebrand, W.; Zgierski, M. Z.; Rios, M. A. J. Am. Chem. Soc. 1999, 121, 6280.
(27) Bicerano, J.; Schaefer, H. F., III; Miller, W. H. J. Am. Chem. Soc. 1983, 105, 2550.
(28) Siebrand, W.; Wildman, T. A.; Zgierski, M. Z. J. Am. Chem. Soc. 1984, 106, 4083, 4089.
(29) Smedarchina, Z.; Siebrand, W. Chem. Phys. 1993, 170, 347. (30) Benderskii, V. A.; Goldanskii, V. I.; Makarov, D. E. Phys. Rep. 1993, 233, 195.
(31) Benderskii, V. A.; Makarov, D. E.; Wight, C. A. Chemical Dynamics at Low Temperatures; J. Wiley & Sons: New York, 1994; p 385.
(32) Liu, Y.-P.; Lu, D.-h.; Gonzalez-Lafont, D. G.; Truhlar, D. G.; Garrett, B. C. J. Am. Chem. Soc. 1993, 115, 7806
(33) Bruno, W. J.; Bialek, W. Biophys. J. 1992, 63, 689.
(34) Antoniou, D.; Schwartz, S. D. Proc. Nat. Acad. Sci. U.S.A. 1997, 94, 12360.
(35) Borgis, D.; Hynes, J. T. J. Chem. Phys. 1991, 94, 3619.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009

![TABLE 1: Bond Lengths [Ångstroms] and Angle [Degrees]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8672234.196162/3.918.91.430.73.645/table-bond-lengths-ångstroms-and-angle-degrees.webp)