Figeys, Weiping Jia and Zemin Yao
Zhenghui G. Jiang, Congrong Wang, Daniel
Parks, Pamela J. McFie, Scot J. Stone,
Chang, Sanjay Manhas, Erik F. Yao, Robin J.
Wang, Hu Zhou, Shumei Zhong, Chia-Ching
Wen Qin, Meenakshi Sundaram, Yuwei
WITHIN THE MICROSOMAL LUMEN
FORMATION OF LIPID PRECURSORS
PLAYS A MAJOR ROLE IN THE
ApoC-III
Lipoproteins: EVIDENCE THAT
Triacylglycerol-rich Very Low Density
Assembly and Secretion of
Human ApoC-III Results in Impaired
C-terminal Lipid Binding Domain of
within the
APOC3
Missense Mutation in
doi: 10.1074/jbc.M110.203679 originally published online June 15, 2011 2011, 286:27769-27780.
J. Biol. Chem.
10.1074/jbc.M110.203679 Access the most updated version of this article at doi:
. JBC Affinity Sites Find articles, minireviews, Reflections and Classics on similar topics on the
Alerts:
When a correction for this article is posted •
When this article is cited •
to choose from all of JBC's e-mail alerts Click here
Supplemental material:
http://www.jbc.org/content/suppl/2011/06/15/M110.203679.DC1.html http://www.jbc.org/content/286/31/27769.full.html#ref-list-1This article cites 33 references, 21 of which can be accessed free at
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
Downloaded from
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
Missense Mutation in APOC3 within the C-terminal Lipid
Binding Domain of Human ApoC-III Results in Impaired
Assembly and Secretion of Triacylglycerol-rich Very Low
Density Lipoproteins
EVIDENCE THAT ApoC-III PLAYS A MAJOR ROLE IN THE FORMATION OF LIPID
PRECURSORS WITHIN THE MICROSOMAL LUMEN
*□SReceived for publication, November 16, 2010, and in revised form, June 13, 2011Published, JBC Papers in Press, June 15, 2011, DOI 10.1074/jbc.M110.203679
Wen Qin‡1,2, Meenakshi Sundaram§1, Yuwei Wang§, Hu Zhou§, Shumei Zhong§, Chia-Ching Chang¶, Sanjay Manhas§, Erik F. Yao§, Robin J. Parks储, Pamela J. McFie**, Scot J. Stone**, Zhenghui G. Jiang‡‡, Congrong Wang‡, Daniel Figeys§3, Weiping Jia‡, and Zemin Yao§4
From the‡Department of Endocrinology and Metabolism, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai Diabetes Institute, Shanghai Clinical Center of Diabetes, Shanghai 200233, China, the§Department of Biochemistry,
Microbiology, and Immunology, Ottawa Institute of Systems Biology, University of Ottawa, Ottawa, K1H 8M5 Canada, the储Ottawa Hospital Research Institute, Ottawa K1H 8L6, Canada, the **Department of Biochemistry, University of Saskatchewan, Saskatoon, Saskatchewan S7N 5E5, Canada , the¶Department of Biological Science and Technology, National Chiao Tung University, Hsinchu 300, Taiwan and Institute of Physics, Academia Sinica, Taipei, Taiwan 11529, and the‡‡Department of Physiology and Biophysics,
Boston University School of Medicine, Boston, Massachusetts 02118-2526
Hepatic assembly of triacylglycerol (TAG)-rich very low den-sity lipoproteins (VLDL) is achieved through recruitment of bulk TAG (presumably in the form of lipid droplets within the microsomal lumen) into VLDL precursor containing apolipo-protein (apo) B-100. We determined apolipo-protein/lipid components of lumenal lipid droplets (LLD) in cells expressing recombinant human apoC-III (C3wt) or a mutant form (K58E, C3KE) initially identified in humans that displayed hypotriglyceridemia. Although expression of C3wt markedly stimulated secretion of TAG and apoB-100 as VLDL1, the K58E mutation (located at the C-terminal lipid binding domain) abolished the effect in trans-fected McA-RH7777 cells and in apoc3-null mice. Metabolic labeling studies revealed that accumulation of TAG in LLD was decreased (by 50%) in cells expressing C3KE. A Fat Western lipid protein overlay assay showed drastically reduced lipid binding of the mutant protein. Substituting Lys58with Arg
dem-onstrated that the positive charge at position 58 is crucial for apoC-III binding to lipid and for promoting TAG secretion. On the other hand, substituting both Lys58 and Lys60 with Glu
resulted in almost entire elimination of lipid binding and loss of function in promoting TAG secretion. Thus, the lipid binding domain of apoC-III plays a key role in the formation of LLD for hepatic VLDL assembly and secretion.
Hepatic assembly of very low density lipoproteins (VLDL) rich in triacylglycerol (TAG)5is initiated during and after
trans-lation and translocation of apolipoprotein (apo) B-100 across the endoplasmic reticulum membrane. The nascent VLDL par-ticle is further enlarged in TAG content through a “second step” lipidation process, where a bulk TAG (presumably present within the microsomal lumen in the form of lipid droplet) is incorporated (1). Although much has been learned about the co- and post-translational lipidation of apoB-100, little is known about the nature or the dynamics of these lumenal lipid droplets (LLD) that are utilized as lipid precursor for VLDL assembly. Early studies with the abetalipoproteinemia gene
MTTPsuggested that the microsomal triglyceride transfer pro-tein (MTP) is required for the partitioning of VLDL lipid pre-cursor, mainly TAG, into the microsomal lumen in mouse liver (2), cultured murine primary hepatocytes (3), and the rat hep-atoma McA-RH7777 cells (4). Thus, inactivation of MTP in the liver cells was associated with lack of LLD and invariably resulted in diminished assembly and secretion of VLDL. Recent proteomic and lipid characterization of LLD isolated from murine liver microsomes has shown that these lipid-rich enti-ties are devoid of apoB but contain proteins such as TAG hydrolase, carboxylesterase 1, MTP, and apoE (5). Working with McA-RH7777 cells stably expressing recombinant human apoC-III, we have obtained evidence for the presence of a met-abolically active TAG-rich entity within the microsomal lumen, which has a buoyant density resembling that of intermediate density lipoproteins (IDL) and low density lipoproteins (LDL)
*This work was supported by the Heart and Stroke Foundation of Ontario (NA-7026 (to Z. Y.), China-Canada Health Research Initiative Grants CCI 92211 (to Z. Y.), and 30811120437 (to W. J.), and by the J.-Louis Le´vesque Foundation (to D. F.).
□S
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1–3 and Fig. S1.
1Both authors contributed equally to this work.
2Recipient of Special Funding for Visiting Doctoral Students Program from
Shanghai Jiao Tong University.
3The Tier I Canada Research Chair in Proteomics and Systems Biology. 4To whom correspondence should be addressed: 451 Smyth Rd., Dept. of
Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, ON, K1H 8M5 Canada. E-mail: zyao@uottawa.ca.
5The abbreviations used are: TAG, triacylglycerol; LLD, lumenal lipid droplet;
MTP, microsomal triglyceride transfer protein; IDL, intermediate density lipoprotein; PC, phosphatidylcholine; DAG, diacylglycerol; PE, phosphati-dylethanolamine; SPM, sphingomyelins; P407, Poloxamer 407; DGAT, dia-cylglycerol acyltransferase; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl) ethyl]glycine.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
(6). Interfering with the second step lipidation, either via treat-ment with brefeldin A or by expression of a loss-of-function human apoC-III mutant A23T, resulted in accumulation of metabolically labeled TAG in the form of LLD in microsomes (6). These results suggest that human apoC-III may play a func-tional role in the formation of the VLDL lipid precursors in the liver.
Apolipoprotein C-III is a small (79 amino acids) protein that comprises 40% of the protein mass of plasma VLDL and is also a component of HDL (7, 8). Mouse and human studies have demonstrated a strong positive correlation between plasma apoC-III and TAG concentrations (9) that is attributable to the inhibitory effect of apoC-III exerted on the TAG hydrolysis catalyzed by lipoprotein lipase (10) and the clearance of TAG-rich lipoproteins via receptor-mediated endocytosis (11). Recently, in vivo (12, 13) and in vitro (6, 14) experimental evi-dence has suggested that the apoC-III level is positively linked to hepatic VLDL production, presumably through enhanced assembly and secretion of TAG-rich VLDL1.
The structural elements within apoC-III responsible for pro-moting TAG-rich VLDL1secretion remain to be determined. In vitrostudies suggested that the lipid binding activity of apoC-III resides within the C-terminal region encompassing amino acid residues 41–79 (15). Thus, mutation at the N-terminal region (e.g. A23T) does not appear to affect apoC-III binding to lipid (6). In the current work we tested the effect of the naturally occurring mutation K58E (16) that is located within the lipid binding domain of apoC-III. Clinical data showed that the heterozygous K58E carriers had increased HDL (⬎95th percen-tiles of sex matched controls) and lowered VLDL as well as lowered VLDL-apoC-III (15% of normal) (16). The present data have demonstrated that the K58E mutation entirely abolished the apoC-III stimulatory effect on assembly and secretion of TAG-rich VLDL and resulted in markedly retarded accumula-tion of LLD in transfected cells. Thus, the lipid binding domain of apoC-III plays a key role in the formation of LLD utilized for VLDL assembly and secretion under lipid-rich conditions. EXPERIMENTAL PROCEDURES
Materials—Medium and reagents used for cell culture stud-ies were obtained from Invitrogen. [35S]Methionine/cysteine
(1000 Ci/mmol) was obtained from PerkinElmer Life Sciences, and protein A-SepharoseTMCL-4B beads, horseradish peroxi-dase-linked anti-mouse, and anti-rabbit IgG antibodies were obtained from GE Healthcare. [2-3H]Glycerol (9.6 Ci/mmol)
was obtained from American Radiolabeled Chemicals (St. Louis, MO), and brefeldin A and horseradish peroxidase-linked anti-goat antibody were obtained from Sigma. Oleate, TAG, and phospholipid standards were from Avanti Polar Lipids (Alabaster, AL). Antibody against human apoC-III (used for Western blot analysis) was obtained from Academy Biomedical Co., Inc. (Houston, TX). Polyclonal anti-mouse apoA-I and anti-mouse apoE antisera were obtained from BioDesign Inter-national (Saco, ME). Polyclonal antisera against rat VLDL (used for immunoprecipitation of apoB-100) or human apoC-III (used for immunoprecipitation of apoC-III) were generated in our laboratory. Protease inhibitor mixture and chemilumines-cent substrates were obtained from Roche Diagnostics.
Polox-amer 407 (P407) was a gift from BASF Corp. (Florham Park, NJ).
Preparation of Expression Plasmids and Transfection—The wild type human apoC-III (C3wt) expression plasmid (14) was used to generate cDNAs with site specific mutations such as K58E (C3KE), K58R (C3KR), K58E/K60E, and K58E/K60R within the coding region of human apoC-III cDNA using appropriate forward and reverse primers and QuikChangeTM
mutagenesis kit (Stratagene, Ann Arbor, MI). The coding sequences of all cDNA constructs were verified by sequencing. Sequences of the mutagenesis and sequencing primers are listed in supplemental Table 1. Stably transfected McA-RH7777 cells expressing wild type or the respective mutant apoC-III were generated as described previously (14). The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, 10% horse serum, and 200g/ml G418.
Metabolic Labeling of Lipids—Cells (⬃1.8 ⫻ 106 cells/
60-mm dish, in triplicate) were labeled with [3H]glycerol (5 Ci/ml) in DMEM supplemented with 20% FBS and 0.4-mm oleate for up to 2 h. At the end of labeling, lipids were extracted from cells and conditioned media for lipid analysis as described previously (14).
Subcellular Fractionation—Cells (⬃6 ⫻ 106 cells/100-mm
dish, in duplicate) were labeled with [3H]glycerol (20Ci/ml) in
DMEM supplemented with 20% FBS and 0.4 mMoleate for 30, 60, and 120 min. At each time point, media were collected for lipid analysis. Cells were harvested in 2 ml of Tris-sucrose buffer (10 mMTris-HCl, pH 7.4, and 250 mMsucrose) that was supplemented with protease inhibitor mixture and homoge-nized in a ball-bearing homogenizer (20 passages). The post-nuclear supernatant was obtained by centrifugation (1000⫻ g, 4 °C, 10 min) and was loaded into a quick-seal centrifuge tube. Cytosol and total microsomes were separated by centrifugation using a Beckman TLA-100.3 rotor (500,000⫻ g, 4 °C, 30 min). The total microsomes were rinsed twice with Tris-sucrose buffer to minimize cytosol contamination. The lumenal con-tents were released from total microsomes with 0.1Msodium carbonate, pH 11.3, by gentle mixing using a nutator for 30 min at room temperature. The lumenal contents were sepa-rated from microsomal membranes by ultracentrifugation (500,000⫻ g, 4 °C, 30 min). Lumenal contents were further subjected to cumulative rate floatation ultracentrifugation (4). Lipids were extracted from subcellular fractions (i.e. cytosol, total microsomes, microsomal lumen, and mem-brane), lumenal lipoprotein fractions were separated by TLC, and the radioactivity associated with [3H]TAG,
[3H]DAG, and [3H]phosphatidylcholine (PC) was quantified
by scintillating counting as previously described (14).
Lipid Pulse-chase Experiments—Cells (⬃ 1.8 ⫻ 106 cells/
60-mm dish, in triplicates) were labeled with [3H]glycerol (5Ci/ml) in DMEM supplemented with 20% FBS and 0.4 mM oleate for 1 h. Increasing concentrations of triacsin C (0 – 40 M) were included in the labeling media to determine the opti-mum condition for blocking TAG synthesis. In subsequent experiments, 40Mtriacsin C was included in the chase media (DMEM supplemented with 20% FBS, 0.4 mMoleate) for up to 8 h. Lipids were extracted from cell and media at the indicated
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
chase time and analyzed as described previously (14). The total radioactivity (cell and media) associated with [3H]DAG,
[3H]TAG, and [3H]PC at each chase time was plotted as the percentage of initial counts (i.e. end of 1 h of labeling in the absence of triacsin C).
Metabolic Labeling of Proteins—Cells (100-mm dishes) were labeled with [35S]methionine/cysteine (100Ci/ml) for 60 and
120 min in 5 ml of methionine/cysteine-free DMEM supple-mented with 20% FBS and 0.4 mMoleate. Cells were harvested and homogenized using ball-bearing homogenizer. Micro-somes were obtained by ultracentrifugation, and the lumenal contents were released by treatment with Na2CO3as described
above. The lumenal contents were separated from the micro-some membranes as described above, and lumenal contents were further subjected to cumulative rate floatation ultracen-trifugation. The35S-labeled apoB-100 and apoC-III proteins
were immunoprecipitated from each fraction using appropri-ate antibodies and resolved by SDS-PAGE followed by fluorog-raphy. Radioactivity associated with these apolipoproteins was determined by scintillation counting as previously described (14).
Lipoprotein Fractionation—Cells (100-mm dishes) were labeled for 2 h with [35S]methionine/cysteine (100Ci/ml) in
methionine/cysteine-free DMEM supplemented with 20% FBS and 0.4 mMoleate. The lipoproteins secreted into the condi-tioned media were fractionated by cumulative rate floatation ultracentrifugation to separate VLDL1(Sf⬎100) and VLDL2(Sf
20 –100) from other dense lipoproteins (4). In some experi-ments where better resolution for HDL fractionations was desired, the conditioned media were fractionated by sucrose density gradient ultracentrifugation (17). To this end, the media (4 ml) were centrifuged at 1000 rpm for 3 min to pellet cell debris. The supernatant was transferred to another tube, and the volume was adjusted to 5 ml with PBS. Sucrose density gradient was formed in 12-ml Ultra-Clear centrifuge tubes (Beckman Coulter) by layering from the bottom of the tube with 2 ml of 54% sucrose, 2 ml of 25% sucrose, 5 ml of sample in 12.5% sucrose, and 3 ml of PBS. The gradients were centrifuged at 35,000 rpm in a Beckman SW41Ti rotor for 65 h at 12 °C. Twelve 1-ml fractions were collected from the top of the tube, and the HDL apolipoproteins (e.g. apoE, and apoA-I) from each fraction were recovered by immunoprecipitation using respec-tive polyclonal antibodies and analyzed by SDS-PAGE followed by fluorography. Radioactivity associated with35S-labeled
apo-lipoproteins or3H-labeled lipids was quantified by scintillation
counting.
For lipid labeling, the cells (100-mm dish) were labeled with [3H]glycerol (5Ci/ml) in DMEM for 2 h. The3H-labeled lipids
were extracted from each fraction and separated by TLC as described above. Radioactivity associated with3H-labeled
lip-ids was determined by scintillation counting.
Analysis of Medium Lipoproteins by Size Exclusion Chromatography—Cells (100 mMdishes) were cultured for 2 h in 3 ml of DMEM supplemented with 2% FBS and 0.4 mM oleate. The conditioned media were concentrated centrifugally to 80l using Amicon Ultra 0.5-ml (Mrcut-off, 3000) filters
(Millipore Corp., Billerica, MA), and a 65-l aliquot was applied to a Superose 6 HR 10/30 column (Amersham
Biosci-ences) on a HPLC system (Gilson, Middleton, WI). Samples were eluted with a buffer containing 150 mM NaCl, 10 mM Na2HPO4, 100MEDTA, pH 7.5, at a flow rate of 0.5 ml/min.
Forty 0.5-ml fractions were collected, and lipoproteins present in fractions from 14 – 40 were concentrated using hydrated fumed silica (Cab-O-Sil) as previously described (18). The silica bound proteins were eluted into 100l of SDS-PAGE sample buffer (8Murea, 2% SDS, 10%-mercaptoethanol), resolved by SDS-PAGE (12% gel) using the Tris-Tricine-SDS running buffer system, and detected for apoC-III by immunoblot anal-ysis using anti-human apoC-III antibody as previously described (14). Elution profiling of VLDL (fractions 14 –17), IDL/LDL (fractions 18 through 24), and HDL plus lipid-free proteins (fractions 25– 40) was calibrated using mouse serum as previously reported (19).
Liquid Chromatography-MS/MS and Data Analysis of Pro-teins Associated with Lumenal Lipid Droplet—The LLDs with buoyant density resembling IDL/LDL were isolated from the microsomal lumen of cells expressing apoC-III as described previously (6). The LLD-associated proteins were resolved by SDS-PAGE (3–15% gel), and in-gel digestion of the resolved protein was performed as previously described (20). The result-ing peptides from the gel bands were resolved in 5% formic acid and loaded on a 200-m ⫻ 50-mm fused silica precolumn packed in-house with 5 cm of 5m Magic C18 AQ resins (5 m, 200 Å; Michrom Bioresources, Auburn, CA) using a 1100 micro-HPLC system (Agilent Technologies, Santa Clara, CA). Following a desalting step, the flow was split, and peptides were eluted through a second 75-m ⫻ 50-mm column packed with the same beads at⬃200 nl/min. The peptides were eluted using a 2-h gradient (5– 80% acetonitrile with 0.1% formic acid) into an ESI LTQ linear ion trap mass spectrometer (Thermo Elec-tron, Waltham, MA). MS/MS spectra were acquired in a data-dependent acquisition mode that automatically selected and fragmented the 10 most intense peaks from each mass spec-trum generated. The acquired MS/MS spectra were searched against the rat International Protein Index protein sequence data base (Version 3.52, 39,906 protein sequences; European Bioinformatics Institute) augmented with the reversed sequence of each entry in the forward data base. Mascot 2.2.02 (Matrix Science) was used to search the protein sequence data base. The precursor and fragment mass tolerances for the LTQ data were set at 2.0 and 0.8 Da, respectively. Mascot cutoff scores were set to 30. Peptides ranked with a probability based Mowse (expect) p⬍ 0.05 were accepted. The false positive rate was controlled at⬍1%.
Fat Western Lipid Protein Overlay Assay—The apoC-III-lipid binding assays were performed according to previously published protocol (21) with modifications. Briefly, various amounts (5–100g) of phospholipids and neutral lipids were spotted on Trans-Blot nitrocellulose membranes (Bio-Rad). Blots were blocked with phosphate-buffered saline containing 3% (w/v) fatty acid-free BSA and incubated at 4 °C for 12 h with equal amounts of apoC-III in conditioned media (collected from 100-mm dishes of cells cultured for 16 h in serum-free media). After washing with PBS and blocked with 5% fat-free milk in blotto, the blots were incubated with goat anti-human apoC-III antibody (1:2000) overnight at 4 °C. Blots were washed
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
with PBS and incubated in HRP-conjugated anti-goat IgG sec-ondary antibody (1:10,000) for 1 h at room temperature fol-lowed by washing with PBS.
The apoC-III bound to spotted lipids was visualized by enhanced chemiluminescence as per the supplier’s (Roche Diagnostics) instructions, and the intensity of each spot was quantified by scanning densitometry. The densitometry data were fitted into the Hill equation, ⫽ [L]n/(K
A
n⫹ [L]n), by the
least square fitting method to calculate apoC-III lipid binding kinetics. In the Hill equation, represents the relative amount of apoC-III that binds to lipids, L is the amount of lipid (in moles), K1ais the amount of lipid to which apoC-III displayed
half-maximum binding, which is also the microscopic dissoci-ation constant, and n is the Hill coefficient.
Real-time RT-PCR—Isolation of RNA from cells, reverse transcription, and relative mRNA concentration determination by real-time RT-PCR were performed as described previously (14). Cyclophilin A was used as the RT-PCR control. The prim-ers used for RT-PCR were obtained from Sigma, and sequences of the primers are listed insupplemental Table 2.
In Vivo Hepatic VLDL Production Assay—apoc3-null mice (B6.129-apoc3tm1Unc/J) obtained from The Jackson Laboratory (Bar Harbor, ME) were maintained and bred at the University of Ottawa animal care facility according to approved protocols. Male mice (8 –12 weeks old) were fed with high fat diet (TD.88137, Harlan Laboratories, Madison, WI) for 2 weeks and injected with adenovirus encoding either wild type human apoC-III (adv-C3wt) or the K58E mutant (adv-C3KE) via tail vein (1 ⫻ 109 plaque-forming units/mouse). Two days after
infection, mice were fasted for 16 h. Blood samples were col-lected via saphenous vein to determine expression of the recombinant apoC-III. Accumulation of newly synthesized apoB100 and apoB48 in the plasma was determined by injection of P407 (1 mg/g of body weight) to block VLDL lipolysis, and plasma samples were collected at 1 and 2 h after P407 injection. Plasma lipoproteins were separated by cumulative rate flota-tion centrifugaflota-tion. An aliquot (100l) of each lipoprotein frac-tion was mixed with 150 l of the TAG reagent (InfinityTM
Triglycerides, Thermo Scientific, Middletown, VA). Samples were incubated at 37 °C for 15 min, and the absorbance was measured at 520 nm. The apoB-100 and apoB-48 mass in TAG-rich lipoprotein fractions was semiquantified by immunoblot-ting after SDS-PAGE.
Histology of the Liver—The livers were fixed in 4% paraform-aldehyde for 24 h after, sliced into 1-mm specimens, and treated with 1% osmium tetroxide (in PBS) for 1 h. The specimens were washed with PBS (10 min, two times), placed in 70% alcohol, and processed for paraffin embedding using ATP1 automatic histoprocessor (Triangular Biomedical Sciences, Durham, NC). Paraffin sections (2.5-m thickness) were obtained, dried, deparaffinized, and stained with 1% nuclear fast red for 5 min. Sections were dehydrated in alcohol, cleared with toluene, and mounted on glass slides using Permount (Fisher). Slides were scanned using Zeiss Mirax Midi microscope scanner, and the images were analyzed using Mirax Viewer software (Carl Zeiss Canada Ltd., Toronto, Canada).
Quantification of TAG Mass in the Liver—The livers (100 – 120 mg by weight) were homogenized using Polytron
Homog-enizer (Kinematica AG, PT 10-35) in chloroform:methanol (2:1, v/v), and the homogenate was mixed overnight on a shaker. After centrifugation, the organic solvent was recovered, dried under N2(60 °C water bath), resuspended in 6 ml of
chlo-roform:methanol (2:1, v/v), and mixed with 1.2 ml of H2SO4
(0.05%, v/v). The aqueous and organic solvent phases were sep-arated by centrifugation, and an aliquot (250l) of the solvent phase was dried under N2(60 °C) and resuspended in 2 ml of
chloroform and 1% Triton X-100. The samples were dried under N2(60 °C) and solubilized in 1 ml of distilled water. An
aliquot (10l) of the sample was diluted 5-fold and mixed with 150l of TAG reagent to quantify TAG as described above.
Other Assays—The in vitro diacylglycerol acyltransferase (DGAT) activity assay in cell lysate was determined by mea-suring the formation of [14C]TAG from [14C]oleoyl CoA as
described previously (22). Briefly, cells were lysed by passing the cells suspended in lysis buffer (50 mMTris, pH 7.6, 250 mM sucrose) through a 27-gauge needle 10 –15 times. Cell debris was pelleted by centrifugation 600⫻ g for 5 min. Supernatant lysate was used for DGAT assays. The reaction contained 100 mMTris-HCl, pH 7.5, 20 or 100 mMMgCl2, 0.625 mg/ml BSA,
200 mM1,2-dioleoylglycerol, 25M[14C]oleoyl-CoA (18 mCi/ mmol), and 50 mg of protein (cell lysate) in a final volume of 200 l. The assay was incubated at 37 °C for 10 min. The reaction was terminated by the addition of chloroform:methanol (2:1 v:v) followed by 800l of H2O. Lipids were extracted and
sep-arated by TLC using hexane:ethyl ether:acetic acid (80:20:1 v:v:v) as a solvent system. Radioactivity in the [14C]TAG band
was quantified by liquid scintillation counting.
The in vitro MTP activity in the cell lysate of apoC3wt or apoC3KE cells was determined as described previously (23, 24). Protein concentration was quantified by the Bradford method (25).
RESULTS
K58E Is a Loss-of-function Mutation—The human apoC-III is composed of six␣-helices (26) of which helix 5, where the K58E mutation resides, assumes a typical type A amphipathic configuration (Fig. 1A). In this structure, Lys58and Lys60situate
at the plane of water-lipid interface, with hydrophobic residues on one side of the helix and acidic residues on the opposite side (27). Intuitively, substitution of Lys58 with Glu would be
expected to destroy the type A amphipathic configuration of helix 5 and impair lipid binding. To determine the effect of K58E mutation, we contrasted expression of the mutant with that of wild type apoC-III on VLDL-TAG secretion. From three stable clones expressing different levels of the mutant apoC-III (designated C3KE; Fig. 1B), secretion of [3H]glycerol-labeled
TAG was invariably decreased as compared with wild type apoC-III expressing cells (C3wt) regardless of the level of mutant expression (Fig. 1C). Fractionation of lipoproteins secreted from C3KE or C3wt cells showed that whereas [3H]TAG secretion as VLDL
1was markedly stimulated by C3wt
expression, this stimulatory effect was lost in C3KE-expressing cells (Fig. 1D). The lack of a stimulatory effect of C3KE on VLDL1secretion was further confirmed by metabolic labeling
of apoB-100. From C3wt cells, the secreted35S-apoB-100 was
predominately associated with VLDL (i.e. VLDL1and VLDL2),
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
whereas from C3KE cells the 35S-apoB-100 was absent in VLDL1and present in VLDL2and IDL/LDL fractions (Fig. 1E,
fluorograms for 35S-apoB-100 are shown in the insets).
Together, these results are the first indication that K58E is a loss-of-function mutation that abolishes the stimulatory effect of apoC-III on TAG-rich VLDL1 secretion under lipid-rich
conditions.
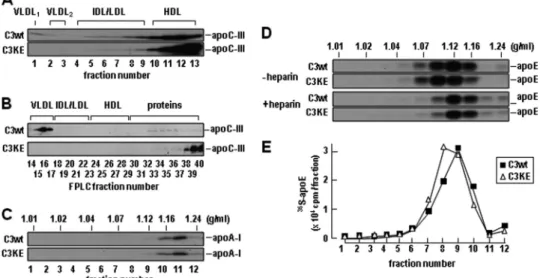
Determination of lipoprotein-association of secreted 35
S-apoC-III by density ultracentrifugation showed that C3wt was associated with VLDL in addition to HDL-like fractions, whereas C3KE mutant was present only in HDL-like fractions and the bottom fractions (Fig. 2A). To rule out that the lack of C3KE association with VLDL was not attributable to ultracen-trifugation, we fractionated the conditioned media using FPLC that minimizes detachment of apoC-III from lipoproteins. The FPLC profiling showed that although the wild type apoC-III was predominantly associated with VLDL fractions, the C3KE mutant was devoid in VLDL fractions and eluted almost exclu-sively in HDL and lipoprotein-free fractions (Fig. 2B). These results together with the density ultracentrifugation data (Fig. 1E),indicate that the K58E mutation resulted in decreased bind-ing of apoC-III to VLDL (i.e. VLDL1or VLDL2).
It was reported that the heterozygote carriers of the K58E mutation had increased HDL in the face of decreased VLDL
(16). Thus, we also determined the effect of C3wt or C3KE expression on the secretion of apoA-I and apoE, two HDL-associated apolipoproteins. To this end, the conditioned media were fractionated by sucrose density gradient ultracentrifuga-tion to better resolve the HDL particles (ranging from d⫽ 1.07 through d⫽ 1.24 g/ml). Secretion and density distribution of
35S-apoA-I from C3KE cells was comparable with that from
C3wt cells (Fig. 2C). Likewise, secretion of35S-apoE from C3KE
cells did not show major changes as compared with that from C3wt cells either under conditions of minus or plus heparin (to prevent reuptake of apoE-containing lipoproteins) (Fig. 2,
Dand E). Thus, the current cell culture model does not offer evidence for the hyperalphalipoproteinemia phenotype observed in heterozygotes carriers of the K58E mutation (16).
However, the hypotriglyceridemia phenotype could be recapitulated in apoc3-null mice infected with adenovirus encoding either C3wt or C3KE mutant (Fig. 3A). Infection of the mice, fed with a high fat diet for 2 weeks, with C3wt virus (adv-C3wt) resulted in marked accumulation of apoB-100 and apoB-48 in VLDL1 fractions under conditions where lipolysis was inhibited (i.e. with P407) (Fig. 3B). In contrast, expression of C3KE mutant (adv-C3KE) under identical con-ditions failed to stimulate VLDL1 production (Fig. 3C). Quantification of TAG mass in the fractionated medium FIGURE 1. The K58E is a loss-of-function mutation that abolished TAG-rich VLDL1secretion. A, the six␣-helices of human apoC-III in helix wheel diagram
are shown. The secondary structure assignment was based on the apoC-III structure (PDB ID 2jq3) in SDS-bound state as determined by NMR (26). Hydrophobic residues are denoted in green, acidic residues are in red, basic residues are in blue, hydrophilic residues are in gray, and Lys58is marked with a red arrow. B, shown
is an immunoblot of recombinant human apoC-III expressed in McA-RH7777 cells. C3wt, wild type apoC-III; C3KE, the K58E variant. C, cells (1.8⫻ 106cells/60 m M
dish, in triplicate) were labeled with [3H]glycerol (5Ci/ml) for 2 h in DMEM containing 20% FBS and 0.4 m
Moleate. Lipids were extracted from media and cells at the end of labeling and resolved by TLC. Radioactivity associated with [3H]TAG was quantified by scintillation counting. Radioactivity associated with the
[3H]TAG in the media is shown. ***, p⬍ 0.001 (Student’s t test of C3wt versus neo or C3KE versus neo; n ⫽ 3). D, cells were labeled with [3H]glycerol for 2 h in the
presence of 20% serum and 0.4 mMoleate. At the end of labeling, media were subjected to cumulative rate flotation ultracentrifugation. Lipids were extracted from each fraction and separated by TLC. Radioactivity associated with [3H]TAG was quantified by scintillation counting. E, cells were labeled with [35
S]me-thionine/cysteine for 2 h in the presence of 20% serum and 0.4 mMoleate. At the end of labeling, media were subjected to cumulative rate flotation ultracentrifugation. ApoB-100 was recovered from each fraction by immunoprecipitation and resolved by SDS-PAGE and visualized by fluorography (top two
panels). Radioactivity associated with35S-apoB100 was quantified by scintillation counting and plotted (bottom panel).
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
lipoproteins showed that the vast majority of TAG was asso-ciated with VLDL1(Fig. 3D), and the VLDL1-TAG
accumu-lated in the plasma of adv-C3wt-infected mice was⬎4-fold higher than that of adv-C3KE-infected mice (Fig. 3D). There was no significant difference in TAG mass associated with VLDL2or IDL/LDL fractions between C3wt- and
adv-C3KE-infected mice. Thus, the K58E mutation specifically impaired VLDL1production in both the McA-RH7777 cell culture model and the apoc3-null mouse model.
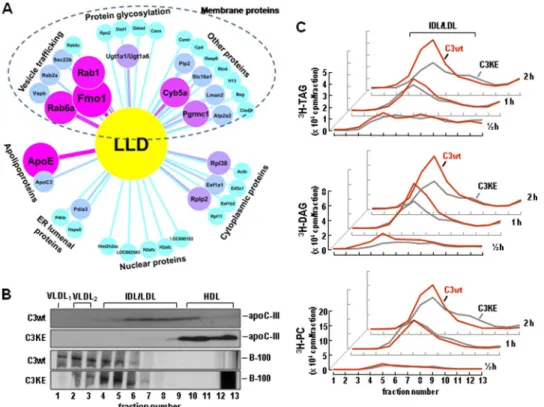
Impaired Lumenal Lipid Droplets Formation in Cells Expressing C3KE Mutant—Our previous studies with McA-RH7777 cells stably expressing human apoC-III have suggested that LLD presented in the microsomes (with buoyant density resembling IDL/LDL) may represent the bulk TAG precursor utilized for VLDL1assembly/secretion (14). Analysis of
pro-teins associated with LLD by liquid chromatography-MS/MS revealed that the LLD fractions were devoid of apoB-100 and contained apoC-III and apoE in addition to 40 other proteins, FIGURE 2. Impaired VLDL1-apoC-III secretion from C3KE cells. A, C3wt and C3KE cells were labeled with [35S]methionine/cysteine for 2 h in the presence of
20% serum and 0.4 mMoleate. The conditioned media were fractionated by cumulative rate flotation ultracentrifugation, and immunoprecipitated35S-apoC-III
was resolved by SDS-PAGE and visualized by fluorography. B, conditioned media were fractionated by size exclusion chromatography using FPLC and apoC-III in fractions 14 – 40 were recovered by immunoprecipitation, resolved by SDS-PAGE, and visualized by immunoblotting. C, cells were treated as in A, and the conditioned media were subjected to sucrose density gradient ultracentrifugation. The immunoprecipitated35S-apoA-I proteins were resolved by SDS-PAGE
and visualized by fluorography. D, the35S-labeled conditioned media (⫺heparin, top panels or ⫹heparin (100 units/ml), bottom panels) were subjected to
sucrose density gradient ultracentrifugation, and the immunoprecipitated apoE proteins were resolved by SDS-PAGE and visualized by fluorography. E, the radioactivity associated with35S-apoE was quantified using scintillation counter (only data were obtained under minus heparin conditions are shown). The
density (g/ml) of each fraction, which is the mean value of seven density gradients determined gravimetrically, is shown on top of the fluorograms in panels C and D. Repetition of the experiments yielded similar results.
FIGURE 3. Hepatic VLDL1production was impaired in apoc3-null mice expressing apoC3KE protein. The apoc3-null mice (10 weeks old) fed with a
high fat diet for 2 weeks were infected with adenovirus encoding C3wt or C3KE. Three days after infection, mice were fasted for 16 h. Blood samples were collected before or 1 and 2 h after P407 injection. A, immunoblots of apoC-III in plasma of infected mice are shown. B and C, shown are immunoblots of apoB-100 and apoB48 in fractionated plasma lipoproteins (only the top six fractions of cumulative rate flotation centrifugation are shown). The plasma samples were pooled from three mice infected with adv-C3wt (B) or adv-C3KE (C). Samples were collected before (top two panels) and 1 h (middle two
panels) or 2 h (bottom two panels) after P407 injection. D, TAG mass in the top six fractions of cumulative rate flotation centrifugation (i.e. VLDL1, VLDL2,
and IDL/LDL) of plasma samples pooled from three adv-C3wt- and three adv-C3KE-infected mice before (top panel), 1 h (middle panel), or 2 h (bottom
panel) after P407 injection.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
including those involved in vesicle trafficking, protein glycosy-lation, endoplasmic reticulum lumenal proteins, and some pro-teins normally present in nucleus or cytoplasm (Fig. 4A). Whether or not all of these membrane or cytoplasmic proteins are functional components of LLD remains to be determined experimentally (seesupplemental Table 3).
Determination of metabolically labeled apoC-III distribution within the microsomal lumen confirmed that apoC-III indeed was a component of LLD. The majority of lumenal35S-C3wt was associated with the isolated LLD fractions, whereas35
S-C3KE was unable to bind to LLD in the microsomes and, rather, was sedimented to HDL and the bottom fractions after ultra-centrifugation (Fig. 4B, top two panels). Determination of lume-nal 35S-apoB-100 distribution under the same conditions
showed that although apoB-100 was readily detectable in the VLDL1fractions in C3wt cells, it was mainly associated with
IDL/LDL fractions in C3KE cells (Fig. 4B, bottom two panels), These results suggest that the loss of C3KE binding to LLD was associated with decreased VLDL1assembly within the
micro-somal lumen.
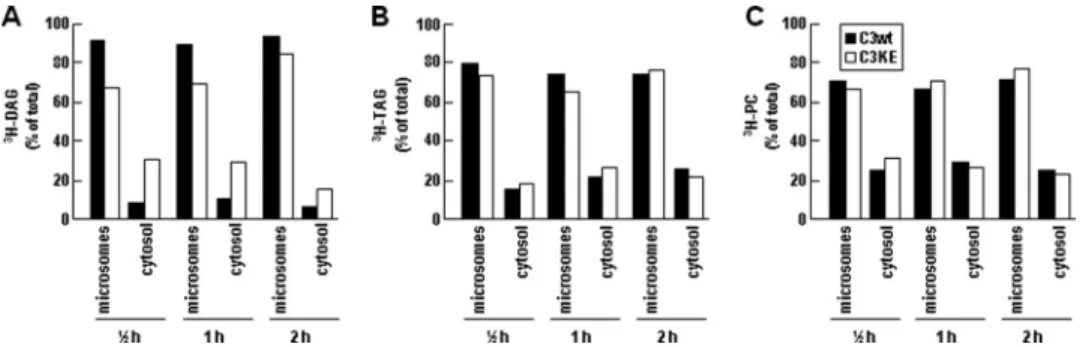
The reduced lipid binding activity of C3KE mutant was also associated with impaired accumulation of metabolically labeled lipids in the LLD fractions. Thus, although accumulation of [3H]glycerol-labeled TAG and PC in LLD fractions was readily
detectable in C3wt cells, the magnitude of LLD-associated [3H]TAG in C3KE cells was markedly reduced (by⬎2-fold)
(Fig. 4C, top panel). Similarly decreased accumulation of LLD-associated [3H]DAG in C3KE cells was also observed (Fig. 4C, middle panel). The effect of K58E mutation on LLD-associated [3H]PC was less pronounced (Fig. 4C, bottom panel). These
results, in accordance with previous data (14), suggest a strong correlation between formation of LLD-associated [3H]TAG
and assembly/secretion of VLDL1-[ 3
H]TAG under lipid-rich conditions.
Lipid Binding of ApoC-III Is Compromised by K58E Mutation— The above results suggested a close link between the dynamic biosynthesis of LLD and VLDL-TAG secretion, the establish-ment of which is facilitated by apoC-III. To gain an insight into mechanism by which apoC-III facilitates LLD formation, we compared the binding of C3wt and C3KE toward different lipid groups, including glycerolipids (e.g. TAG and diacylglycerol, DAG), glycerophospholipids (e.g. PC and phosphatidylethano-lamine (PE), sphingolipids (e.g. sphingomyelin (SPM)), and ste-rol lipids (e.g. cholesteste-rol and cholesteryl ester) using Fat West-ern lipid protein overlay assay (Fig. 5). The wild type apoC-III showed robust binding to PE, PC, and SPM and, to a lesser degree, to cholesterol and DAG (Fig. 5A, left). The calculated
KAvalues (nmol) for PE, PC, cholesterol, DAG, and SPM were
FIGURE 4. Impaired LLD formation in C3KE cells. A, shown is a proteomics analysis of LLD-associated proteins. C3wt cells were cultured in DMEM supple-mented with 20% serum and 0.4 mMoleate. The cells were homogenized to isolate the microsomes, and the lumenal contents were released from the microsomes by Na2CO3treatment. The content was fractionated by cumulative rate flotation centrifugation to obtain LLD fractions, and the identity of the
LLD-associated proteins was determined by liquid chromatography-MS/MS as described under “Experimental Procedures.” The identified proteins are visu-alized using Cytoscape software (Version 2.6.0) (34). The line width, size, and color intensity of each individual protein is related to its identified peptides. The detailed information of each identified protein is shown insupplemental Table 3. B, apoC-III and apoB-100 distribution within microsomal lumen is shown. C3wt and C3KE cells were labeled with [35S]methionine/cysteine for 60 min in the presence of 20% serum and 0.4 m
Moleate. Microsomal content was fractionated as in A. The35S-apoC-III (top two panels) or35S-apoB-100 (bottom two panels) in each fraction was recovered by immunoprecipitation, resolved by
SDS-PAGE, and visualized by fluorography. C, C3wt and C3KE cells (⬃ 6 ⫻ 106cells/100-mm dish, in duplicates) were labeled with [3H]glycerol (20Ci/ml) in
DMEM supplemented with 20% FBS and 0.4 mMoleate for 0.5, 1, and 2 h. At the end of labeling, cells from two dishes were combined, and lumenal content was isolated from the microsomes followed by fractionation as in A. Lipids were extracted from each fraction and resolved by TLC. Radioactivity associated with [3H]TAG (top), [3H]DAG (middle), and [3H]PC (bottom) was quantified and plotted.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
15.17, 15.60, 56.78, 62.50, and 118.80, respectively. The KA value denotes the lipid concentrations at which apoC-III dis-played half-occupation in the lipid protein overlay assay.
The C3KE mutant, at equivalent amounts of protein used, displayed drastically decreased binding toward the lipids tested (Fig. 5A, right). The calculated KAvalues (nmol) for PE, PC, and SPM were 26.55, 32.01, and 71.83, respectively (lack of C3KE mutant binding to cholesterol and DAG precluded calculation of the corresponding KAvalues). Thus, the binding activity to PE or PC was decreased by⬃50% by the K58E mutation as compared with wild type apoC-III. Neither C3wt nor C3KE showed binding to the neutral lipids of lipoprotein core, such as TAG or cholesteryl ester. These in vitro data suggested strongly that the K58E mutation within helix 5 of apoC-III diminishes its binding toward the surface lipids of lipoproteins (presumably the surface lipids of LLD as well), most likely through destroyed amphipathicity of the helix.
To further test the importance of type A amphipathic con-figuration of helix 5, we generated two additional mutations within this region. First, Lys58residue was substituted with Arg
(designated K58R) to preserve the charge of the side chain. Sec-ond, both Lys58and Lys60residues were mutated into Glu
(des-ignated K58E/K60E) to entirely destroy the type A amphipathic helix 5 (see structure in Fig. 1A). Data from the lipid protein overlay assay showed, as expected, that the K58R mutation had no discernible effect on lipid binding as compared with the wild type apoC-III (Fig. 5B, left), suggesting that the charge of the side chain at position 58 is critical for lipid binding. On the other hand, the K58E/K60E mutation almost entirely abolished lipid binding of apoC-III under identical experimental condi-tions (Fig. 5B, right).
The effect of K58R or K58E/K60E mutation on apoC-III function in stimulating hepatic VLDL-TAG secretion was ver-ified by metabolic labeling experiments using cells stably
expressing the respective variants. Thus, although expression of the K58R mutant enhanced [3H]TAG secretion as effectively
as that of the wild type apoC-III, expression of the K58E/K60E mutant failed to exert an effect on [3H]TAG secretion as
com-pared with neo control cells (Fig. 5C). Taken together, these data highlight the importance of amphipathic helix 5 of apoC-III in mediating the formation of LLD that are utilized for VLDL assembly/secretion.
Expression of K58E Mutant Impairs DAG-to-TAG
Conversion—Finally we inquired a potential effect of C3KE mutant expression on hepatic TAG biosynthesis. Metabolic labeling experiments with [3H]glycerol showed that the
incor-poration of radiolabel into TAG was significantly decreased in C3KE cells as compared with C3wt cells (Fig. 6A). On the other hand, incorporation of radiolabel into PC was significantly increased in C3KE cells (Fig. 6B). These results indicate that the lowered incorporation into cell-associated TAG in C3KE cells was not attributable to impaired uptake of [3H]glycerol. Recent
studies with nonalcoholic fatty liver disease patients have impli-cated APOC3 variants and hepatic DAG levels in the pathology (28, 29). Thus, we determined [3H]glycerol-labeled DAG in
cells expressing the recombinant human apoC-III and observed markedly increased [3H]DAG during metabolic labeling in
C3KE cells as compared with C3wt cells (Fig. 6C). We also measured secretion of [3H]glycerol-labeled lipids during the
labeling period. As expected, secretion of [3H]TAG was
mark-edly increased from C3wt cells as compared with C3KE cells (Fig. 6D), whereas secretion of [3H]PC showed a significant
increase at 1 h (but not 2 h) labeling time from C3KE cells (Fig. 6E). Secretion of [3H]DAG showed no significant difference
between the two cell lines (Fig. 6F).
We next determined whether the apparently lowered TAG synthesis in C3KE cells was attributable to impaired DAG-to-TAG conversion or else due to rapid hydrolysis of newly syn-FIGURE 5. Amphipathicity of helix 5 is important for apoC-III function. A and B, increasing quantities (5–100g) of the indicated lipid groups spotted on nitrocellulose membranes were incubated with an equal amount of apoC-III collected from conditioned media of stable cell lines expressing the indicated apoC-III variants. The bound apoC-III was detected by immunoblotting, and the intensity of each spot was semiquantified by scanning densitometry (for calculating KAvalues for each lipid group). Chol, cholesterol. CE, cholesteryl ester. C, cells (1.8⫻ 106cells/60 mMdish, in triplicate) expressing the indicated
apoC-III variants were labeled with [3H]glycerol (5Ci/ml) in DMEM supplemented with 20% FBS and 0.4 m
Moleate, and secretion of [3H]TAG and [3H]PC was
determined as in Fig. 1C. * p⬍ 0.05; *** p ⬍ 0.001 (Student’s t test of C3 variants versus neo; n ⫽ 3).
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
thesized [3H]TAG. To this end, we determined TAG synthesis
enzyme and TAG-transfer proteins known to play a role in VLDL assembly/secretion. Quantitative RT-PCR analysis of the expression of two key genes involved in de novo TAG biosyn-thesis, namely Lpin1 and Dgat1/2, showed that their expression was not diminished but, rather, augmented in C3KE cells as compared with C3wt cells (supplemental Fig. S1A). In vitro DGAT1 and DAGT2 activity assay, however, did not show a significant difference between C3wt and C3KE cells ( supple-mental Fig. S1B). Quantification of the level of newly synthe-sized 35S-lipin-1 between C3wt and C3KE cells also did not
show significant changes (supplemental Fig. S1C). Pulse-chase experiments showed that the turnover of [3H]TAG (in the
pres-ence of triacsin C during chase) was not faster but was, rather, slower in C3KE cells as compared with that in C3wt cells (Fig. 6G). Thus, the decreased incorporation of3H label into TAG in C3KE cells was unlikely attributable to decreased TAG synthe-sis machinery or rapid turnover of newly synthesized TAG. The turnover of [3H]PC (Fig. 6H) or [3H]DAG (Fig. 6I) also did not
show major changes between C3wt and C3KE cells.
We performed additional subcellular fractionation experi-ments to define mechanisms responsible for the decreased DAG-to-TAG conversion in C3KE cells. Separation of newly
synthesized cell [3H]DAG into microsomes and cytosol
frac-tions revealed that in C3wt cells⬎90% of total [3H]DAG was
associated with microsomes and the remainder in cytosol. In contrast, less than 70% of total [3H]DAG was present in
micro-somes, and the cytosolic [3H]DAG accounted for 30% in C3KE
cells (Fig. 7A). The microsome/cytosol distribution of [3H]TAG
(Fig. 7B) or [3H]PC (Fig. 7C) between C3KE and C3wt cells was
comparable. These results together indicate that the reduced [3H]TAG synthesis observed in C3KE cells might be
attributa-ble to a skewed partitioning of [3H]DAG toward cytosol (and
thus less available for TAG synthesis), The decreased partition-ing of [3H]DAG into microsomes in C3KE cells was, however,
unlikely to have resulted from impaired activity of MTP, because quantitative RT-PCR analysis (supplemental Fig. S1A) and activity assay (supplemental Fig. S1D) showed no decrease but, rather, an increase in the MTP mRNA and activity.
Currently, the mechanism by which expression of C3KE mutant results in decreased hepatic TAG synthesis in McA-RH7777 cells remains unclear. Histological analysis of liver samples (Fig. 8, A–C) and quantification of TAG mass (Fig. 8D) of mice infected with adv-C3wt or adv-C3KE showed no signif-icant difference in TAG mass in the liver between the two groups.
DISCUSSION
The Lipid Binding Domain of ApoC-III—By unraveling the functional lipid-binding within the amphipathic helix 5 of human apoC-III, the current study extends our previous dem-onstration for an intracellular role of apoC-III in promoting hepatic VLDL assembly/secretion. Disruption of type A amphipathic configuration of helix 5 through K58E mutation results in the lack of association with LLD in the microsomes of transfected cells (Fig. 4B) as well as impaired binding to surface lipids in vitro (Fig. 5A). The impaired lipid binding of the mutant apoC-III was intimately linked to its inability to pro-mote TAG-rich VLDL assembly and secretion in both trans-fected McA-RH7777 cells under lipid-rich conditions (Fig. 1, D and 1E) and in high fat diet-fed apoc3-null mice reconstituted with hepatic apoC-III expression (Fig. 3). Thus, the present cell culture and in vivo studies with the naturally occurring mis-sense mutation in APOC3 within the lipid binding domain of human apoC-III provide strong evidence for an intrahepatic cellular function of this apolipoprotein in promoting hepatic TAG-rich lipoprotein assembly/secretion.
What is the structural element within helix 5 that mediates its lipid binding to LLD? The current available structural infor-mation of human apoC-III was obtained from NMR analysis of the protein in association with SDS micelles (26). Early in vitro studies suggested that binding of apoC-III to phospholipids was mediated by residues 41–79 at the C terminus of the protein (15). Furthermore, site-specific mutagenesis experiments showed that two hydrophobic residues (Phe64and Trp65) near
the carboxyl end are crucial for initiating and maintaining lipid binding of apoC-III (30). Helical wheel modeling suggests a perfect type A amphipathic␣-helix for helix 5, with the nonpo-lar and pononpo-lar faces separated by positively charged residues (e.g. Lys58, Lys60) at the interface (27). The type A amphipathic
␣-helix provides an optimal structural motif for binding to FIGURE 6. Synthesis, secretion, and turnover of lipids in C3wt and C3KE
cells. C3wt and C3KE cells (1.8⫻ 106cells/60 m
Mdish, in triplicate) were labeled with [3H]glycerol (5Ci/ml) for up to 2 h in DMEM containing 20% FBS
and 0.4 mMoleate. Lipids were extracted from media and cells, respectively, at the end of labeling and resolved by TLC. Radioactivity associated with3
H-la-beled lipid was quantified by scintillation counting. A-C, at each time point, the radioactivity of cell-associated [3H]DAG, [3H]TAG, and [3H]PC are shown. D–F, at each time point, the radioactivity of [3H]DAG, [3H]TAG, and [3H]PC
from the conditioned media is shown. G–I, cells were labeled with [3
H]glyc-erol (5Ci/ml) for 1 h in DMEM supplemented with 20% FBS and 0.4 mM
oleate. The cells were replaced with DMEM supplemented with 20% FBS, 0.4 mMoleate, and 40Mtriacsin C and cultured (chase) for up to 8 h. Lipids were extracted from cells and media at the indicated chase time and separated by TLC, and radioactivity associated with [3H]DAG, [3H]TAG, and [3H]PC was
quantified. The total counts (media and cell) are expressed as percent of counts at the end of initial 1 h labeling. ***, p⬍ 0.001; **, p ⬍ 0.01; *, p ⬍ 0.05 (Student’s t test of C3wt versus C3KE; n⫽ 3).
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
phospholipid surface, as the positively charged patches interact with negatively charged phospholipid headgroups, whereas the hydrophobic moment of the helix buries part of the helix among the phospholipid acyl chains. The present protein lipid overlay assay has verified the specific interaction between apoC-III with amphipathic lipids, showing that the binding affinity for apoC-III toward lipids is in the order of PE⬎ PC ⬎ SPM ⬎ cholesterol ⬎ DAG. The overlay assay also yielded insight into the effect of mutations that destroyed the type A amphipathic configuration of helix 5. For instance, K58E muta-tion effectively abolished binding of apoC-III with cholesterol, DAG, and SPM and significantly weakened its binding with PE and PC. However, the K58R mutation exhibited lipid binding as avidly as the wild type apoC-III, further validating amphipathic helix 5 model in which the positive charge of the side chain confers lipid binding (Fig. 5B). Moreover, experiments with the K58E/K60E double mutation provided further evidence for the amphipathic helix 5 model and suggested that that the two interfacial Lys residues may exert a synergistic effect on lipid binding (Fig. 5B).
It was reported that the heterozygote K58E carriers exhibited increased plasma HDL-cholesterol and HDL-apoA-I in the face of decreased VLDL-TAG and VLDL-apoC-III (16). The reason for this apparent hyperalphalipoproteinemia phenotype in the K58E carriers was not explained. Our cell culture experiments showed that the decreased VLDL secretion (both VLDL-apoB-100 and VLDL-TAG) from the C3KE cells was not associated with significant changes in the secretion of apoE or HDL-apoA-I (Fig. 2, C and D). Thus, the current cell culture study did not recapitulate the hyperalphalipoproteinemia phenotype in human K58E carriers. The binding of apoC-III toward HDL lipids requires further characterization.
A “Two-domain” Model for ApoC-III Action—Our studies with the naturally occurring missense mutations in APOC3, previously with A23T (6) and at present with K58E variants, led us to postulate a two-domain model for apoC-III action in pro-moting VLDL assembly/secretion under lipid-rich conditions. Although expression of both A23T and K58E displayed loss-of-function of apoC-III in promoting VLDL assembly/secretion, the underlying mechanisms for the deficiency were different. FIGURE 7. Distribution of lipids in microsomes and cytosol are different in C3wt and C3KE cells. The C3wt and C3KE cells (100 mMdishes, in duplicate) were labeled with [3H]glycerol (20Ci/ml) for up to 2 h in DMEM supplemented with 20% FBS and 0.4 m
Moleate. The cells were harvested, and the intracellular components were separated into post-nuclear supernatant and nucleus-mitochondria (pellet) by centrifugation (1000⫻ g, 10 min, 4 °C). Post-nuclear super-natant was further separated into microsomes (pellet) and cytosol (supersuper-natant) by ultracentrifugation (500,000⫻ g, 30 min, 4 °C). Lipids were extracted from the conditioned media as well as from all intracellular fractions and separated by TLC, and the radioactivity associated with [3H]DAG (A), [3H]TAG (B), and [3H]PC
(C) was quantified. The counts in microsomes and cytosol fractions were plotted as a percent of total (cellular fractions and media).
FIGURE 8. Lipid staining of apoc3-null mice infected with adv-C3wt or adv-C3KE. The experiments were performed as in Fig. 3. Two hours after P407 treatment, the mice were sacrificed, and the livers were retrieved and processed for lipid staining (panels A–C) and TAG mass quantification (panel D). A, liver of three control mice (apoc3-null mice infected with empty adenovirus vector) is shown. B, liver of three mice expressing C3wt is shown. C, liver of three mice expressing C3KE is shown. Scale bar, 50m. CV, central vein. D, TAG mass in the liver is shown. Error bars indicate S.D. of average TAG values of liver samples obtained from three independent mice per group.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
Although the present study with K58E mutation indicated a deficiency in binding to surface lipids presumably surrounding the LLD, studies with the A23T mutation showed no impair-ment in lipid binding or LLD formation (6). We have recently verified that the A23T mutant exhibited enhanced binding to LLD in the microsomes of transfected cells and that the A23T mutation had no negative effect on apoC-III lipid binding as determined by Fat Western lipid protein overlay assay.6Thus,
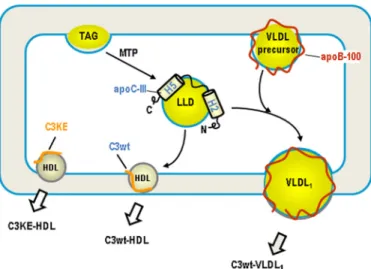
our cell culture and in vitro results are in agreement with pre-viously reported thrombin cleavage studies (31) that excluded the N terminus of apoC-III being responsible for lipid binding. Remarkably, binding of A23T variant to LLD resulted in accu-mulation of TAG in the microsomal lumen (6). We interpreted this result as a blockage of fusion of LLD with the VLDL pre-cursor by A23T variant and postulated that the N-terminal region of apoC-III might possess a fusogenic function in facili-tating incorporation of bulk TAG (derived from LLD) into VLDL (6). On the other hand, the C-terminal␣-helix 5 confers apoC-III lipid binding and promotes formation of LLD. The binding of apoC-III to LLD may represent a docking step that is the prerequisite for a subsequent fusion event. A model depict-ing the two-domain hypothesis for apoC-III action in promot-ing LLD formation and subsequent VLDL1assembly is shown
in Fig. 9.
LLD Represents the TAG Precursor for VLDL1Maturation—
VLDL maturation describes a process whereby the bulk TAG is incorporated, presumably in the form of metabolically active LLD. Technically, it has been challenging to determine the bio-synthetic origin or the metabolic fate of LLD, because of the lack of a proper protein marker(s) for this metabolically dynamic entity. It is conceivable that formation of LLD only becomes apparent under lipid-rich conditions, through which the VLDL assembly/secretion apparatus is activated to purge
excess hepatic lipid and prevent endoplasmic reticulum stress associated with lipotoxicity. In this regard, up-regulated apoC-III expression represents a hepatic defense mechanism in pro-tecting hepatic steatosis as a consequence of the insult of excess fatty acid influx, a common feature associated with hepatic insulin resistance under diabetic conditions. The current bio-chemical experiments in conjunction with mass spectrometry analysis not only ascertained that apoC-III is a bona fide protein component of LLD but also revealed a dynamic link between the apoC-III domain structure and LLD metabolism. Thus, apoC-III is a suitable functional marker for LLD.
Notably, the impaired secretion of VLDL1-TAG from cells
expressing the mutant apoC-III proteins (either A23T or K58E variants) was not accompanied with TAG accumulation in transfected McA-RH7777 cells (Fig. 6A) or in the infected livers (Fig. 8D). Rather, metabolic labeling experiments with C3KE cells showed decreased [3H]DAG-to-TAG conversion (Fig. 6, A–C), and histology studies revealed low lipid content in the liver of high fat diet-fed mice infected with the C3KE mutant. Data obtained from pulse-chase experiments with stable cell lines suggested that the decreased [3H]TAG in C3KE cells was
not attributable to rapid turnover of the newly synthesized TAG (Fig. 6, G–I). Recent studies with McA-RH7777 cells have shown that overexpression of lipin-1 (encoding phosphatidate phosphatase 1) (32) or DGAT2 (33), two key enzymes involved in the de novo TAG biosynthesis, resulted in increased VLDL assembly/secretion. However, expression of C3KE mutant in McA-RH7777 cells did not alter DGAT activity or lipin-1 expression (supplemental Fig. S1). Thus, the decreased TAG synthesis in C3KE cells was unlikely attributable to an altered biosynthetic enzyme machinery. Data obtained from subcellu-lar fractionation experiments have offered some clues to a potential role of apoC-III in the partitioning DAG between the cytosol and microsomes (Fig. 7A). Mechanisms by which apoC-III expression facilitates the channeling of substrate DAG into TAG synthesis for VLDL assembly/secretion remain to be defined.
How apoC-III can facilitate the partitioning of lipid substrate toward microsomes as well as formation of LLD is unclear. It is known that partitioning of VLDL lipid substrates into micro-somal lumen requires the activity of MTP (2– 4). We have shown previously that although apoC-III could not substitute the MTP activity required for VLDL assembly/secretion, it facilitates TAG incorporation into VLDL, a process that is inde-pendent of MTP activity (14). Neither the A23T (6) nor the K58E mutation (supplemental Fig. S1) drastically altered the MTP activ-ity in transfected cells. Thus, it is likely that the action of apoC-III is downstream of MTP-mediated TAG partitioning into microsomes (Fig. 9).
In conclusion, the present cell culture and in vivo mouse studies have identified the C-terminal amphipathic␣-helix of apoC-III as an important functional structural element in pro-moting LLD formation (probably through facilitated TAG bio-synthesis) and VLDL assembly/secretion. Mutations that destroy the amphipathicity of helix 5 lead to impaired forma-tion of LLD and diminished VLDL assembly/secreforma-tion. Because active assembly and secretion of TAG-rich VLDL represents a critical component in hepatic lipid homeostasis, particularly in
6M. Sundaram, Y. Wang, and Z. Yao, unpublished observation.
FIGURE 9. A model for the two-domain hypothesis of apoC-III action. The C-terminal lipid binding domain, encompassing the amphipathic␣-helix 5, facilitates TAG synthesis and binding to LLD, probably through its strong interaction with glycerophospholipids. The N-terminal domain, near␣-helix 2, possesses fusogenic activity in promoting fusion between LLD and VLDL precursors to form VLDL1. Some apoC-III molecules remain associated with
the resultant VLDL1after secretion, and the others dissociate from LLD and
are secreted as HDL. The K58E mutant (C3KE) is unable to bind LLD or partic-ipate in VLDL1maturation and is, therefore, secreted as HDL.
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/
protecting the liver from lipid overload, up-regulating apoC-III expression may represent a defense mechanism against lipo-toxicity under stress conditions.
Acknowledgments—We thank Vincent Ngo for generating the K58E expression plasmid, Michele Geoffrion (University of Ottawa Heart Institute) for assistance in FPLC, Dr. Jahangir Iqbal and Dr. M. Mah-mood Hussain (SUNY Downstate Medical Center) for measuring the MTP activity, and Louise B. Pelletier (University of Ottawa) for his-tology studies of mouse liver.
REFERENCES
1. Sundaram, M., and Yao, Z. (2010) Nutr. Metab. 7, 35
2. Raabe, M., Ve´niant, M. M., Sullivan, M. A., Zlot, C. H., Bjo¨rkegren, J., Nielsen, L. B., Wong, J. S., Hamilton, R. L., and Young, S. G. (1999) J. Clin.
Invest. 103,1287–1298
3. Kulinski, A., Rustaeus, S., and Vance, J. E. (2002) J. Biol. Chem. 277, 31516 –31525
4. Wang, Y., Tran, K., and Yao, Z. (1999) J. Biol. Chem. 274, 27793–27800 5. Wang, H., Gilham, D., and Lehner, R. (2007) J. Biol. Chem. 282,
33218 –33226
6. Sundaram, M., Zhong, S., Bou, Khalil, M., Zhou, H., Jiang, Z. G., Zhao, Y., Iqbal, J., Hussain, M. M., Figeys, D., Wang, Y., and Yao, Z. (2010) J. Lipid
Res. 51,1524 –1534
7. Jong, M. C., Hofker, M. H., and Havekes, L. M. (1999) Arterioscler.
Thromb. Vasc. Biol. 19,472– 484
8. Talmud, P. J., Hawe, E., Martin, S., Olivier, M., Miller, G. J., Rubin, E. M., Pennacchio, L. A., and Humphries, S. E. (2002) Hum. Mol. Genet. 11, 3039 –3046
9. Pollin, T. I., Damcott, C. M., Shen, H., Ott, S. H., Shelton, J., Horenstein, R. B., Post, W., McLenithan, J. C., Bielak, L. F., Peyser, P. A., Mitchell, B. D., Miller, M., O’Connell, J. R., and Shuldiner, A. R. (2008) Science 322, 1702–1705
10. Ginsberg, H. N., Le, N. A., Goldberg, I. J., Gibson, J. C., Rubinstein, A., Wang-Iverson, P., Norum, R., and Brown, W. V. (1986) J. Clin. Invest. 78, 1287–1295
11. Sehayek, E., and Eisenberg, S. (1991) J. Biol. Chem. 266, 18259 –18267 12. Zheng, C., Khoo, C., Ikewaki, K., and Sacks, F. M. (2007) J. Lipid Res. 48,
1190 –1203
13. Zheng, C., Khoo, C., Furtado, J., Ikewaki, K., and Sacks, F. M. (2008) Am. J.
Clin. Nutr. 88,272–281
14. Sundaram, M., Zhong, S., Bou, Khalil, M., Links, P. H., Zhao, Y., Iqbal, J., Hussain, M. M., Parks, R. J., Wang, Y., and Yao, Z. (2010) J. Lipid Res. 51, 150 –161
15. Sparrow, J. T., Gotto, A. M., Jr., and Morrisett, J. D. (1973) Proc. Natl.
Acad. Sci. U.S.A. 70,2124 –2128
16. von Eckardstein, A., Holz, H., Sandkamp, M., Weng, W., Funke, H., and Assmann, G. (1991) J. Clin. Invest. 87, 1724 –1731
17. McLeod, R. S., Wang, Y., Wang, S., Rusin˜ol, A., Links, P., and Yao, Z. (1996) J. Biol. Chem. 271, 18445–18455
18. Vance, D. E., Weinstein, D. B., and Steinberg, D. (1984) Biochim. Biophys.
Acta 792,39 – 47
19. Rogers, L., Burchat, S., Gage, J., Hasu, M., Thabet, M., Willcox, L., Wilcox, L., Ramsamy, T. A., and Whitman, S. C. (2008) Cardiovasc. Res. 78, 167–174
20. Zhong, S., Magnolo, A. L., Sundaram, M., Zhou, H., Yao, E. F., Di, Leo, E., Loria, P., Wang, S., Bamji-Mirza, M., Wang, L., McKnight, C. J., Figeys, D., Wang, Y., Tarugi, P., and Yao, Z. (2010) J. Biol. Chem. 285, 6453– 6464 21. Dowler, S., Kular, G., and Alessi, D. R. (2002) Sci. STKE 2002, l6 22. Cases, S., Stone, S. J., Zhou, P., Yen, E., Tow, B., Lardizabal, K. D., Voelker,
T., and Farese, R. V., Jr. (2001) J. Biol. Chem. 276, 38870 –38876 23. Athar, H., Iqbal, J., Jiang, X. C., and Hussain, M. M. (2004) J. Lipid Res. 45,
764 –772
24. Chang, T. Y., Limanek, J. S., and Chang, C. C. (1981) Anal. Biochem. 116, 298 –302
25. Bradford, M. M. (1976) Anal. Biochem. 72, 248 –254
26. Gangabadage, C. S., Zdunek, J., Tessari, M., Nilsson, S., Olivecrona, G., and Wijmenga, S. S. (2008) J. Biol. Chem. 283, 17416 –17427
27. Segrest, J. P., De Loof, H., Dohlman, J. G., Brouillette, C. G., and Anan-tharamaiah, G. M. (1990) Proteins 8, 103–117
28. Petersen, K. F., Dufour, S., Hariri, A., Nelson-Williams, C., Foo, J. N., Zhang, X. M., Dziura, J., Lifton, R. P., and Shulman, G. I. (2010) N. Engl.
J. Med. 362,1082–1089
29. Samuel, V. T., Liu, Z. X., Wang, A., Beddow, S. A., Geisler, J. G., Kahn, M., Zhang, X. M., Monia, B. P., Bhanot, S., and Shulman, G. I. (2007) J. Clin.
Invest. 117,739 –745
30. Liu, H., Talmud, P. J., Lins, L., Brasseur, R., Olivecrona, G., Peelman, F., Vandekerckhove, J., Rosseneu, M., and Labeur, C. (2000) Biochemistry 39, 9201–9212
31. Sparrow, J. T., Pownall, H. J., Hsu, F. J., Blumenthal, L. D., Culwell, A. R., and Gotto, A. M. (1977) Biochemistry 16, 5427–5431
32. Bou Khalil, M., Sundaram, M., Zhang, H. Y., Links, P. H., Raven, J. F., Manmontri, B., Sariahmetoglu, M., Tran, K., Reue, K., Brindley, D. N., and Yao, Z. (2009) J. Lipid Res. 50, 47–58
33. Stone, S. J., Levin, M. C., Zhou, P., Han, J., Walther, T. C., and Farese, R. V., Jr. (2009) J. Biol. Chem. 284, 5352–5361
34. Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., Amin, N., Schwikowski, B., and Ideker, T. (2003) Genome Res. 13, 2498 –2504
at National Chiao Tung University on April 24, 2014
http://www.jbc.org/