!
國立臺灣大學醫學院分子醫學所 碩士論文
Institute of Molecular Medicine College of Medicine
National Taiwan University Master Thesis
!
探討一去泛素 酶藉由調控VPS34複合物 進⽽促進細胞⾃噬之機轉
A Deubiquitinase Promotes Autophagy through Regulating VPS34 Complex
!
蕭湘蓉
Hsiang-Jung Hsiao
!
指導教授:陳瑞華 博士 Advisor: Ruey-Hwa Chen, Ph.D.
!
中華民國105年7月
July 2016
誌謝
感謝陳瑞華老師,雖然這兩年來在實驗上常遇到瓶頸,但您總是耐心地引導 我們,藉由不斷地討論訓練我們溝通、表達與邏輯思考的能力,帶領我們探索、
面對許多不同的挑戰。感謝兩位口試委員陳光超老師與徐立中老師的悉心指導,
兩位老師給予諸多重要建議,亦點出細節讓我更仔細地檢視並闡釋實驗結果。
感謝RHC lab的成員們,沒有你們我是無法完成這本論文的!有你們的日子 充實且充滿歡笑,能與你們共事是我的幸運。YU學姊和欣潔學姊,感謝妳們帶著 我入門且給予我關於未來的建議與鼓勵,在處事及態度上你們樹立了很好的模範
;感謝毛毛學長與忻怡學姊,你們總是不吝分享實驗經驗並給予忠實的建議,並 在生活上也給我許多幫助;洲維學長和文鑫學長,謝謝你們時常分享在學術界打 滾數年的各種實用經驗,並不時展現你們的幽默風趣;感謝雅葶學姊辛苦的管理 並協調實驗室事務,很欣賞妳做自己的態度,每每和妳一起自在地大笑總是能獲 取滿滿的正能量;懷萱學姊,謝謝妳給予我人生規劃上的關心與實際的建議,並 讓我更有鞭策自己運動的決心;感謝飛澐學長,你總是向我提點各個實驗的眉角 並幫助我消滅壞情緒,聽你分享特別的人生觀與生活體悟總是特別有趣;鈺軒學 長,非常謝謝你總是毫不保留地和我討論實驗與project的走向,和你一起腳踏實 地地推論或分享天馬行空的點子讓我受益良多,也很敬佩你在實驗上的衝勁與認 真的態度,期待你帶著project起飛的那天;方怡學姊與尚殷學長,跟隨你們的腳 步向前減低了我的擔心與不安,謝謝你們在課業、實驗、生活上給予的幫助和照 應,你們是我最好的典範;謝謝同學其寰在碩班每個的環節互相提醒且在面對挑 戰時互相打氣,並一起回憶清華的各種美好;丁丁與閔棋,謝謝貼心的你們協助 我報告前的準備工作及實驗室的大小事,有你們在總是能感受到青春的氣氛。實 驗室的助理們,謝謝你們除了處理實驗室的事務與實現自己的人生規劃外也抽空 協助我們。阿徹與雅鈞,感謝妳們對我的關心與照顧,和妳們相處總是像冬日的 陽光一樣溫暖;謝謝璿安在留學路上給予的鼓勵與經驗、資訊的分享,收到你的 佳節祝福或關心總是感到特別暖心;俊民和Mia,謝謝你們總是慷慨地分享,為 實驗室創造熱情與友善的氛圍;資彧與黨皓,感謝你們在完成份內的工作後仍不 遺餘力地幫助大家,並為實驗室注入許多活力與歡笑;謝謝阿姨將實驗室的環境 打理得那麼好使我們能更專注於實驗,您開朗的笑容也感染了我們。
親愛的家人與男朋友,感謝你們給我最多的愛、包容和最大的自由,讓我能 無後顧之憂地追尋自己的夢想。你們在我遇到挫折時用愛與鼓勵填補我的心;在 我想家時寄來充滿家鄉味的食物填飽我的肚子!雖然相聚的時間並不多,但我知 道你們永遠是我的後盾。老朋友們,謝謝你們的相伴與傾聽,告訴我不論流淌在 臉上的是雨水或是淚水都得向前衝的道理。期待在未來共同努力,一起完成約定 好的目標。
!
摘要
細胞⾃噬為⼀演化上具保守性的細胞作⽤。藉由分解及回收細胞質中的物質,
細胞⾃噬在維持細胞恆定及對抗環境壓⼒上扮演了不可或缺的⾓⾊。⽬前已有許 多研究證實了泛素連接系統於調控細胞⾃噬作⽤的重要性,然⽽去泛素酶如何參 與這條路徑尚未被透徹了解。在這篇研究中,我們發現⼀去泛素化酶可透過對細 胞⾃噬中的關鍵因⼦VPS34脂質激酶執⾏去泛素化進⽽促進細胞⾃噬。藉由⽔解 VPS34上所帶有以離胺酸29/48連接的異質泛素鏈,此去泛素酶能使VPS34免於被 降解並穩定VPS34-VPS15-Beclin1複合物。更重要的是,我們的研究顯⽰了此去泛 素酶不論在細胞養分充⾜狀態或養分缺乏所引發的壓⼒下與VPS34都有持續的交 互作⽤並皆能促進細胞⾃噬,因⽽暗⽰了此去泛素酶在維持基礎⽔平的細胞⾃噬
⽅⾯有⼀定的⾓⾊。此外,我們亦發現此去泛素酶和兩種分別與ATG14L或UVRAG 結合的VPS34複合物皆有交互作⽤,且也調控了胞吞作⽤。綜觀來說,我們的研 究指出此去泛素酶藉由去泛素化並穩定VPS34蛋⽩進⽽正向調控細胞⾃噬及胞吞 作⽤。
! !
! !
! !
! !
! !
! !
! !
關鍵字:細胞⾃噬、泛素、去泛素酶、VPS34複合物
Abstract
Autophagy, an evolutionary conserved process, is pivotal for maintaining cell homeostasis and adapting to environmental stresses through degrading or recycling cytoplasmic components. While the involvement of E3-ubiquitin conjugation system in regulating autophagy was unveiled by various studies, how deubiquitinases (DUBs) participate in this process receives less attention.
Here, we discovered that a promising deubiquitinase mediates autophagy induction through deubiquitinating VPS34, a lipid kinase that functions at nucleation stage. Through hydrolyzing Lys29/Lys48-linked heterotypic ubiquitin chains on VPS34, this deubiquitinase prevents degradation of VPS34 and concomitantly stabilizes VPS34-VPS15-Beclin1 complexes. Importantly, this deubiquitinase constitutively interacts with VPS34 to promote autophagy at both nutrient-rich and starved conditions, indicating its potential role in maintaining basal level of autophagy. In addition, further explorations indicate that this deubiquitinase interacts with both ATG14L and UVRAG-containing VPS34 complexes to modulates both autophagy and endocytic pathway. Together, this study revealed that this deubiquitinase positively regulates autophagic and endocytic pathways via deubiquitinating and stabilizing VPS34.
!
!
!
!
!
Keywords: autophagy; ubiquitin; deubiquitinase; VPS34 complex
Contents
誌謝 ---i
摘要 ---ii
Abstract ---iii
Contents ---iv
I. Introduction ---1
1. Autophagy ---1
2. Ubiquitination ---5
3. The Role of Ubiquitin Signal in Autophagy ---10
4. VPS34 complex ---13
II. Preface ---20
III. Material and Methods ---21
Plasmids ---21
Antibodies and Reagents ---21
Antibodies Production ---21
Cell Culture and Transient Transfection ---23
Lentivirus Production and Infection ---23
Western Blot ---24
Immunofluorescence ---25
Immunoprecipitation ---25
In vitro Deubiquitination Assay ---26
Cycloheximide-Chase Assay ---27
Mass PI3P ELISA ---27
EGFR Degradation Assay ---28
IV. Results ---29
V. Discussion ---36
VI. References ---42
VII. Figures ---51
VIII. Appendixes ---62
I. Introduction
1. Autophagy
Autophagy is an evolutionarily conserved intracellular degradative process which maintains cell homeostasis and protects cells from stress through degrading or recycling intracellular organelles and protein aggregates under basal and stressed- induced conditions. To date, at least three types of autophagy are characterized, including macroautophagy, microautophagy, and chaperon-mediated autophagy (CMA). Macroautophagy, usually referred to as autophagy, is distinct from the other autophagy types by sequestering substrates into a double-membrane structure termed autophagosome. Followed by fusing with lysosome, the contents of autophagosome are degraded by lysosomal enzymes. Although macroautophagy is generally considered as a non-selective process, there are also specialized types of autophagy, named selective autophagy, which sequester specific cargos by cargo receptors and adaptors (Lamb et al., 2013). Both microautophagy and CMA directly deliver cargo into lysosomes in different ways. Microautophagy imports cargo into the lumen through an inward budding mediated by lateral segregation of lipids and transmembrane-proteins, whereas CMA is conducted by HSP70, which targets proteins by recognizing specific motifs, and LAMP-2A, which combines HSP70 and transfers both chaperon and substrate into the lumen of lysosome (Bejarano and Cuervo, 2010; Li et al., 2012). As the main character of this thesis, macroautophagy will be referred to as autophagy hereafter.
!
1.1 Molecular Mechanism
In response to diverse environmental cues, one of the morphological features of autophagy is the formation of autophagosome. Autophagosome originates from extended compartment on endoplasmic reticulum (ER) membrane, named omegasome, and expends with plasma membrane, Golgi, mitochondria and endosomes serving as the reservoir of membrane source (Axe et al., 2008; Hailey et al., 2010; Knaevelsrud et al., 2013; Longatti et al., 2012; Ravikumar et al., 2010). Autophagosome formation can be defined into several stages: initiation, nucleation, expansion, maturation and termination (Lamb et al., 2013) .
In the initiation stage, ULK1 complex, consisting of Ser/Thr kinases ULK1/2 and accessory proteins ATG13, FIP200, and ATG101, plays a major role. Under nutrient-rich condition, ULK1 kinase activity is repressed by mTOR, a negative regulator of autophagy, through phosphorylating ULK1 at Ser757 (Ganley et al., 2009;
Hosokawa et al., 2009). Upon nutrient starvation, mTOR is inactivated to relieve its repression effect on ULK1. In addition, a key energy sensor AMPK is activated in this condition. AMPK contributes to ULK1 activation by several mechanisms. First, it relieves mTOR-mediated ULK1 repression by activating mTOR inhibitor TSC complex, or by inhibiting mTOR activity via directly phosphorylating one of it subunit Raptor (Gwinn et al., 2008; Shaw et al., 2004). Furthermore, AMPK phosphorylates ULK1 complex at Ser317 and Ser777, which is required for ULK1 activation (Kim et al., 2011). Thus, through the combinatory effect of mTOR inactivation and AMPK activation, ULK1 is activated which in turn phosphorylate its partners ATG13L and FIP200 (Dorsey et al., 2009). Activated ULK1 complex translocates from cytosol to certain domains on ER or closely attached membrane structure that would contribute to the autophagosome formation. At these places, ULK1 passes down the signal by
phosphorylating components of the VPS34 complex, composed of class III PI3K VPS34 and its partners VPS15, Beclin1 and ATG14L/UVRAG, thereby enhancing the lipid kinase activity of VPS34 (Egan et al., 2015; Russell et al., 2013; Russell et al., 2014).
Followed by its redistribution to omegasome, VPS34 complex drives the autophagosome nucleation process through producing phosphatidylinositol-3-phosphate (PI3P). Autophagy-specific PI3P binding effectors such as DFCP1 and WIPI family proteins are recruited to the membrane platform where they facilitate the development of isolation membrane (Itakura and Mizushima, 2010; Polson et al., 2010).
During the elongation stage, two ubiquitin-like (UBL) conjugation systems are required. Analogous to the ubiquitination machinery, conjugation of ubiquitin-like molecule ATG12 to ATG5 is catalyzed by E1-like enzyme ATG7 and E2-like enzyme ATG10 (Mizushima et al., 1998). Subsequently, ATG5-ATG12 conjugates associate ATG16L on the isolation membrane, forming ATG5-ATG12-ATG16L complex (Mizushima et al., 2003). In the second UBL system, ubiquitin like protein Atg8 or its mammalian orthologs LC3, GATE16, GABARAP and ATG8L are proteolytically processed by ATG4B, then subjected to ATG7 (E1-like), ATG3 (E2-like), ATG5- ATG12-ATG16L (E3-like) for subsequent phosphotidylethanoamine (PE) conjugation, converting LC3 from unlipidated I form to the lipidated II form (Geng and Klionsky, 2008; Kabeya et al., 2004; Tanida et al., 2004). Among these ATG8 homologs, LC3 has been best characterized and is widely used as a marker of autophagy. The lipidated LC3- II decorates on the membrane of autophagosome and contributes to cargo selection and autophagosome maturation via interacting with LC3-interacting region (LIR) motif- containing proteins (Birgisdottir et al., 2013; Pankiv et al., 2007). Besides the UBL systems, Atg9, a multi-spanning transmembrane protein, also assists in formation and
expansion of autophagosome by shuttling between the membrane sources and autophagosome as a membrane supplier (Yamamoto et al., 2012).
Before the closure of autophagosome, autophagy core proteins such as ATG5- ATG12-ATG16L complex dissociate from the membrane, whereas a portion of LC3-II and cargo receptors remain attached and are retained inside the autophagosome (Lamb et al., 2013). In the maturation process, autophagosome fuses with lysosome to form autolysosome, which is aided by SNAREs and tethering complexes such as STX17- HOPS complex (Jiang et al., 2014). Contents inside the autolysosome are degraded and can be recycled back to the cytosol.
Interestingly, amino acid released from autolysosome can reactivate mTORC1 at the lysosomal membrane, which contributes in part to the termination of autophagic process during prolonged starvation (Yu et al., 2010). A recent work in our lab also demonstrated that starvation-induced autophosphorylation of ULK1 facilitates its interaction with CUL3-KLHL20, which promotes Lys48-linked ubiquitination and subsequent proteasomal degradation. Additionally, CUL3-KLHL20 also governs the turnover of Beclin1, VPS34, ATG14L and ATG13 directly or indirectly, thereby contributing to autophagy termination (Liu et al., 2016).
!
1.2 Physiological Role
Basal level of autophagy takes place in all eukaryotic cells, participating in quality control of proteins and organelles and maintaining cell homeostasis. The primordial function of autophagy also includes response to metabolic, genotoxic or hypoxic stress by acting as an adaptive mechanism essential for cell survival.
Autophagy is vital in a range of physiological processes including developmental regulation and aging. Dysregulation of autophagy may lead to a broad range of diseases such as neurodegenerative diseases, infectious or inflammatory diseases, metabolic diseases, tumorigenesis, and myopathy (Choi et al., 2013; Levine and Kroemer, 2008).
Although autophagy has been regarded as a protective process, it also alternatively directs cells to cellular demise through facilitating the activation of apoptosis or necrosis in some cases (Marino et al., 2014). Thus, only by better understanding the fundamental mechanism of the process can we discriminate its character in diseases pathogenesis and eventually provide useful therapies.
!
2. Ubiquitination
Protein post-translational modifications (PTMs) widely contribute to the complexity and functional diversity of the proteome. These dynamic chemical modifications on proteins, which modulate the activity, localization, and ability to interact with other molecules, enable cells to swiftly response to environmental stimuli and delicately regulate intracellular signaling cascades. Ubiquitination is one of the key regulatory modifications among PTMs.
!
2.1 Ubiquitin Machinery
Ubiquitin is a highly conserved 76-amino-acid-residue protein that ubiquitously exists in eukaryotic cells. Through a three-step sequential conjugation reaction, ubiquitin can be appended to the ε-amino group of a Lys within the substrate protein via its C-terminal Gly residue. Ubiquitin activating enzymes, the E1s, activate ubiquitin
through conjugating the C-terminal Gly of ubiquitin to their own Cys residue in an ATP- requiring manner. In the second step, activated ubiquitin is then transferred to catalytic Cys residue of the E2 ubiquitin conjugating enzymes, which could mediate the transfer of ubiquitin to substrate directly or indirectly. The last step is conducted by E3 ubiquitin ligases, which recognize specific protein substrates and catalyze the formation of a covalent amide isopeptide linkage between C-terminal Gly of ubiquitin and ε-amino group of a Lys within the substrate (Hershko and Ciechanover, 1998).
Different topologies of ubiquitination exist in cells which grant the system with versatility. Protein substrates can be modified with a single ubiquitin on a single Lys residue or multiple Lys residues, which is defined as monoubiquitination and multi- monoubiquitination respectively. As there are seven Lys residues and the first methionine residue within ubiquitin itself (K6, K11, K27, K29, K33, K48, K63, M1), each of them can also serve as an acceptor for subsequent ubiquitin moieties, leading to the formation of polyubiquitination chain. During the elongation of ubiquitination chains, different linkages between each ubiquitin moiety result in formation of homotypic or heterotypic chains with distinct conformations and functions in the cells.
Homotypic chains are constructed with homogenous linkages, through one of the seven Lys residues and N-terminal Met residue of ubiquitin. Heterotypic chains, on the other hand, can be classified into mixed chains, chains with alternating linkage types, or branched chains, in which more than one ubiquitin moieties extended from the preceding one (Komander et al., 2009a; Komander and Rape, 2012).
Proteins marked by ubiquitin were initially thought to be delivered and destructed by proteasome (Ciehanover et al., 1978; Wilkinson et al., 1980). After years of research, “canonical ubiquitin chains” are termed, which represent the Lys48-linked
chains and Lys63-linked chains that contribute to degradation of modified proteins through ubiquitin-proteasome system (UPS) and assembly of protein interaction complexes respectively (Chau et al., 1989; Deng et al., 2000). With the application of proteomic techniques like quantitative mass spectrometry, it was revealed that all ubiquitin linkages indeed coexist in cells (Peng et al., 2003; Xu et al., 2009). Apart from extensively studied Lys48-linked chains, Lys6, Lys11, Lys29-linked and branched chains are implicated to play roles in proteasomal degradation as well (Meyer and Rape, 2014). In addition, many non-proteolytic functions of ubiquitination, including regulating activity, localization, or interactions of ubiquitinated proteins, have also been reported, showing diverse functions of ubiquitin signals (Komander and Rape, 2012).
However, with only limited knowledge about atypical ubiquitin chains and heterotypic chains, biological functions and physiological roles of them still remain to be explored.
!
2.2 Deubiquitinases (DUBs)
About 100 DUBs are predicted to exist in human genome. Based on the mechanism of catalysis, DUBs can be subdivided into two groups: the Cys proteases and the zinc-dependent metalloproteases. Among six DUB families, only the JAMM/
MPN+ DUBs are metalloproteases while members of USP, OTU, UCH, Josephine, MINDY are Cys proteases (Abdul Rehman et al., 2016; Komander et al., 2009a).
With only one-fifth of the quantity of E3 ligases, it is reasonable that the possible substrates of each DUB may surpass that of each E3 ligase. In accordance with the complexity and versatility of ubiquitin modification, DUBs must display numerous layers of specificity. It was revealed that many DUBs are able to distinguish between
different ubiquitin chain linkages, although the ability is not determined by family.
During the hydrolysis of ubiquitin chain, catalytic domains of DUBs interact with distal ubiquitin by its enzymatic S1 site and proximal ubiquitin by S1’ site. Given that the proximal ubiquitin contributes its Lys to the isopeptide bond, it is elicited that linkage specificity of DUBs is characterized by position and orientation of the proximal ubiquitin (Kulathu and Komander, 2012; Mevissen et al., 2013). Based on the concept, structure of enzymatic active site, accessory ubiquitin-binding domains (UBDs), specific sequences surrounding the ubiquitinated Lys on the proximal ubiquitin are thought to be involved in the determination of linkage specificity as well (Mevissen et al., 2013). The other layer of specificity lies to the recognition of substrates. Most of the DUBs contain additional protein-interaction domains that directly interact with the substrates or indirectly target them to a specific localization within the cells (Komander, 2010; Mevissen et al., 2013).
The general functions of DUB fall into three categories. First, DUBs are required for generating free cellular mono-ubiquitin since the precursors encoded by four human ubiquitin genes are produced into linear-fused poly-ubiquitins or ribosomal protein-fused ubiquitins. Second, DUBs remove the ubiquitin chains of the modified proteins to reverse the ubiquitin signals and to contribute to ubiquitin homeostasis through releasing reusable free ubiquitin. Third, DUBs may participate in the editing of ubiquitin chains by trimming the chains (Komander et al., 2009a).
!
2.3 TRABID
TRABID, also named ZRANB1, is a member of the second largest DUB family - OTU family. Similar to most of the members in OTU family which display an intrinsic linkage specificity toward a particular subset of ubiquitin chain types, TRABID shows a preference in hydrolyzing Lys29- and Lys33-linked ubiquitin chains 40-fold more efficiently than Lys63-linked chains in vitro (Licchesi et al., 2012; Virdee et al., 2010).
The human TRABID protein contains 708 amino acids and consists of three N-terminal Npl4-like zinc finger domains (NZFs), two ankyrin repeat ubiquitin binding domains (AnkUBD), and a C-terminal A20-like OTU domain. Although the OTU domain of TRABID and A20 are closely related, the presence of evolutionarily conserved ankyrin repeats sequence upstream of TRABID’s OTU domain implies a different catalytic preference between TRABID and A20 (Komander and Barford, 2008; Licchesi et al., 2012). Consistent with this notion, the accessory ubiquitin binding domains, NZFs and AnkUBD, were reported to provide extra contributions to linkage specificity (Abdul Rehman et al., 2016; Komander et al., 2009b; Licchesi et al., 2012; Michel et al., 2015).
AnkUBD functions to enhance the specificity and activity of the enzyme. The S1 ubiquitin binding site within the OTU domain binds distal ubiquitin, whereas the AnkUBD interacts with hydrophobic Ile44 patch of proximal ubiquitin, serving as a S1’
ubiquitin binding site (Kulathu and Komander, 2012; Licchesi et al., 2012). Based on the general function of NZF domain, three N-terminal NZFs are thought to provide additional ubiquitin binding sites via binding to the hydrophobic Ile44 patch of ubiquitin (Hurley et al., 2006). The tandem NZFs were shown to recognize K63-linked or linear ubiquitin chains for their equivalent conformations (Komander et al., 2009b).
However, further studies implicate that NZF1 specifically interacts with K29/33-linked di-ubiquitin and captures K29-linked heterotypic polyubiquitin chains containing K48
linkages present in cells (Kristariyanto et al., 2015; Michel et al., 2015). Interestingly, catalytic inactive TRABID (C443S) but not wild-type TRABID has been reported to form highly dynamic puncta in cells depending on their NZF domains. Colocalization of mutant TRABID with K29 only, K33 only, K27 only and K63 only ubiquitin mutants also indicates the possibility that inactive TRABID might be trapped by its substrates bearing these atypical ubiquitin chains (Licchesi et al., 2012; Tran et al., 2008).
Unlike A20, the physiological role of TRABID has not been completely characterized. TRABID has been reported as a positive regulator of Drosophila and mammalian Wnt-β-catenin signaling pathway through cleavage of Lys63-linked ubiquitin chain on APC, resulting in stabilization of β-catenin, which then associates with TCF/LEF to facilitate transcription of Wnt target genes (Tran et al., 2008).
However, the other study that aimed to identify small molecule TRABID inhibitors failed to confirm the role of TRABID in Wnt signaling pathway, leaving this function controversial (Shi et al., 2012). Later, TRABID was found to be involved in Drosophila immune-deficiency (IMD) pathway by directly removing Lys-63 ubiquitin chain from dTAK1 thereby reducing immune signaling output and lifespan (Fernando et al., 2014).
More recently, TRABID was identified to function as an innate immunological regulator through reducing Lys29- and Lys11-linked ubiquitin chains on demethylase Jmjd2d.
Stabilized Jmjd2d mediates demethylation of histone H3, allowing transcription of genes encoding IL-12 and IL-23 to contribute to inflammatory T cells responses (Jin et al., 2016).
!
3. The Role of Ubiquitin Signal in Autophagy
As a tightly controlled process, autophagy is known to be regulated sophisticatedly by various post-translational modifications, one of which is ubiquitination (Kuang et al., 2013).
3.1 Ubiquitin-Dependent Regulation of Autophagy
Studies have unveiled that many E3-ubiquitin ligases are involved in different steps of autophagic process, and can positively or negatively regulate autophagy.
Ubiquitin-proteasome system (UPS), usually coordinated with Lys48-linked ubiquitin chains, controls the stability of upstream regulators and crucial components in autophagy machinery. Fluctuation of abundance of these proteins may impact on the cellular autophagy amplitude or the autophagic flux (Kuang et al., 2013). For instance, UPS system plays a role in the regulation of mTOR. SCF (Skp1, Cul1, and F-box protein)-type E3 ubiquitin ligase complex, coordinating with substrate recognition subunit β-TRCP, acts on phosphorylated mTOR inhibitor, DEPTOR, and drives its subsequent destruction. The decreased abundance of DEPTOR causes enhanced activity of mTOR, leading to the inhibition of autophagy (Wang et al., 2012; Zhao et al., 2011).
VPS34 complex is the other target frequently modified by ubiquitin. Beclin1, the member with the most reported post-translational modifications in the complex, hence serves as the hub within autophagic pathway. RNF216 and NEDD4 were reported to regulate Beclin1 through conjugating Lys48- and Lys11-linked ubiquitin chains, respectively. Both types of modification target Beclin1 for degradation to negatively regulate autophagy (Platta et al., 2012; Xu et al., 2014). Besides, VPS34, ATG14L and Ambra1 are also modified and targeted to proteasomal degradation by Cul1-FBXL20,
Cul3-ZBTB16 and RNF2, respectively (Xia et al., 2014; Xiao et al., 2015; Zhang et al., 2015).
In addition to the degradative function, non-degradative ubiquitination also plays a crucial role in autophagy regulation. Lys63-linked ubiquitin chains preserve the ability to convey downstream signals through acting as scaffolds that trigger the assembly of protein complex. For instance, Ambra1 takes the center stage in different steps of autophagy with its diverse functions, including acting as a scaffold for protein complex formation and an adapter protein for linking Cul4-DDB1 E3 ligase complex to substrates (Jin et al., 2006). Under nutrient-rich condition, mTOR restrains autophagy through inducing inhibitory phosphorylation on Ambra1 at Ser52. Upon autophagy induction, Ambra1 associates with TRAF6 and bridges it to ULK1. This leads to Lys63- linked polyubiquitination of ULK1 which facilitates the stabilization and self- association of ULK1 to enhance its kinase activity (Nazio et al., 2013). The stabilized ULK1 then phosphorylates and activates Ambra1, releasing Ambra1-associated VPS34 complex from cytoskeleton for subsequent ER relocation (Di Bartolomeo et al., 2010;
Nazio et al., 2013). Moreover, Ambra1 was demonstrated to associate with Cul4-DDB1 E3 ligase complex during autophagy induction and to promote the conjugation of Lys63-linked ubiquitin chain on Beclin1. Consequently, Beclin1 displays enhanced interaction with VPS34 and elevated PI3P production activity of VPS34 (Xia et al., 2013).
!
3.2 DUBs Involved in Autophagy
Compared to E3 ligases, knowledge about how DUBs participating in autophagic process has been rather limited. In recent years, several groups have demonstrated the general effects of DUBs in autophagy through applying DUB inhibitors to cells, highlighting the tight correlation between DUBs and autophagy (Driessen et al., 2015; Liu et al., 2011; Seiberlich et al., 2013). In the case of spautin1 (specific and potent autophagy inhibitor 1), autophagy is blocked via inhibiting the activity of USP10 and USP13. The impairment of USP10/13 deubiquitinating activity results in increased ubiquitination and destabilization of components in the VPS34 complex including Beclin1, VPS15, VPS34, and ATG14L. Since direct interaction is only detected between USP13 and Beclin1, whether USP10 and USP13 act directly on other VPS34 complex components or indirectly through a Beclin1-dependent stabilization of other members in the complex, is still unknown (Liu et al., 2011).
Although accumulating evidence indicates the correlation between DUBs and autophagy, our knowledge about molecular targets and physiological activity of DUBs is incomplete. Since alterations in functionality and amplitude of autophagy are often associate to occurrence of diseases. Understanding how DUBs are integrated in the regulation of autophagy can offer new therapeutic strategies by moderating its activity (Heideker and Wertz, 2015; Magraoui et al., 2015).
!
4. VPS34 complex
VPS34 (vacuolar protein sorting 34) gene was first discovered in a screening performed in Saccharomyces cerevisiae and was demonstrated to be essential for intracellular sorting of various vacuolar proteins (Herman and Emr, 1990). However, its
function as a lipid kinase was not elucidated until the significant homology between VPS34 and mammalian Class IA PI3K, p110α was uncovered (Schu et al., 1993).
VPS34 is classified as Class III PI3K, which produces PI3P from phosphatidylinositol (PI), and is highly conserved from yeast to mammalian cells. Its critical role in regulating endocytic trafficking and autophagy has been demonstrated in mouse model, in which VPS34 acts indispensably in sensory neuron, heart and liver, and T cells (Jaber et al., 2012; Parekh et al., 2013; Zhou et al., 2010).
4.1 Complex Composition and Cellular Function
VPS34 forms diverse multiple-subunits complexes in which VPS34-VPS15- Beclin1 composes the “core complex.” It is the additional proteins that combine with the core complex determine the cellular function of each VPS34 complex. UVRAG and ATG14L, homologs of VPS38 and ATG14 in mammalian cells, are two additional proteins that exist in distinct VPS34 complexes and constitute PI3K complex I and II, respectively (Matsunaga et al., 2009). Although studies performed in yeast reported that PI3K complex I is mainly involved in autophagy while complex II plays a role in vacuolar protein sorting pathway, both complexes are implicated in autophagy in mammalian cells (Kihara et al., 2001; Sun et al., 2010). The finding that depletion or ectopic expression of key members, such as VPS15 or VPS34, affects the protein level of other subunits suggests the interdependency of these subunits to maintain complex stability (Liu et al., 2011; Platta et al., 2012; Thoresen et al., 2010; Yan et al., 2009).
Currently, structure of yeast PI3K complex II was solved, which shed a light on the organization and differential activity of the two distinct complexes. The crystal structure
of PI3K complex II in yeast appears in a Y-shaped organization, with VPS15/VPS34 forming one arm in an antiparallel manner and the parallel VPS30/VPS38 heterodimer consisting of the other arm. In addition, VPS34 C2 domain stands as a central hub in the complex to engage all subunits. Though the structure of PI3K complex I remained unsolved, it is suggested that structure of these two PI3K complexes are very similar (Rostislavleva et al., 2015). The details of VPS34 core complex components, together with several peripheral binding proteins, will be discussed below:
!
VPS15
VPS15, a putative Ser/Thr protein kinase also known as p150, is often regarded as the regulatory subunit of Class III PI3K (Herman et al., 1991). Studies have revealed that VPS15 is required for both PI3P production activity and interaction with additional proteins of VPS34 complex (Stack et al., 1993; Yan et al., 2009). Since the kinase activity of VPS15 has never been proofed, it is possible that VPS15 regulates VPS34 by interacting and modulating the active conformation of its kinase domain or stabilizing the protein but not phosphorylation (Backer, 2008; Rostislavleva et al., 2015). Besides, VPS15 may assist VPS34 targeting to a specific localization of cellular membrane via two mechanisms. First, an N-terminal myristoylation allows VPS15 to anchor on a membrane structure. Second, it possesses a WD40 domain that can interact with various proteins such as activated Rab5 on early endosomes, Rab7 on late endosomes, or even bridge between VPS15/34 and VPS30/VPS38 pairs in yeast (Christoforidis et al., 1999;
Rostislavleva et al., 2015; Stein et al., 2003).
!
Beclin1
Beclin1, homolog of ATG6/VPS30 in mammalian cells, is another component of VPS34 core complex and contributes to the interaction with additional set of proteins.
In PI3K complex I and II, the central CCD domain of Beclin1 serves as a docking site for ATG14L or UVRAG (Liang et al., 2006; Sun et al., 2008). Furthermore, some other proteins may regulate autophagy by loosely or transiently interacting with Beclin1.
Among these peripheral proteins, the most famous example is a negative regulator of autophagy - anti-apoptotic Bcl2 family proteins. Through binging to the BH3 domain of Beclin1, Bcl2 proteins inhibit autophagy by interfering the interaction between Beclin1 and VPS34 (Pattingre et al., 2005). Ambra1, on the other hand, competes Bcl2 for binding Beclin1 (Strappazzon et al., 2011). Additionally, Ambra1 promotes Beclin1 ubiquitination, which increases Beclin1 interaction with VPS34 and VPS34 catalytic activity (Fimia et al., 2007).
!
UVRAG
The UVRAG-containing VPS34 complex, mainly localized to endocytic compartments (Itakura et al., 2008), is the relatively dominant population and is known to participate in endocytic and autophagic pathways (Funderburk et al., 2010).
Endosomal fusion and autophagosome maturation are accomplished by two different populations of UVRAG that collaborate on regulating Rubicon, which is a negative regulator of autophagy and endocytic pathway and also a binding partner of UVRAG- VPS34 containing complex (Sun et al., 2010). While a population of UVRAG interacts with VPS34 complex, the other associates with Class C VPS complex, which is required for vesicle tethering and fusion (Itakura et al., 2008; Liang et al., 2008) . UVRAG-C- VPS complex on endosomes facilitates endosomal fusion and activates Rab7 by acting
as its GEF. GTP-loading enables Rab7 to compete for Rubicon binding with UVRAG- containing VPS34 complex, which finally abolish Rubicon’s sequestration of UVRAG- containing VPS34 (Sun et al., 2010).
!
ATG14L
Although being regarded as a perquisite component that only exist in autophagy- specific VPS34 complex, the mammalian ortholog of ATG14 has not been identified until 2008 (Sun et al., 2008). Despite the functions of ATG14L have not been well characterized, it was shown that ATG14L resembles its yeast homolog by directing VPS34 complex to the site where autophagosomes originally form (Obara and Ohsumi, 2011). To recruit VPS34 complex onto the omegasome, both the N-terminal conserved cysteine repeats and C-terminal Barkor/Atg14(L) autophagosome targeting sequence (BATS) domain are essential (Fan et al., 2011; Matsunaga et al., 2010). The cysteine repeats determine the localization of VPS34 complex whereas BATS domain senses membrane curvature and preferentially incorporates to the PI3P-enriched membrane through the hydrophobic surface of an intrinsic amphiphilic helix, leading to enhanced binding and stabilization of membrane curvature (Fan et al., 2011; Matsunaga et al., 2010). Interestingly, several proteins, such as RACK1, Dapper1, PAQR3 and SLC35D3, were reported to promote autophagy by enhancing ATG14L-associated PI3K complex formation under various conditions (Ma et al., 2014; Wei et al., 2016; Xu et al., 2016;
Zhao et al., 2015), while Nrbf2 modulates complex formation conversely (Zhong et al., 2014).
!
4.2 Regulation of VPS34
Compared to other components such as Beclin1 and ATG14L in the complex, regulatory process on VPS34 by post-translational modification is relatively rare. To date, studies have revealed that VPS34 could be regulated by phosphorylation, ubiquitination, and sumoylation.
Through phosphorylation, kinase activity and complex composition of VPS34 could be fine-tuned under various conditions. The key energy sensor AMPK was demonstrated to regulate distinct VPS34 complexes upon glucose starvation. Among multiple VPS34 complexes, non-autophagy VPS34 complex is suppressed by AMPK through phosphorylating VPS34 at Thr163/Ser165 to protect cells from starvation (Kim et al., 2013). During mitosis, the mitotic kinase CDK1 phosphorylates VPS34 at Thr159, which reduces VPS34 lipid kinase activity through disrupting its interaction with Beclin1 (Furuya et al., 2010). The other member of CDK family - CDK5, a neuronal CDK that functions during neuronal development as well as neurotransmitter signaling in mature nervous system, phosphorylates VPS34 at Thr668, leading to the inhibition of VPS34 lipid kinase activity (Furuya et al., 2010). Moreover, the interplay between phosphorylation and ubiquitination also plays a role in regulating VPS34.
When cells encounter DNA damage, level of FBXL20, an adaptor of Cul1-Skp1 E3 ligase complex, is induced in a p53-dependent manner and the following mitotic arrest leads to the accumulation of cyclin B1 that activates CDK1. Phosphorylation of VPS34 by CDK1 facilitates its recognition by Cul1-FBXL20, which targets VPS34 for ubiquitination and proteasomal degradation, thereby downregulates autophagy (Xiao et al., 2015).
Ubiquitination, as mentioned, is a complicated modification for its competence to form various chain types. However, VPS34 has only been reported to be modified
with the degradative ubiquitin chains. In addition to Cul1-FBXL20, previous work of our lab also discovered that another E3 ligase, Cul3-KLHL20, displays an enhanced binding with VPS34 upon prolonged starvation and mediates its ubiquitination and degradation (Liu et al., 2016). Deubiquitination and stabilization of VPS34 has also been addressed in the study referring a small molecular autophagy inhibitor Spautin-1, which interferes with the deubiquitinating function of USP10/13 and causes destabilization of VPS34 and other components in the complex (see section 3.2) (Liu et al., 2011).
Sumoylation is also involved in VPS34 regulation. Under stress-induced condition, increased intracellular acetylated HSP70 binds to VPS34-Beclin1 complex and recruits SUMO E3 ligase - KAP1. KAP1 mediates Lys840 sumoylation on VPS34, which strengthens the interaction between VPS34 and Beclin1 (Yang et al., 2013).
!
!
!
II. Preface
To gain insights into the potential role of DUBs in autophagic pathway, we intended to identify and characterize DUBs that regulate autophagic process. For this purpose, we collaborated with Dr. Guang-Chao Chen’s lab to conduct a shRNA-based screen. An lentivirus-based shRNA library targeting 91 human DUB genes was used to transduce HeLa cells that stably expressed GFP-LC3. The systematic screening was accomplished with automatic image analysis, in which the number and intensity of LC3 of each clone were recorded and normalized with control cells. Among the possible candidates, TRABID was revealed to be one of the potential modulators of autophagy.
The effect of TRABID on autophagy was validated by observing the autophagy marker LC3 of two TRABID knockdown clones. Depletion of TRABID hindered formation of GFP-LC3 puncta (Appendix 1A, Yu-Hsuan Chen, unpublished results) and inhibited LC3-II conversion with Bafilomycin A1 treatment, the autophagy inhibitor, under both basal and starved conditions (Appendix 1B, Yu-Hsuan Chen, unpublished results), suggesting that TRABID positively regulates autophagy.
In this thesis, we aim to study the detail mechanism of TRABID in autophagy regulation and to identify TRABID substrate that is responsible for this effect.
!! !
III. Material and Methods
Plasmids
Plasmid encoded His-Ubiquitin, Flag-WT-TRABID, Flag-C443S-TRABID, V5- WT-TRABID, 3xFlag-VPS34 were described in previous study (Liu et al., 2016; Yuan et al., 2014). Two TRABID fragments—3NZF and ankyrin repeat—were generated by PCR and cloned into pET32a vector. EGFP-ATG14L and EGFP-UVRAG were kindly provided by Dr. Guang-Chao Chen. For establishing stable overexpression clone, Flag- TRABID were subcloned into the lentivirus-based vector pLAS5W.Pneo.
Antibodies and Reagents
Mouse anti-flag (M2; Sigma), goat anti-TRABID (GeneTex), rabbit anti- GAPDH (GeneTex), rabbit anti-LC3B (Abcam), rabbit anti-LC3B (Cell Signaling), rabbit anti-VPS34 (PI3 Kinase Class III; Cell Signaling), rabbit anti-Beclin1 (Novus), rabbit anti-Ambra1 (Novus), rabbit anti-Flag (DDDDK, GeneTex), rabbit anti-β-actin (GeneTex), mouse anti-6xHis (Clontech), rabbit anti-V5 (Millipore), rabbit control IgG (Abcam), rabbit anti-VPS34 for IP (Echelon), rabbit anti-VPS34 (life technologies), rabbit anti-VPS15 (PIK3R4; Novus), rabbit anti-Beclin1 (Santa Cruz), EasyBlot anti- rabbit IgG (GeneTex), mouse anti-α-Tubulin (Millipore), mouse anti-GFP (Santa Cruz) antibodies were purchased from commercial sources. Rabbit anti-Rubicon and Rabbit anti-EGFR were kindly provided by Dr. Guang-Chao Chen. Coomassie Brilliant Blue R-250 and lysozyme was purchased from USB and BSA (Bovine serum albumin) was obtained from NEB. IPTG (Isopropyl β-D-thiogalactoside), DTT (1,4-Dithiothreitol), Bafilomycin A1 and cycloheximide were bought from Sigma-Aldrich, whereas recombinant human EGF protein was purchased from R & D systems.
Antibodies Production
Two TRABID fragments—3NZF and ankyrin repeats—were generated PCR and cloned into pET32a vector, which carries a 6xHis tag, followed by transformation into Rosetta competent cells. Cells were inoculated at 37℃ in LB containing 100 µM/ml of Ampicilin until OD600 reached 0.6, then induced with 0.5mM IPTG at 16℃ for 16-18 hours. For ankyrin repeats, recombinant proteins were purified under native condition.
Cells were pellet down and lysed with lysis buffer (100mM NaCl, 20mM Tris-HCl [pH=8.0], 1% NP40, adjust to pH=7.5) supplemented with protease inhibitor, 5mM DTT, 10mg/ml lysozyme. After six cycles of sonication (sonicate 20 sec, rest 20 sec as one cycle) and centrifugation, supernatants were transferred to 15 ml tube and incubated with Ni sepharose (GE Healthcare) at 4℃ for 2 hours. Beads were washed with wash buffer (300mM NaCl, 50mM Tris-HCl [pH=8.0], 20mM imidazole, adjust pH to 6.3) for three times and eluted with elution buffer (300mM NaCl, 50mM Tris-HCl [pH=8.0], 250mM imidazole, adjust pH to 7.5) at 4℃ for 30 min with agitation, which were repeated for two times. In the other hand, 3NZF were purified under denaturing condition. Cells were lysed with lysis buffer (300mM NaCl, 50mM Tris-HCl [pH=8.0], 1mM EDTA, adjust to pH=8.0) supplemented with protease inhibitor, 5mM DTT, 2%
Triton X-100, 10mg/ml lysozyme. Followed by sonication and centrifugation, insoluble pellet were washed with denaturing wash buffer (300mM NaCl, 50mM Tris-HCl [pH=8.0], 2M urea, 1mM EDTA, adjust pH to 6.3) supplemented with 1% Triton X-100 and 5mM DTT for three times then washed for another three times with wash buffer free from urea and Triton (300mM NaCl, 50mM Tris-HCl [pH=8.0], 1mM EDTA, adjust pH to 6.3). Insoluble proteins were extracted from the wash pellet with warm extraction buffer (300mM NaCl, 50mM Tris-HCl [pH=8.0], 6M guanidine hydrochloride, adjust to pH=8.0) supplemented with 20mM β-mercaptoethanol and
1mM PMSF rocking at 37℃ for 30min. Supernatants containing extracted proteins were collected after centrifugation and the denaturing binding buffer (300mM NaCl, 50mM Tris-HCl [pH=8.0], 2M urea, 20mM imidazole, adjust pH to 7.5) were added then subjected to incubation with Ni sepharose at 4℃. Beads were washed with denaturing wash buffer (300mM NaCl, 50mM Tris-HCl [pH=8.0], 20mM imidazole, adjust pH to 6.3) for three times and eluted with denaturing elution buffer (300mM NaCl, 50mM Tris-HCl [pH=8.0], 2M urea, 500mM imidazole, adjust pH to 7.5) at 4℃.
Purified antigens were provided to LTK BioLaboratories for immunization followed by 7 times of boosting. For antibody purification by affinity chromatography, soluble antigens were required for column packaging. To remove urea presenting in eluted antigens that were purified under denaturing conditions, eluted antigens were loaded into dialysis cassettes (ThermoFisher) and dialyzed in three different dialysis buffer with decreasing concentration of urea (1M urea and 1mM DTT in 1x PBS, 0.5M urea in 1x PBS, 1xPBS).
Cell Culture and Transient Transfection
293T, 293FT cells were cultured in Dulbecco’s Modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/
streptomycin (P/S). HeLa cells were maintained in Minimum Essential Medium (MEM) containing 10% FBS, 1% P/S and 1mM sodium pyruvate. For starvation, cells were cultured in Earle’s Balanced Salt Solution (EBSS; Sigma-Aldrich). Transient transfection of 293T was performed using calcium phosphate method. Cell medium was refreshed after 16-18 hours of incubation, and the cells were harvested 48 hours after transfection.
Lentivirus Production and Infection
Recombinant lentiviruses were packaged in 293FT cells by transiently transfecting 14 µg packing plasmid (pCMV△8.91), 2 µg envelope VSV-G plasmid (pMD.G) together with 14 µg Flag-TRABID cloned in pLAS5w.Pneo vector using calcium phosphate method. Cell medium was refreshed after 8-12 hours of incubation, and the supernatant containing packaged viruses were harvested after 42-48 hours of transfection then filtered by 0.45 µm pore-size syringe filter.
For infection, the supernatant were supplemented with 8 µg/ml polybrene and the infected cells were selected with 800 µg/ml neomycin. The constructs mentioned above were obtained from National RNAi Core Facility (NRC), Academia Sinica, Taiwan.
Western Blot
Cells were lysed by 1x RIPA buffer (150 mM NaCl, 20 mM Tris-HCl [pH=7.5], 1% NP40, 0.1%SDS, 1% sodium deoxycholate) with protease inhibitors (1mM PMSF, 1 µg/ml aprotinin and 10 µg/ml leupeptin). After sonication and centrifugation, supernatants were quantified by Bradford (Bio-Rad, Hercules, CA) and prepared with sample buffer (50mM Tris-HCl [pH=6.8], 2% SDS, 10% glycerol, 0.02% bromophenol blue, 2.5% β-mercaptoethanol) that is 1/4 volume of the sample. Protein samples were resolved by SDS-PAGE then transferred onto PVDF membranes (Millipore) that are activated by methanol beforehand. Membranes were blocked with 1-5% non-fat dry milk or 1% BSA in 1x TBST (Tris-buffered saline with 0.1% Tween-20) at room temperature for at least 20 min then incubated with indicated primary antibodies, which is diluted in blocking buffer, at 4℃ overnight. Next, membranes were washed 5 min for three times with 1 x TBST then incubated with indicated HRP-conjugated secondary antibody at room temperature for an hour. After washed 10 min for three times with 1x TBST and rinse with ddH2O, membrane were subjected to detection by immersing in
Western HRP substrates purchased from Millipore (LuminataTM Crescendo) or PerkinElmer, Inc. (Western Lightning® Plus-ECL)
Immunofluorescence
2.3x105 of HeLa cells stably expressing Flag-TRABID were seeded on glass coverslips, which was placed at the bottom on the 6-well plates, one day before the experiment. For serum starvation, cells were washed with 1x PBS (phosphate-buffered saline) for two times and EBSS for one time then cultured with EBSS for the indicated time. Cells were washed with 1x PBS for two times and fixed with 4% formaldehyde for 20-25min on ice. After washed with 1xPBS for three times, cells were permeabilized at room temperature with -20℃ methanol for 10 min and washed another three times with 1x PBS. Cells were then blocked with blocking reagents (1% BSA, 10% goat serum in 1xPBS) at 4℃ for 1hr, and incubated with anti-LC3B antibody diluted with blocking reagents at 4℃ overnight . After washed with 1xPBS for three times, cells were rinsed with blocking reagents and subjected to fluorescent dye-conjugated secondary antibodies (life technologies) and DAPI (Sigma-Aldrich) at room temperature for 1 hour. Stained cells were washed with 1x PBS for three times, mounted onto microscope slides with fluorescence mounting medium (Dako), then examined by confocal microscope (Zeiss LSM510) with 63x oil objective lens. A total of approximately 100 cells were analyzed in each group and the average puncta/cell was quantified.
Immunoprecipitation
For Fig. 4 and Fig. 8A, cells were lysed with 1x RIPA supplemented with protease inhibitors and phosphatase inhibitors (1mM Na3VO4, 2mM NaF, 200µM NaPPi). Followed the above-mentioned protocols, cell lysates containing equal amount
of proteins were incubated at 4℃ for 2 hours with anti-Flag agarose beads (M2; Sigma- Aldrich) or anti-V5 agarose beads (Sigma-Aldrich) respectively. After washed three times with lysis buffer and prepared with sample buffer, immunoprecipitants were subjected to Western blot analysis. While in Fig. 8B, same protocol was applied except replacing the lysis buffer with 1x NP40 buffer (150mM NaCl, 50mM Tris-HCl [pH=7.5], 1% NP40) and the beads with GFP-Trap®_A (chromotek).
For endogenous co-immunoprecipitation in Fig. 6, the other kind of lysis buffer, named “Lysis Buffer-C” (100mM NaCl, 10mM Tris-HCl [pH=7.5], 2mM EDTA, 1%
NP40), supplemented with protease and phosphatase inhibitors were used. Equal amount of cell lysates were first pre-cleared at 4℃ for 40-60min with protein A sepharose (GE Healthcare). Then, the supernatants were incubated with 5 µg of control IgG or anti-VPS34 antibody at 4℃ overnight followed by 2 hours of incubation with protein A. Immunoprecipitants were subjected to Western blot analysis followed the standard protocols, except for replacing the secondary antibody with EasyBlot anti- rabbit IgG.
In vitro Deubiquitination Assay
For substrate purification, 293T cells were transfected with His-Ubiquitin together with 3xFlag-VPS34. After 48 hours of transfection, cells were lysed with 2x RIPA and incubated with anti-Flag agarose beads for 2 hours. Purified ubiquitinated substrates were washed with 2x RIPA for five times and 2x elution buffer (300mM NaCl, 20mM Tris-HCl [pH=7.5]) for one time. Ubiquitinated substrates were eluted with 2x elution buffer containing 150 ng/ml of 3xFlag peptides by agitating at 4℃ for 30 min, which were repeated for two times. Concentration of eluted proteins were quantified by SDS-PAGE relative to BSA. Purified substrates could be stored at -80℃.
For enzyme preparation, 293T cells were transfected with V5-WT-TRABID. After 48 hours of transfection, cells were lysed with 1x RIPA and incubated with anti-V5 agarose beads for 2 hours. Purified enzymes were washed with 1x RIPA for three times and 2x reaction buffer (300mM NaCl, 20mM Tris-HCl [pH=7.5], 0.4% NP40, 0.2% Triton X-100) for one time. After draining the washing reagents, same volume of 2x reaction buffer containing 20 mM DTT was added to the beads for resuspension and incubated at 23℃ for 10 min to restore the full activity of enzymes. Purified TRABID proteins were incubated with 500-1000 ng of substrates at 37℃ for 4 hours with agitation. After the reaction was terminated by preparing with sample buffer, samples were resolved by Bis- Tris Gel using 1x MOPs running buffer (50 mM MOPs, 50mM Tris-HCl, 1mM EDTA, 0.1% SDS, 5mM sodium bisulfate) then followed by standard protocol of Western blot analysis.
Cycloheximide-Chase Assay
293T cells that stably expressing two different TRABID shRNAs were seeded on 6-well plates a day before the experiment, whereas 293T cells that transiently transfected with Flag-TRABID were subjected to cycloheximide treatment 36-42 hours after transfection. Cells were incubated with medium containing 100 µg/ml of cycloheximide for indicated time and the lysates were subjected to Western blot analysis. VPS34 protein level was first quantified relative to β-actin using image J and then normalized to the amount of untreated cells.
Mass PI3P ELISA
HeLa cells stably expressing empty vector or Flag-TRABID were seeded on 10 cm plates a day before the experiment. To extract lipid from cells, cells were scraped with ice cold 0.5M TCA followed by centrifugation. Pellet were collected and wash two
times with 5% TCA containing 1mM EDTA. Pellets were washed another two times with MeOH:CHCl3(2:1) to eliminate neutral lipids. Acidic lipids were extracted by applying MeOH:CHCl3:12M HCl (80:40:1). Supernatants were subjected to phase spit step through adding CHCl3 containing 0.1N HCl and the organic phase (lower phase) were collected to eppendorfs for 30 min of vacuum drying by SpeedVac. Extracted lipids were then assayed by using PI3P Mass ELISA kit (Echelon, K-3300). Extracted lipids were rehydrated and the samples and PI3P standards were transferred to incubation plate for following incubation on shaker with PI3P detector at room temperature for an hour. Mixture of each well was transferred to the corresponding well of detection plate and incubated for another hour. After washing three times with PBS- T, secondary detectors were added to each well and incubate at room temperature for an hour. Followed by three times of washing, TMB solution were added and incubated for 30 min in dark to develop the color. The reaction was stopped by adding 1N H2SO4, and the plate were presented to Beckmen Paradigm for absorbance measurement.
EGFR Degradation Assay
HeLa cells stably expressing empty vector or Flag-TRABID and 293T cells stably expressing control or TRABID shRNA were seeded on 6-well plates a day before the experiment. When the confluency reached 80% the next day, cells were washed two times with PBS and serum-starved overnight in medium containing only MEM or DMEM. On the third day, cells were stimulated with medium (MEM or DMEM supplemented with 20mM HEPES, 0.2% BSA) containing 100 ng/mL EGF for the indicated time. EGFR protein level was quantified relative to β-actin using image J and normalized to the amount of untreated cells.
!
IV. Results
TRABID antibody production
Owing to the poor quality of commercially available TRABID antibodies, we intended to generate recombinant TRABID protein for antibody production. To this end, we cloned two fragments of TRABID, the 3 NZF domain and Ankyrin-repeat domain, into pET32 vector, which carries a 6xHis tag, and transformed them into Rosetta competent cells. After inoculation and IPTG induction, recombinant proteins of 3NZF and ankyrin repeats were purified with Ni-sepharose under denaturing and native conditions, respectively, and quantified by SDS-PAGE (Figure 1A). The result indicated the recovery of recombinant proteins with high purity and yield. Purified antigens were provided to a biotech company for polyclonal antibody production. To test the resulting antisera, cell lysates obtained from TRABID knockdown clones, transiently expressing Flag-TRABID cells and several nontransfected cell lines were used. Although the antisera showed higher sensitivity to ectopically expressed Flag-TRABID compared with commercial Flag and TRABID antibodies, endogenous level of TRABID still could not be detected (Figure 1B). Hence, we tried to purify antibodies by affinity chromatography using the soluble antigens. Western blot analysis revealed that purified antibodies were able to recognize both overexpressed and endogenous TRABID proteins. Furthermore, the reduced Western blot signal in TRABID knockdown cells indicated the specificity of the antibodies (Figure 1C). These antibodies were then used in the following studies presented in this thesis.
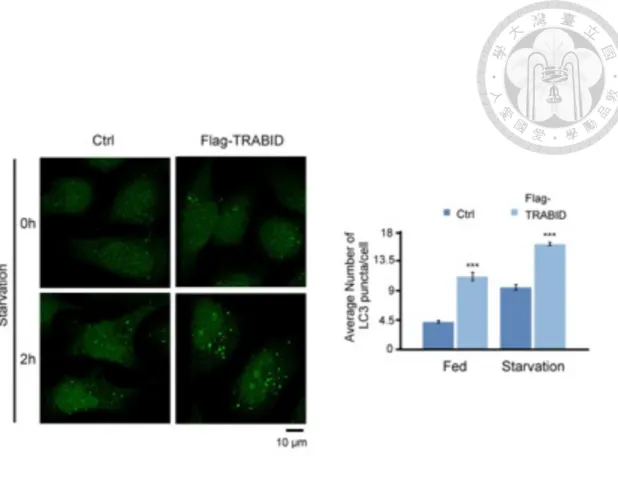
TRABID promotes autophagy induction under both basal and starved conditions
To further study the role of TRABID in autophagy regulation, we established a TRABID overexpression HeLa cell line and assayed for the abundance of autophagosomes in fed and starved cells. As shown in Fig. 2, the LC3 puncta (a marker of autophagosome) were increased upon TRABID overexpression in both fed and starved cells. However, increased autophagosome levels could result from either an increased onset of autophagy or a decreased autophagic flux. To discriminate between the two possibilities, we treated cells with Bafilomycin A1, which blocks autophagic flux by preventing the fusion of autophagosome with lysosome. Under this circumstance, TRABID overexpression significantly enhanced LC3 lipidation (LC3-II), at both basal state and upon starvation (Figure 3). Together, our data are consistent with the results of previous study in our laboratory using TRABID knockdown strategy and confirm a positive regulatory role of TRABID in autophagy.
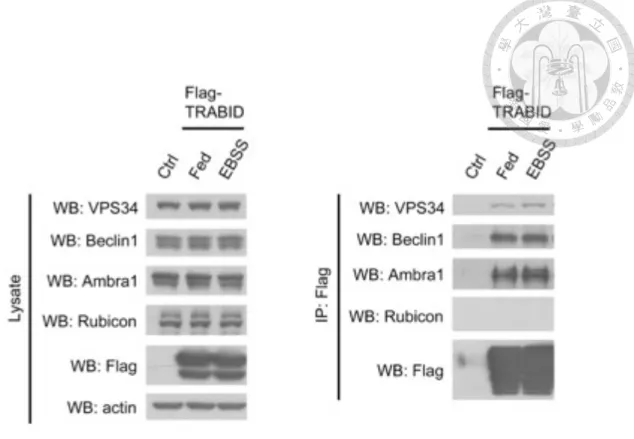
TRABID interacts with several components within VPS34 complex
Studies in our laboratory revealed that TRABID knockdown attenuated ATG16 puncta and DFCP-1 puncta (markers of forming autophagosome) without affecting mTOR activity (Yu-Hsuan Chen, unpublished data). These findings suggest a functional place of TRABID at the autophagosome nucleation stage. We therefore tested whether VPS34 complex serves as a target of TRABID. To this end, interaction between transiently expressed Flag-WT-TRABID and endogenous VPS34 complex subunits under fed and starved conditions was examined by co-immunoprecipitation, in which Flag-TRABID was pulled down with M2 beads. Owing to the availability of antibodies, only VPS34, Beclin1, Ambra1 and Rubicon were analyzed by Western blot. The result showed that VPS34, Beclin1, Ambra1, but not Rubicon, the negative regulator of
autophagy, were co-immunoprecipitated with TRABID (Figure 4). Furthermore, the association between TRABID and these proteins was not altered upon starvation, indicating a constitutive interaction.
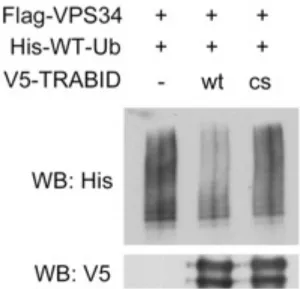
TRABID promotes VPS34 deubiquitination
Next, we explored whether TRABID can influence on the ubiquitination status of VPS34 complex components. To this end, 293T cells were transfected with His-WT- ubiquitin, Flag-VPS34 together with empty vector, V5-WT-TRABID or the catalytically dead mutant V5-C443S-TRABID. Ubiquitinated proteins were pulled down with Ni sepharose under denaturing condition and followed by Western blot analysis.
Remarkably, WT-TRABID significantly reduced ubiquitination level of VPS34 while the catalytic dead mutant failed to remove ubiquitin from VPS34 (Appendix 2).
Subsequently, direct deubiquitination of VPS34 by TRABID was tested by in vitro deubiquitination assay, in which ubiquitinated Flag-VPS34 and V5-TRABID were separately purified from 293T cells and then were incubated together. Similarly, WT- TRABID, but not C443S TRABID mutant, was capable of deubiquitinating VPS34 (Figure 5). These results indicate that VPS34 as a substrate of TRABID.
Notably, most of the DUBs in the OTU family display intrinsic linkage specificity. TRABID was also reported to preferentially cleave Lys29/33-linked di- ubiquitin in vitro and Lys11/29-linked or Lys63-linked polyubiquitin chains in vivo (Afonina and Beyaert, 2016; Fernando et al., 2014; Licchesi et al., 2012; Tran et al., 2008). To examine the types of VPS34 polyubiquitin chains affected by TRABID, we conducted in vivo deubiquitination assay by introducing 3xFlag-VPS34, V5-WT- TRABID together with a set of ubiquitin mutants with individual Lys residues are
mutated to Arg. Ubiquitinated VPS34 was pulled down with Ni sepharose under denaturing condition and subjected to Western blot analysis. Overexpression of WT- TRABID was less able to remove ubiquitin molecules lacking Lys29 and Lys48, suggesting that TRABID tends to hydrolyze Lys29 and Lys48-linked polyubiquitin chains on VPS34 (Appendix 3). Although TRABID’s ability to cleave Lys48-linked polyubiquitin chain in vivo has never been demonstrated, its first NZF domain was reported to capture Lys29-linked heterotypic polyubiquitin chains containing Lys48 linkages from cell extracts (Kristariyanto et al., 2015), which is consistent with our finding. Collectively, these data indicate that catalytic activity of TRABID is essential for VPS34 deubiquitination through Lys29/48 linkages.
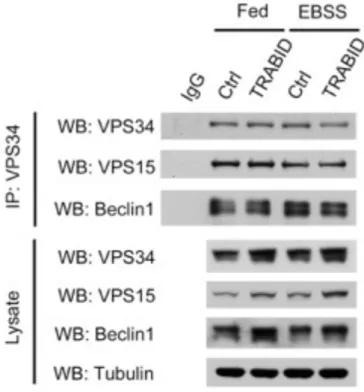
TRABID does not facilitate complex formation but regulates stability of VPS34
After identifying VPS34 as TRABID’s substrate, we next sought to elucidate how TRABID promotes autophagy by interacting and deubiquitinating VPS34. Here, two possible downstream events were proposed: deubiquitination of VPS34 may lead to impacts on either its interaction with other components in the complex or its lipid kinase activity. We first tested whether TRABID affects the formation of VPS34 complex by conducting endogenous co-immunoprecipitation. Cell lysates of control or stably expressing Flag-TRABID HeLa cells under fed or starved conditions were subjected to IgG or anti-VPS34 antibody. Western blot analysis revealed that overexpressing TRABID did not alter the amount of endogenous VPS15 and Beclin1 that co- precipitated with endogenous VPS34 regardless of serum starvation (Figure 6), implying that the complex assembly was not influenced by TRABID.
Although overexpression of TRABID did not modulate the formation of VPS34 complex, we noticed a remarkably elevated protein abundance of endogenous VPS34, VPS15 and Beclin1 in cell lysates, pointing to a potential role of TRABID on stabilizing VPS34. To assess whether TRABID regulates the stability of VPS34, we examined the half-life of endogenous VPS34 by cycloheximide (CHX) treatment, which blocks de novo protein synthesis. Endogenous VPS34 protein displayed a prolonged half-life in
stably expressing Flag-TRABID HeLa cells compared with control HeLa cells (Figure 7A), whereas the accelerated turnover rate was observed in two TRABID knockdown clones (Figure 7B). Considering that the stability of components within the VPS34 complex is interdependent on each other (Liu et al., 2011; Platta et al., 2012; Thoresen et al., 2010), we therefore deduced that increased level of VPS15 and Beclin1 might attribute to the stabilization of VPS34, which possibly increases the population of VPS34-VPS15-Beclin1 complex in cells.
TRABID interacts with both ATG14L and UVRAG-containing VPS34 complexes and increases intracellular PI3P production
ATG14L and UVRAG are two additional proteins that exist in distinct VPS34 complex in a mutually exclusive manner. While ATG14L is reported to facilitate autophagy process, UVRAG participates in both endocytic and autophagic pathways.
Since our previous data suggested that TRABID regulates VPS34, we were also interested in investigating the pool of VPS34 complexes that TRABID interacts with.
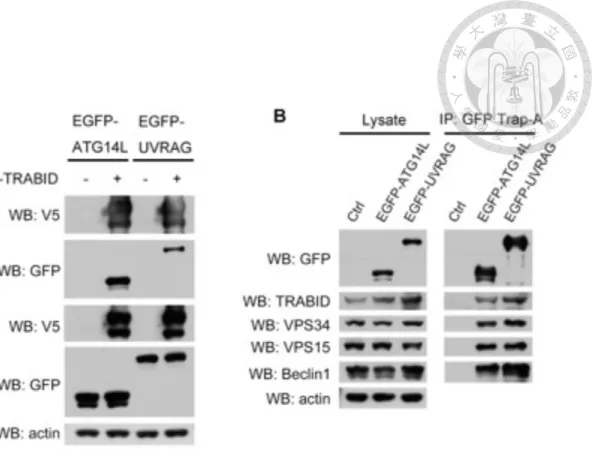
First, interaction between ectopically expressed V5-WT-TRABID and EGFP-ATG14L or EGFP-UVRAG in 293T cells was examined by co-immunoprecipitation, in which V5-TRABID was pulled down with V5 beads and followed by Western blot analysis.
Both EGFP-fused ATG14L and UVRAG could be detected in the immunoprecipitants (Figure 8A). The association between transiently expressed EGFP-ATG14L or EGFP- UVRAG and endogenous TRABID were also determined by using GFP-Trap_A.
Consistently, endogenous TRABID could associate with endogenous VPS34, VPS15, Beclin1 and both EGFP-fused ATG14L and UVRAG (Figure 8B), indicating that TRABID is a binding partner of both ATG14L and UVRAG-containing VPS34 complexes.
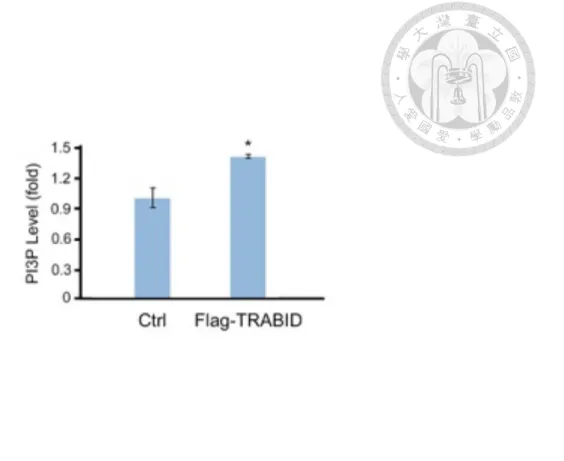
Localized production of PI3P after membrane targeting of VPS34 complex is believed to be essential to both endocytic and autophagic pathways for passing down the signals via recruiting effector proteins, which contain PI3P-binding domains. Our aforementioned data indicate that TRABID could deubiquitinate and stabilize VPS34, which also led to stabilization of VPS15 and Beclin1 and possible increase of VPS34- VPS15-Beclin1 complexes. Therefore, we postulated that TRABID may also have influence on PI3P production. Intracellular PI3P of control and stably expressing Flag- TRABID HeLa cells were extracted and subjected to quantitative mass PI3P ELISA analysis. As expected, overexpression of TRABID elevated intracellular PI3P level under nutrient rich condition (Figure 9), supporting our hypothesis that TRABID increases the population of functional VPS34 complexes through deubiquitinating and stabilizing VPS34.
TRABID also regulates endocytic pathway
Given that TRABID interacts with UVRAG-containing VPS34 complex, we wonder if TRABID also modulates endocytic pathway via regulating VPS34. PI3P signaling, occurred on endosomal membrane, controls sorting of internalized receptor
molecules, including recycling of transferrin receptor (Tfr) to plasma membrane or targeting epidermal growth factor receptor (EGFR) to multivesicular bodies (MVB) for later degradation (Jaber et al., 2012; Raiborg et al., 2003). Therefore, we carried out EGFR degradation assay to monitor TRABID’s effect on endocytic trafficking. After serum starvation for overnight, EGF was employed for indicated time to stimulate EGFR activation and subsequent degradation. In stably expressing Flag-TRABID HeLa cells, EGFR degradation rate was accelerated compared with that in control cells (Figure 10A). In contrast, EGFR degradation was drastically attenuated in TRABID knockdown clones (Figure 10B) in accordance with previous results described in VPS34-null MEFs (Jaber et al., 2012). Thus, our findings implied that regulation of VPS34 by TRABID also has an impact on endocytosis in addition to autophagy.