行政院國家科學委員會專題研究計畫 成果報告
光斷裂 RNAi 配位基及其在細胞週期控制上的應用
計畫類別: 個別型計畫
計畫編號: NSC94-2311-B-041-002-
執行期間: 94 年 08 月 01 日至 95 年 07 月 31 日 執行單位: 嘉南藥理科技大學醫藥化學系
計畫主持人: 林敬涵
計畫參與人員: 林敬涵
報告類型: 精簡報告
處理方式: 本計畫涉及專利或其他智慧財產權,2 年後可公開查詢
中 華 民 國 95 年 10 月 30 日
UV-Inducible RNA Interference
RNA interference (RNAi) has become a powerful and widely adopted tool for gene-specific knockout experiments1. RNAi is initiated by the presence of long double-stranded RNA or hairpin RNA molecules processed into discrete 21-23 nucleotide RNA duplexes known as small interfering RNA (siRNA). These siRNAs are then incorporated into the RNA-induced silencing complex (RISC), which in turn directs the cleavage of messenger RNAs (mRNAs) that are complementary to the antisense strand of the siRNA2-5. By rationally designing the siRNA sequence, one can knock out genes selectively. One feature contributing to the widespread adoption of RNAi technology is the availability of commercial synthetic facilities capable of producing siRNAs that enable biologically oriented laboratories that are lacking organic synthesis capability to adopt this technology.
In this communication, we have developed a strategy that allows external control of the RNAi process. The strategy involves incorporating photolabile monomers consisting of 1,3-(2- nitrophenyl)-1,3-propanediol into a siRNA-based small hairpin RNA6 (shRNA) using standard machine-assisted RNA synthesis.
The same photolabile monomers are designed to be compatible with incorporation at both the 5' and 3' ends of the shRNA in such a manner as to sterically hinder the siRNA-RISC interaction.
Upon UV irradiation at 365nm, the photolabile moiety is cleaved off leading to the release of the formerly “caged” siRNA to enter the RNAi pathway and inhibit the expression of the target gene (Figure 1).
Figure 1. UV-inducible RNAi using short hairpin (sh) RNA with photolabile groups (yellow ovals) covalently attached at its 3'-and 5'-ends.
Upon irradiation, caged shRNA gets released to enter the RNAi pathway resulting in endonucleolytic cleavage of the target mRNA.
Our strategy differs from that published by Friedman7 where caging is effected by reaction of the precursor hydrazone so as to form the corresponding diazo compound, which can react with phosphate groups found on the RNA. Our strategy creates a single shRNA with two photolabile groups while the Friedman strategy creates a mixture of protected siRNAs, and depending on the amount of reagents used, the number of phosphate modifications can be increased7. Additionally, our strategy is compatible with machine-assisted solid-phase RNA synthesis, hence does not require additional synthetic chemical steps. The photolabile moiety named Linker-365 (L365) was synthesized as previously described8 with the slight modification that LDA was used instead of the reported K2CO3 to deprotonate 2-nitroacetophonene (Scheme 1).
NO2O +
NO2 H O
LDA 41%
NO2O OH NO2
89%
NO2OH OH NO2 DMTCl, CH2Cl2
Et3N,75%
NO2OH OTs NO2
Ethylene Glycol 40%
1 2 3 4
NO2OH O NO2
DMTCl, CH2Cl2 Et3N,75%
NO2OH O NO2
5 6 7
POCH2CH2CH2CN TEA, CH2Cl2, 85%
O NO2
O2N P O
OR2 O
365 nm pH 7
+
8 9
10
OH ODMT
NO2O O NO2 ODMT P
O N NC
Cl N
O P O O R1O
O P OH O R1O NaBH4
P OR2 HO
O O
Scheme 1. Synthesis of Linker-A (1-8), and photocleavage (9 and 10).
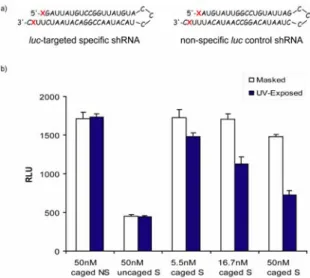
To analyze the efficiency of our UV-inducible RNAi system, we designed a shRNA composed of the sense and antisense strands of a siRNA targeting firefly luciferase, as well as a non- specific shRNA with scrambled sequence9. They were synthesized using standard solid-phase chemistry with L365 attached at both their 3'- and 5'-ends (Figure 2a). Compared to monomer attachment at only one end, modification at both ends of the shRNA ensures an added level of control over the activation of RNAi, ostensibly because photo-deprotection of both ends is required for function (data not shown).
The ability of the caged shRNAs to silence gene expression in an externally controllable fashion was investigated in CHO-AA8- Luc Tet-Off cells that stably expressed luciferase. Cells were seeded at 105 per well in black 96-well plates the day prior to transfection to achieve about 70% confluency after overnight incubation at 37ºC. shRNAs were transfected with Lipofectamine (Invitrogen) for a period of 8 hr and then changed to fresh culture medium. The plate was covered on one side to protect the caged shRNAs, and exposed on the other side to 365nm from a Blak- Ray UV lamp at a distance of 10cm for 15 min. The cells were returned for an additional 48 hr incubation followed by analysis using the Promega Luciferase Assay System.
Upon irradiation, caged target-specific shRNAs were activated and brought about approximately 5, 35, and 50% knock-down of luciferase activity at 5.5, 16.7, and 50nM respectively (Figure 2b).
Controls suggested a regulated RNAi effect, as intended. No effect was observed on the non-specific shRNA transfected cells.
In addition, uncaged target-specific shRNA showed no difference in its gene silencing activity between the masked and the UV- exposed cells. However, a much more potent gene silencing effect was produced by the uncaged shRNA compared to caged shRNA post irradiation. Longer UV exposure times up to 30 min (data not shown) did not improve the potency of the silencing effect from the caged shRNA, possibly due to the inefficiency of the photocleavage of L365 as illustrated in a photocleavage experiment using a L365-incorporated 43-mer DNA where only about 85% of cleavage was achieved after 30min of irradiation (see supporting information). Another possible explanation could be that there is some degree of cell membrane protection against UV light.
Figure 2. (a) Target-specific (S) and non-specific (NS) shRNA sequences with L365 covalently attached at both 3'- and 5'-ends. X denotes L365. (b) Effect of the two types of L365-modified shRNA on RLU (relative light units) in the absence of UV (light bars) and with UV exposure (dark bars). Where indicated, "caged" means that the RNA molecules had L365 covalently attached; "uncaged" refers to shRNA specifically targeted to luc but lacking attached L365.
To further investigate the potential for caged shRNA to manipulate gene expression in an adjustable fashion, a time- course experiment was performed to evaluate the effect of UV exposure for periods up to 15 min on the degree of gene suppression (Figure 3). A gradual decrease in luciferase activity was observed, indicating time-dependent UV-activation of caged shRNAs.
An effective inducible system requires that gene expression be tightly regulated such that “leaky” expression does not obscure the experimental results; therefore, the basal level of RNAi prior to UV activation must be minimized. In our system, the L365 modification at both ends of the shRNA requires double deprotection reactions to take place before the shRNA becomes competent to induce RNAi. Introducing this extra deprotection step successfully blocks more than 80% of RNAi activity at concentrations of the L365-modified shRNAs as high as 50nM (see supporting information). The residual background RNAi activity is likely due to auto-cleavage by UV from the environment during the experimental procedure.
Figure 3
.
Decrease in RLU (relative light units) with irradiation time in cells transfected with caged shRNA, but not in cells transfected with uncaged shRNA.Another valuable feature of our system is that when the inducer (UV) is applied there is no apparent toxicity nor interference with
the protein complexes involved in the RNAi process.
Additionally, RNAi can be induced to levels that are sufficient to generate biological responses very specifically against the target gene. The simple application of UV as inducer affords excellent flexibility for controlling the degree of RNAi at a desired level by varying the time of exposure.
The results from this work establish the potential for control of gene expression in both spatial and temporal fashion. Desired levels of gene expression can also be attained by exposure to selected doses of UV. The properties of our UV-inducible RNAi system make it a very attractive tool to study a wide range of biological activities, particularly in areas such as developmental biology where many genes in the course of development are spatially restricted in animals10 as well as in plants11. This temporal external control of RNAi is also ideal for studying the eukaryotic cell cycle as the cycle is regulated by the periodic synthesis and destruction of proteins such as cyclins that associate with and activate cyclin-dependent kinases12-13. These experiments are currently under way and the results will be reported in due course.
Acknowledgments. We are grateful to Prof. Michael Waring for helpful comments and Dr. David Ann for providing access to instruments and assistance with cell culture. This work was aided by grant #IRG-58-007-45 from the American Cancer Society to CCCW and by grant #NSC94-2311-B-041-002 from the National Science Council of the Republic of China to CHL.
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References:
1. Hannon, G.J.; Rossi, J.J. Nature 2004, 431, 371-8.
2. Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Nature 1998, 391, 806-11.
3. Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. Cell 2000, 31, 25-33.
4. Yu, J.Y.; DeRuiter, S.L.; Turner, D.L. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 6047-52.
5. Mello, C.C.; Conte, D. Jr. Nature 2004, 431, 338-42.
6. Paddison, P.J.; Caudy, A.A.; Bernstein, E.; Hannon, G.J.; Conklin, D.S.
Genes Dev. 2002, 16, 948-58.
7. Shah, S; Rangarajan S.; Friedman, S. H. Angew. Chem. Int. Ed. 2005, 44, 1328-32.
8. Zhang, K.; Taylor, J.S. Biochemistry 2001, 40, 153-9.
9. Ui-Tei, K.; Zenno, S.; Miyata, Y.; Saigo, K. FEBS Lett. 2000, 479, 79-82.
10. Gittes, G.K.; Rutter, W.J. Proc. Natl. Acad. Sci. U.S.A., 1992, 89, 1128- 32.
11. Yu, B.; Yang, Z.; Li, J.; Minakhina, S.; Yang, M.; Padgett, R.W.;
Steward, R.; Chen, X. Science. 2005, 307, 932-5.
12. Morgan, D.O.; Roberts, J.M. Nature 2002, 418, 4495-6.
13. Ubersax, J.A.; Woodbury, E.L.; Quang, P.N.; Paraz, M.; Blethrow, J.D.;
Shah, K.; Shokat, K.M.; Morgan, D.O. Nature 2003, 425, 859-64.
Supporting Information
Photocleavage experiment
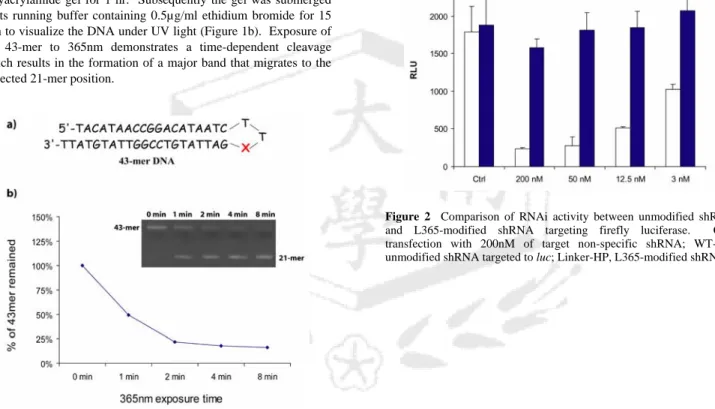
Photosensititivity of the monomer L365 was tested by incorporation at the 22nd position (Figure S1a) of a 43-mer hairpin oligonucleotide. Photocleavage of L365 was monitored by the disappearance of the 41-mer hairpin and the appearance of two co- migrating 21-mer oligonucleotides. For reasons of economy this experiment was conducted using DNA instead of RNA. The 43-mer DNA was dissolved in water at 0.88µM in an 1.5mL Eppendorf tube and was exposed to 365nm light from a Blak-Ray UV lamp (XX-15L, 15W) at a distance of 10cm at room temperature.
Aliquots of 5µl were taken at time 0, 1, 2, 4, and 8 minutes of irradiation and electrophoresed on a denaturing 15%
polyacrylamide gel for 1 hr. Subsequently the gel was submerged in its running buffer containing 0.5µg/ml ethidium bromide for 15 min to visualize the DNA under UV light (Figure 1b). Exposure of the 43-mer to 365nm demonstrates a time-dependent cleavage which results in the formation of a major band that migrates to the expected 21-mer position.
Figure 1. (a) A 43-mer DNA with L365 incorporated at the 22nd position. X denotes L365. (b) Cleavage of the L365-containing 43-mer at various UV exposure time points. Relative amounts of 43-mer remaining (%) are plotted against time at 0, 1, 2, 4, and 8 min. The inset is a 15% polyacrylamide denaturing gel showing the starting 43-mer DNA at time 0 min, and its 21-mer cleavage product at various times.
Cell culture and validation of L365-modified shRNA
CHO-AA8-Luc Tet-Off cells (BD Biosciences, Clontech) that stably expressed luciferase were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 units/ml penicillin, 100 µg/ml streptomycin, 100 µg/ml G418, and 100 µg/ml hygromycin B. 105 cells per well were seeded in black 96-well plates (BD Falcon) to achieve ~70% confluency at the time of the transfection after an overnight incubation at 37ºC in a humidified atmosphere containing 5% CO2. To test our hypothesis that the L365-modified shRNA can impede the RNAi pathway, shRNA that targets firefly luciferase was generated with
L365 covalently attached to its 3'- and 5'-ends. A range of concentrations of the modified and unmodified shRNAs were tested in the luciferase assay. shRNAs were delivered into the CHO-AA8- Luc Tet-Off cells by transfection using Lipofectamine (Invitrogen) as directed by the manufacturer. The results are summarized in Figure 2, demonstrating that the unmodified shRNA can effectively silence luciferase expression in a dose-dependent manner. In contrast, when the shRNA was modified at its 3'- and 5'-ends, no gene silencing effect was observed and all RNAi activity was abolished. This indicated that covalent attachment of L365 to the ends of shRNA can successfully block its RNAi activity at concentrations as high as 50nM, which can therefore be used as the starting concentration to test the effect of the L365 modified shRNA on luciferase activity after UV irradiation.
Figure 2 Comparison of RNAi activity between unmodified shRNA and L365-modified shRNA targeting firefly luciferase. Ctrl, transfection with 200nM of target non-specific shRNA; WT-HP, unmodified shRNA targeted to luc; Linker-HP, L365-modified shRNA.