行政院國家科學委員會專題研究計畫 成果報告
化妝品與養髮製品中毛果芸香鹼測定方法之研究
計畫類別: 個別型計畫
計畫編號: NSC94-2113-M-041-001-
執行期間: 94 年 08 月 01 日至 95 年 07 月 31 日 執行單位: 嘉南藥理科技大學醫藥化學系
計畫主持人: 王來好
報告類型: 精簡報告
處理方式: 本計畫涉及專利或其他智慧財產權,2 年後可公開查詢
中 華 民 國 95 年 10 月 12 日
政院國家科學委員會專題研究計畫成果報告 化妝品與養髮製品中毛果芸香鹼測定方法之研究
計畫編號:NSC 94-2113-M-041-001 計畫主持人:王來好
中文摘要
利用汞膜修飾金與碳纖維電極,研究 金屬(銅、鈷、鎳與鋅)錯合物之伏安行 為。將汞膜修飾金絲電極當做電化學 偵測器( electrochemical detector, ECD ) 之電池的工作電極連接高效能液相層 析儀,測定藥物毛果芸香鹼含量。
Abstract
The voltammetric behaviors of metal chelate namely copper, cobalt, nickel and zinc in the presence of pilocarpine on thin-film mercury modified gold and carbon fiber electrodes have been studied. Cobalt (II) forms a labile chelate with pilocarpine in0.1 M lithium perchlorate, which subjected to
differential pulse voltammetry at mercury coat–ed carbon fibre and gold wire microelectrodes, respectively, give rise to a catalytic phenomenon which can be utilized for the determination of pilocarpine in the pharmaceuticals. The gold with surface deposit of mercury was used as sensor in liquid
chromatography-electrochemical detection (LCEC) to enhance the sensitivity and selectivity of analytical methods for the determination of trace concentration of pilocarpine.
報告內容 前言
Introduction
Imidazole compounds are of
considerable biomedical important [1], for example, pilocarpine
(2(3H)-Furanone, -ethyldihydro-4- [(1-methyl-1H-imidazol-5-yl) methyl]-) is a cholinergic parasympathomimetic agent belonging to the group of
imidazole alkaloids [2]. Recently it has been widely used as a topical agent for the treatment of glaucoma [3] and for the management of patients with xerostomia [4]. The transition metal complexes aromatic N-containing ligands such as imidazole and their derivatives to antitumor agents, is drawing attention [5]. The imidazole nucleus is polarographically inert and the lactone ring of pilocarpine is not reducible at a mercury electrode down to –2.5 V [6]. Although Kolthoff [7]
reported a catalytic wave for pilocarpine at –1.8 V (pH 8), it was found to be of little value. The polarographic
(voltammetric) effects of the evolution of hydrogen catalysed by ligands in solutions containing cobalt and nickel salts are very important for a sensitive analytical determination [8-10]. Review the literature concerns to the authors’
recent research on the reduction of Ni (II) and Co (II) at mercury electrodes
catalysed by imidazole compounds [11-14]. Complexation methods employing copper, iron, nickel and cobalt for electroactive pharmaceuticals have been utilized [15-17]. However, up to the present, there have been relatively few reports of analytical methods for determining polarographically inert substances such as pilocarpine, which complex with metal ions [18-19]. In addition to polarography, the survey of the literature for pilocarpine
determination include ion-selective electrode [20], spectrophotometry [21-23], gas-chromatography with mass spectrometric detection [24], liquid chromatography using fluorescence derivation [25], with UV 214 nm [26-27], with tandem with mass spectrometric detection [27], and micellar electrokinetic capillary chromatrography [29-30].
研究目的
The poly (vinyl chloride) pilocarpine membrance electrodes only suited in the analysis of ophthalmic preparations by direct potentiometry. The determination of pilocarpine by conventional
spectrophotometry without prior
separation procedures is not possible due to the overlapping of their UV spectra and the presence of interfering
excipients. The lactone ring of pilocarpine is known to be highly
unstable in high temperature, and should be acylated using heptafluorobutyric anhydride. The trifluoroacetylated derivative was monitored using GC/MS.
The analysis of pilocarpine by LC-UV is complicated, since it lack chromophoric group and is, thus, determined at low wavelengths (typically around 214 nm).
The pre-column derivatization of pilocarpine with
4-bromomethyl-7-methoxycoumarin and subsequent LC with fluorescence
detection, but the derivatization needed 12-48 h for completion. Capillary electrophoresis can be achieved high separation efficiencies and resolution and performed in different modes.
However, it combined with UV absorbance detection, does not extract the more polar pilocarpine, the
determination of which is essential for the investigation of metabolism of pilocarpine. Merbel et al [25] used LC-MS to determine pilocarpine but this method requires costly instrumenation.
Due to the existence of the imidazole moiety enables pilocarpine to form complexes with divalent metal ions namely copper (II), cobalt (II), nickel (II) and zinc (II) so that this paper is
concerned with a study of the chelation of pilocarpine with metal ions using votammetric techniques such as cyclic voltammetry (CV) and differential pulse voltammetry (DPV) to develop of sensitive methods for the determination of pharmaceuticals (pilocarpine).
文獻探討
1. De Luca, L., Naturally occurring and synthetic imidazoles: Their chemistry and their biological activities, Curr.
Med. Chem. 2006,13 (1) 1-23.
2. J. Scheerer, I.G.P. Vieira, G. Jacob, Extraction and analysis of the natural active ingredient pilocarpine,
Bioforum Int. 1, 1997, 38-40.
3. J. Vandervoort, A. Ludwing, Evaluation of pilcarpine loaded gelatin particles for topical
ophthalmic use. J. Pham. Belg. 54(3), 1999, 85-86.
4. R. Cecanho, M. Anaya, A. Renzi, J.V.
Menani, L.A.J. De Luca, Sympathetic mediation of salivation induced by intracerebroven tricular piloarpine in rats, J. Auton. Nerv. Syst. 76(1), 1999, 9-14.
5.G. Zhao ., H.Lin,.Curr. Metal complexes with aromatic N-containing ligands as otential agents in cancer treatment Med.
Chem., 2005, 5 (2),137-147.
6. G. C. F.Clark, G. J. Moody, J.D.R.
Thomas, The polarographic behavior of copper complexes of pilocarpine and some related imidazoles, Anal.
Chim. Acta. 98, 1978, 215-220.
7. I.M Kolthoff., J. J. Lingane, Polarography, Vol. 2,1952, Interscience Publishes New York, p.836.
8. B. K. Puri, A. Kumar,
Electrochemical behavior cobalt and nickel complexes with
epsilon –caprolactam at dropping mercury electrode, Electrochim. Acta, 1984, 29 (3),345-347.
9. U. Kucharska, Complexing influence of cobalt and nickel ions on the electrochemical reduction of pterin
and related compounds at a mercury drop
Talanta, 1997,44 (1)85-96.
10. L. A.M. Baxter, A. Bobrowski, A. M.
Bond, G. A. Heath, R. L. Paul, R.
Mrzljak, J. Zarebski, , Electrochemical and Spectroscopic Investigation of the Reduction of Dimethylglyoxime at Mercury Electrodes in the Presence of Cobalt and Nickel
Anal. Chem., 1998, 70 (7), 1313-1322.
11.M. L. F. Juan , L. M. A. Jesus ., G. C.
Josefa ., J. M. O. Arturo ., F. Dolores ., Electrocatalytical reduction of metal ions by adsorbed organic nitrogen bases on mercury electrodes, Recent.
Res. Devel. In Electrochem. 1998, 1, 281-293.
12. M. Aslanoglu, M. Ulusoy, M. Guler, E.
Tas, Synthesis, characterization and electrochemical studies of nickel (II) and cobalt (II) complexes with novel
bidentate salicylaldimines, Polish J.
Chem.2004, 78 (7),903-909.
13.U. Ray,D. Banerjee,S..Chantrapromma, H. K.Fun, J. N..Lin, T. Lu, H. C. Sinha,.
CobaltII)-azoimidazole complexes:
Structures of [Co(MeaaiMe) 4](ClO4)2 ? H2O and [Co(HaaiMe)2(NCS)2]
(MeaaiMe = 1-methyl-2-(p-tolylazo) imidazole; HaaiMe =
1-methyl-2-(phenylazo)imidazole) ,Poly hedron, 2005,24( 9) 1071-1078.
14. M. Hirotsu, M. Kojima, K. Nakajima,S.. Kashino, Y.Yoshikawa, Y. Stereochemistry and
electrochemistry of cobalt (II) and cobalt (III) complexes containing optically active
tetradentate schiff base ligands
Bull.Chem.Soc.Japan, 1996,69 (9) 2549-2557.
21 J.E. Parkin, The assay of benzalkonium chloride in pilocarpine, hypromellose and polyvinyl alcohol ophthalmic drops by second-order derivative
ultraviolet spectrophotometry. J.
Pharm. Biomed. Anal. 11(7), 1993, 609-11.
15.W. F.Smyth , R. Scannell T. K. Goggin , D. L Hernandes, A spectrometric, separation and voltammetric study of the complexation reactions of
bromazepam with iron (II), copper (II) and cobalt (II), Anal. Chim. Acta.
1982, 141,321-327.
16. R. K. Gilpin , L. A. Pachia ,
Pharmaceuticala and related drugs, Anal. Chem., 1985, 57, 29R-46R.
17.C. Semiha , B. Iclal , B. Ender , C..
Osman. Electrochemical study of the mixed ligand complexes of Co (II) and Ni (II) with acetylsalicylic acid and nicotinamide ,J. Coord. Chem., 2003, 56 (6) 511-521.
18. K. Gromek, Application of
complexation equilibria of pilocarpine with cobalt (II) and nickel (II) for polarographic determination of pilocarpine, Chem. Anal. 1984, 29, 387-394.
19.N. X. Wang, J. M. Chen, R. C. Lu, X. L.
Zhang and J. Q. Deng,
Electroanalytical chemistry studt of pilocarpine, Acta Phar. Sinica, 1990, 25, 362-367.
20. N. M. P. Manuel, L. F. C. Jose, B. S.
M. Conceceicao, Application of poly(vinyl chloride) pilocarpine membrance electrodes in ophthalmic products, Analyst, 119,
1994,2327-2330.
22. M.L. Satuf, J.C. Robles, H.C.
Goicoechea, A.C. Olivieri,
Simultaneous determination of timol maleate and pilocarpine hydrochloride in ophthalmic solutions by first
derivative UV spectrophotometry and PLS-1 multivariate calibration, Anal.
Lett. 32(10), 1999, 2019-2033.
23. A. H. Elsayed, S.P. Agarwal,
Spectrophotometric determination of atropine, pilocarpine and strychnine with chloranilic group, Talanta, 29, 1982, 535-537.
24.K.L. Birk, J.D.Y. Hsieh, J.L.
Demetriades, B.K. Matuszewski, Detemination of pilocarpic acid in human plasma by capillary gas chromatography with mass-selective detection. J. Chromatogr. B, 719, 1998, 93-102.
25. C. Aromdee, J.P. Fawcett, L. Robin, Sensitive high-performance liquid chromatographic assay for pilocarpine in biological fluids using fluorescence derivatisation. J. Chromatogr. 677(2), 1996, 313-318.
26.L. Weaver, J.M. Tanzer, P.A. Kramer, High-performance liquid
chromatographic determination of pilcarpine in plasma. J. Chromatogr.
581(2), 1992, 293-6.
27. D. Suketu, B. James, A simplified and rapid high-performance liquid
chromatographic assay for pilocarpine hydrochloride, J. Chromatogr. Sci. 30,
1992, 149-153.
28.N.C. van de Merbel, A.P. Tinke, B.
Oosterhuis, J.H.G. Jonkman, J.F. Bohle, Determination of pilcarpine,
isopilocarpine, pilocarpic acid and isopilocarpic acid in human plasma and urine by high-performance liquid chromatography with tandem mass spectrometric detection, J. Chromatogr.
708(1+2), 1998, 103-112.
29 W.N. Charman, A.J. Humberstone, S.A.
Charman, Analysis of pilocarpine and its degradation products by micellar electrokinetic capillary
chromatography, Pharm. Res. 9(9), 1992, 1219-1223.
30.W. Baeyens, G. Weiss, W. Van Der, W.V.D. Bossche, Analysis of pilocarpine and its trans epimer, isopilocarpine by capillary chromatography, J. Chromatogr.
638(2), 1993, 319-326.
31 L. H. Wang 1, *, H..C. Hsia 1, Y.Z. Lan, Design of a Flow – Through
Polarographic Sensor Based on Metal Films for Determination
N-nitrosodiethanolamine in Rabbit Biological Fluids, Sensor has accepted for publication on July 15, 2006.
32. A.M. Bond, Modern polarographic methods in analytical chemistry, Marcel Dekker, New York 1980, p.195.
研究方法
1.Preparation of chelate of pilocarpine A 100 mg (0.408 m mole) amount of pilocarpine hydrochloride dissolved in aqueous sodium hydroxide and 5.30 mg (0.0408 m mole) metal chlorides
dissolved in deionized water, then transferred both into a10 ml reaction flask and made up to10 ml volume with deionized water. This mixture was heated for 5, 10, 15, 20, 30, 40 and 50 min in a constant temperature water bath maintained at 50 ± 2℃.
3.Determination of the chelate of pilocarpine by voltammetry
CV and DPV experiments were performed using an EG&G Princeton Applied Research (Princeton, NJ) Model 394 connected to an EG&G325 Faraday cage with Smart stir and KO269 A Faraday cage. The in situ formulation of mercury –coated carbon fibre and gold wire microelectrodes, respectively, as working electrodes, were achieved according to our published paper [31].
Comparative tests of supporting electrolytes and pH such as Britton-Robinson buffer (pH 2.56-8.33), phosphate buffer (pH 2.12 and 6.60), acetate buffer (pH 4.50) and an aqueous containing 0.1 M tetrabutylammonium hydroxide (pH 12.01), lithium perchlorate (pH 6.01), sodium perchlorate, tetraethylammonium perchlorate, and tetraethylammonium tetrafluorobrate supporting electrolyte were conducted. A 1.0 g amount of ophthalmic solution and treatment of dry mouth (xerostomia) tablet sample was accurately weighed, dissolved in 10 ml of 0.1 M sodium bicarbonate aqueous and extracted three times with 6 ml chloroform. The aqueous phase and organic phase, respectively, were
collected, evaporated and transferred into a 10 ml calibrated flask and make up to a volume with methanol and deionized water (1:1 v/v).
. In order to obtain calibration graph for the chelation of pilocarpine, 10 ml of supporting electrolyte was pipetted into a voltammetric cell and de-aerated with nitrogen for 4 min before votammetric measurement. By micropipette, aliquots of 40.8 mM pilocarpine solution were added. After each addition
voltammograms were obtained; the solution de-aerated for 1min after each addition before obtaining the
voltammogram. Quantitative analyses were performed in the differential pulse mode. The potential was set at –0.4 to –2.0 V versus Ag/AgCl reference electrode for reduction. The pulse height was 50 mV, and the scan rate 10 mVs-1 with a film electrode. For sample solution analysis, 1ml of the solution was pipetted to volume with 0.1 M lithum perchlorate solution. This
solution was analysed by DPV using the same condition as for calibration graph.
3.Determination of chelate of pilocarpine by HPLC
Working standard solutions were prepared from a stock standard solution in methanol in the range 2- 40 mg/ml.
RP- HPLC was performed on a Phenomenex Luna C18 5mm (250 × 4.6 mm) column. The isocratic mobile phases were 10:90, 20:80, 30:70 and 40:60 v/v) methanol - phosphate buffer
(pH 6.86) and methanol –lithium perchlorate (0.1-0.2 mM), respectively;
the mobile phase flow rate was 0.3 ml/min. Construction of a flow-through polarographic detector combined with an ultraviolet detector (HPLC-UV) has been previously described [31] Detection after separation on the Phenomenex Luna C18
column was carried out using ultraviolet detector set at 235 nm. The polarographic detector was operated at –1.4 V. Using the injection value, 20 μl of the prepared sample solution and standard solution were chromatographed under the operating conditions described above.
Quantitation was based on the peak area of the sample.
結果與討論
1.Choice of analytical method
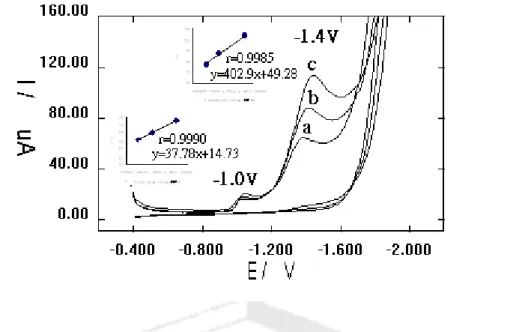
To compare the electroanalytical utility of Hg/Au and Hg/GCE (glassy carbon electrode) modified electrodes, we studied the electrochemical experiments, in cyclic voltammograms (CV) on chelate of pilocarpine (Fig.1), and it was found that Hg/Au film gave a better performance than Hg /GCE, and was chosen for use in the determination of pilocarpine in pharmaceuticals. Cyclic voltammograms (CV) of the Co (II), Zn (II), Cu (II), and Ni (II) chelates with pilocarpine at the Hg/Au electrodes were shown in Fig. 2. Due to the Co (II) chelate was reduced to a less negative potential than the others that this complex was chosen to study the voltammetric behaviour, and to
determine the concentration of pilocarpine. CV of Co (II)-pilocarpine in Britton-Robinson buffer (5.01), phosphate buffer (pH 6.60) and lithium perchlorate (pH 6.01) solution with a mercury-modified gold electrode showed two well-defined reductions.
Britton-Robinson buffered was peak current more higher than on the other, but peak potential more negative than the other. Therefore, the lithium perchlorate (pH 6.01) solution was used to Co (II)-pilocarpine determine levels in pharmaceuticals.
2. Voltammetric behaviors of Co (II)-pilocarpine
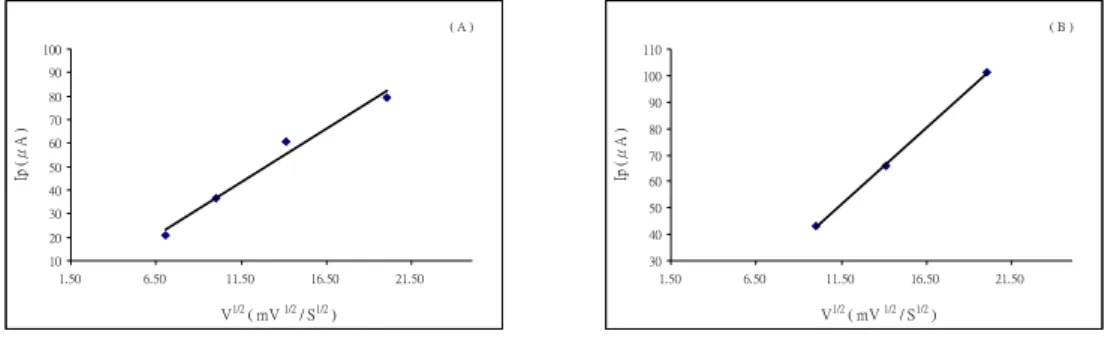
CV obtained using the standard (Co (II)-pilocarpine) addition method on an Hg/Au electrode (Fig. 3, the regression equations used was y = 37.78 x +14.73, the correlation coefficient was r =0.9990 for first wave –1.0 V; and y = 402.9 x + 49.28; r = 0.9985 for second wave – 1.40 V, respectively) showed two well-defined reduction peaks. In the reverse scan, no oxidation peak is observed (Fig. 3). The cathodic peak current ip is found to increase with the scan rate (Fig.4) and the concentration of Ca (Fig. 3). Good linearity was observed between the peak height (current) and the square root of the scan rate (v 1/2 Fig.5). The regression equations used was y = 4.56 x - 9.08, the correlation coefficient was r =0.9905 for first wave –0.882 V; and y = 5.83 x-15.6;
r = 0.9997 for second wave – 1.37 V, respectively). The ip is proportional to Ca
and v 1/2. The current function values (Ip
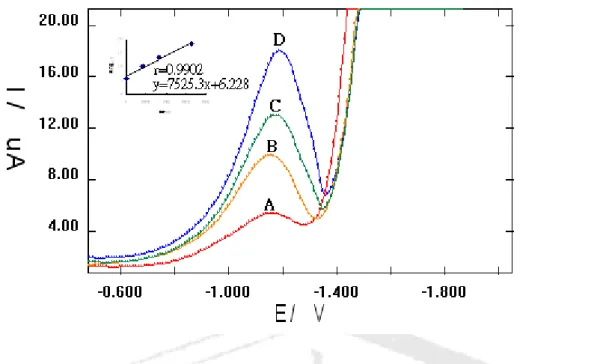
= ip/ACa v 1/2) in lithium perchlorate and Britton-Robinson buffer (where A is area of electrode) were constant. This type of voltammetric behavior should correspond either to diffusion controlled irreversible charge transfer followed by an irreversible fast chemistry reaction [32]. Differential pulse voltammograms (DPV) obtained using the standard (Co (II)-pilocarpine) addition method on an Hg/Au electrode (Fig. 6, the regression equations used was y = 210 x +5.08 the correlation coefficient was r =0.9916 for first wave –0.69V; and y = 287 x +3.15;
r = 0.9992 for second wave –1.20V, respectively) showed two well-defined reduction peaks. A representative DPV voltammogram of a commercial ophthalmic solution is shown in Fig.7
3. Separation of Co (II)-pilocarpine Various ratios of methanol- –lithium perchlorate (0.1-0.2 mM) (10:9 0, 20:80, 30:70, 40:60 v/v) were experimented with on chelate of pilocarpine. After various studies of the retention behavior of the Co (II)-pilocarpine, baseline separation was achieved. Methanol- lithium perchlorate (0.2 mM) (30:70, v/v) was found to be the best mobile phase for a good resolution and for the least peak interference in the matrix. Fig.8 shows representative LC-EC and LC-UV chromatograms for the Co (II)-pilocarpine.
Fig.1 Cyclic voltammograms of pilocarpine (20.8 mM) and Co(0.130 mM) at
different modified electrodes in 0.1 M LiClO4: (-)Hg (2 mM)/GCE and (----)Hg (2 mM)/Au, Scan rate 50 mV/s
Fig. 2 Cyclic voltammograms of 4.17x10-2 M pilocarpine in 0.1 M LiClO4 containing a variety of metal 1.35 x10-4 M (a) Co (II);(b) Zn (II) (c)Cu(II); (d) Ni(II) , Scan rate50 mV/s.
Fig. 3 Cyclic voltammograms of 0.1M LiClO4 solution containing1.30x10-4 M cobalt and pilocarpine (a) 4.17x10-2 M; (b) 8.35 x10-2 M); (c) 16.7 x –2 M. Scan rate 50 mVs-1
Fig.4 Cyclic voltammograms of0.1M LiClO4 solution containing Co (0.336 mM) and pilocarpine (3.27mM) on Hg (2 mM)/Au modified electrode at different scan rates (─) rate100 mV/s; (---) rate 200 mV/s;( ) rate 400 mV/s. Peaks: (a) first wave at -0.882 V; (b) second wave at-1.369 V.
( A )
10 20 30 40 50 60 70 80 90 100
1.50 6.50 11.50 16.50 21.50
V1/2 ( mV 1/2 / S1/2 )
Ip (μA )
( B )
30 40 50 60 70 80 90 100 110
1.50 6.50 11.50 16.50 21.50
V1/2 ( mV 1/2 / S1/2 )
Ip (μA )
Fig. 5 Magnitude of the peak current, ip, for Co-piloacrpine reduction as a function of square root of scan rate (A) first wave at -0.882 V; (B) second wave at-1.369 V.
Fig.6 DPV obtained to produce calibration plot for Co- pilocarpine at a Hg (2mM) / Au .The current peak values were at –0.68 and –1.20 V (1) with 3.23 – 0.336 m mol of Co- pilocarpine, (2) with 6.53 –0.672 m mol of Co- pilocarpine, (3) with
13.1 –1.34 m mol of Co- pilocarpine. Scan rate 10 mV/s; pulse height 50.
Fig. 7 Differential pulse voltammograms traces for pilocarpine of commercial
ophthalmic solution at a Hg/Au. Peaks; A, -1.16 V, 5.44µ A (0 mM pilocarpine added);
B, -1.16 V, 9.94 µ A (0.408 mM pilocarpine added); C, -1.18 V, 13.0 µ A (00.816 mM pilocarpine added); D, -1.19 V, 18.0 µ A (1.632 mM pilocarpine added); Scan rate 10 mV/s; pulse height, 0.05V.
Fig. 8 Chromatograms obtained by (A) LC-ECD (B) LC-UV. Peaks: (a) Co-pilocarpine (b) pilocarpine.