國立臺灣大學生命科學院分子與細胞生物學研究所 碩士論文
Institute of Molecular and Cell Biology College of Life Science
National Taiwan University Master Thesis
突變果蠅支鏈甲型酮酸脫氫複合物導致 肌肉以及神經性損傷
Mutations of the Drosophila branched-chain α-keto acid dehydrogenase complex (BCKDH) result in
muscular and neuronal dysfunctions
蔡惠穎 Hui-Ying Tsai
指導教授:陳俊宏 博士 Advisor: Chun-Hong Chen, Ph.D
中華民國108年7月
July, 2019
中文摘要
楓糖尿症為罕見的代謝性胺基酸異常疾病,由於患者缺乏支鏈胺基酸代謝酵素 -支鏈甲型酮酸脫氫複合物(BCKDH),導致過量的支鏈胺基酸(BCAAs)累積在血 液中,進而對神經系統造成傷害。此病在台灣的發病率為十萬分之一,在嬰兒時 期就會發病,如果未立即處理將會有死亡的風險。目前治療方法有兩種,一為飲 食控制,避免攝入過多的支鏈胺基酸以減少體內過多的累積,二為嚴重者需要進 行肝臟移植手術。本篇研究中,我們以果蠅為模式生物,將參與支鏈胺基酸代謝 路徑中的五個酵素基因分別進行剔除。支鏈甲型酮酸脫氫複合物(BCKDH)包含
四個亞基E1α、E1β、E2 和 E3 分別對應到果蠅基因的 CG8199、CG17691、CG5599
和CG7430,此外路徑上游的支鏈胺基酸轉移酶(BCAT)針對基因則是 CG1673,
也將進行基因剔除。實驗結果顯示,支鏈胺基酸的代謝地點在肌肉,不同於其他
胺基酸在肝臟代謝。表現量在肌肉較高的是E1α、E2、E3,表示著這三個是主要
代謝支鏈胺基酸的亞基,而其中又以E3 表現量最高,被認為是整個代謝路徑中
最主要的基因。另外,確認了在五株基因剔除果蠅中支鏈胺基酸會上升之後,發 現脂肪細胞的脂滴大小也會改變,顯示支鏈胺基酸的累積也會改變著脂肪的代謝。
此外,我們利用視網膜電圖(ERG),紀錄在果蠅視神經在光刺激下電位的變化,
E2 和 E3 的變異株在神經傳導的功能上有明顯的受損,若解開導致神經性退化的 機制,將對解決楓糖尿症嬰兒大腦受到支鏈胺基酸堆積是一大里程碑。此外,我
們利用一種降血糖藥物-二甲雙胍進行測試,並且看到幼蟲體內支鏈胺基酸含量以
及爬行能力有一定程度的恢復,在未來能進行更多的測試,為楓糖尿症帶來藥物治 療的契機。
關鍵字: 楓糖尿症、支鏈胺基酸、支鏈甲型酮酸脫氫複合物缺乏、神經性退化、
果蠅、二甲雙胍
Abstract
Maple Syrup Urine Disease (MSUD) is a rare autosomal recessive inherited disease caused by dysfunctions of the branched-chain α-keto-acid dehydrogenase (BCKDH) complex; this complex is involved in the branched-chain amino acid (BCAA) degradation pathway. Although it has been reported that patients with MSUD suffer
from neuronal injuries, the causes are currently unknown.
In order to investigate the underlying mechanisms of MSUD, we generated Drosophila melanogaster BCKDH enzyme and BCAA degradation pathway mutants using
CRISPR-Cas9 techniques. The BCKDH enzyme consists of three components, each of which was targeted for mutation; alpha-keto acid decarboxylase (E1/ composed of the sub-units α (CG8199) and β (CG17691)), dihydrolipoyl transacylase (E2/CG5599) and dihydrolipoamide dehydrogenase (E3/CG7430). Branched-chain amino acid aminotransferase (BCAT), which is upstream of the BCKDH complex, was also targeted (CG1673).
We found that BCAAs accumulated and lipid droplet (LD) sizes changed in mutant lines as compared to controls. Furthermore, we used GMR Gal4 to drive RNAi for each of the five genes and found that changes to rhabdomere morphology were only observed in CG5599 and CG7430 RNAi lines, indicating these two genes may be the most important for neuronal development. Using a Drosophila electroretinogram (ERG) to
examine mutant physiology, we found that CG5599 and CG7430 RNAi lines lacked on- and off- transients and displayed defective depolarization, indicating that they had a neurodegenerative phenotype. Our results suggest that dihydrolipoyl transacylase and dihydrolipoamide dehydrogenase may be the most promising targets for further research into MSUD associated chronic neuropsychiatric symptoms. Moreover, we
treated Metformin in this Drosophila model by detecting BCAAs level and larval locomotor, finding that an extent of rescuing. These results indicated that Metformin probably could be a potential drug for curing MSUD.
Key words: Maple Syrup Urine Disease, BCKDH deficiency, Drosophila melanogaster, BCAA degradation pathway, Neurodegeneration, Metformin
Contents
Introduction ... 1
Literature review ... 4
1. BCAA degradation pathway ... 4
2. Lipid metabolism ... 6
3. Monomethyl Branched-chain Fatty Acids (mmBCFAs) ... 8
4. mTOR and BCAA metabolic pathways ... 9
5. Neurodegeneration ... 10
6. Autophagy in Drosophila ... 11
7. Maple Syrup Urine Disease (MSUD)... 13
8. Animal models of MSUD ... 15
9. The biguanide antihyperglycemic agent metformin ... 17
Aims of thesis ... 19
Materials and Methods ... 20
1. Fly/larva stocks and genetics ... 20
2. Pupal eclosion rate analysis ... 20
3. GC analysis of mmBCFA ... 21
4. UPLC-MS-Multiple Reaction Monitoring (MRM) analysis ... 22
5. Fat body dissection and imaging ... 22
6. Photoreceptor immunofluorescence (IF) and imaging ... 23
7. Lysotracker staining and imaging ... 24
8. Brain paraffin sectioning ... 24
9. Electroretinogram (ERG) ... 25
10. Larval locomotion analysis ... 25
11. RNA extraction and real-time PCR (qPCR) ... 26
1. RNA extraction. ... 26
2. Reverse transcription -PCR (RNA→cDNA). ... 27
12. Statistical analysis ... 28
Results ... 29
1. Generation of BCKDH knock-out mutants via CRISPR-mediated mutagenesis ... 29
2. Mutant larvae display elevated levels of BCAAs (valine, leucine and isoleucine). ... 29
3. Loss of several BCKDH genes resulted in larval arrested development. ... 30
4. Vacuolar lesions increased in number in mutant brains. ... 30
5. Knockdown of BCKDH genes causes damage to neurons and results in neurodegeneration. ... 31
6. BCKDH complex expression site in WT flies. ... 32
7. Lipid droplets accumulate in larval fat bodies. ... 32
8. BCKDH mutant found increased lipid droplets size but not CG1673 mutant in high protein diet. ... 33
9. Production of mmBCFA increased in MSUD mutants. ... 33
10. Starvation-induced autophagy may be downregulated in the larval fat body. ... 34
11. Larval movement capability was significantly reduced in BCKDH mutants... 35
12. Mitochondrial morphology changes in muscle and fat body ... 36
13. Metformin treatment reduces the accumulation of leucine and isoleucine in E2 mutant larvae. ... 37
14. Metformin treatment rescues locomotor behavior in mutant larvae. ... 37
Discussion ... 39
References ... 45
Figures ... 56
Supplementary information ... 75
Appendix ... 79
List of abbreviations BCAA = Branched-Chain Amino Acid
BCAT = Branched chain aminotransferase
BCKDH = Branched-Chain α-Keto-acid Dehydrogenase mmBCFA = Monomethyl branched-chain fatty acid
C15ISO = 13-methyl myristic acid C17ISO = 15-methyl hexadecanoic acid Drosophila melanogaster = D. melanogaster
GPDH = Glycerophosphate dehydrogenase MSUD = Maple Syrup Urine Disease MUT = methylmalonylCoA mutase PBS = Phosphate Buffered Saline
PBST = Phosphate Buffered Saline with Triton X-100 TAG = triacylglycerol
TCA = Tricarboxylic acid
mTORC = mammalian target of rapamycin complex dTOR = Drosophila target of rapamycin complex WT = Wildtype
ERG = Electroretinogram
Introduction
The branched-chain amino acid (BCAA) degradation pathway provides mammalian muscles with much of their energy supply and is therefore crucial for mammalian well-being1. BCAAs are widespread in mammalian systems, with approximately 35%
of the central amino acids in muscle proteins and 40 % of the total required preformed amino acids being BCAAs2. BCAAs can promote protein synthesis, upregulate
various signaling pathways, increase fatty acid oxidation and are also thought to play a role in the development of obesity3,4,5. BCAAs are degraded in the liver and
peripheral tissues (especially skeletal muscle), with all of the steps of the BCAA catabolic pathway taking place within mitochondria6. Disorders in the BCAA degradation pathway can thus have significant effects on the health of a variety of tissues and can result in the onset of serious disease.
Once such disease, Maple Syrup Urine Disease (MSUD), is specifically caused by dysfunctions of the branched-chain α-keto-acid dehydrogenase (BCKDH) complex and affects around 1 in every 185,000 newborn infants in the general population7.
Despite this, therapeutic options are restricted to dietary restriction and organ transplantation, with even these limited options proving to be unsatisfactory or unattainable for many patients8. Novel therapeutics for MSUD are thus highly desirable; this will in part necessitate the further development of existing, limited
animal models of disease to allow for more advanced methods of drug testing9. These existing models tend to utilize either mice or zebrafish. Potential Drosophila melanogaster (D. melanogaster) models of MSUD have yet to be fully
exploited however, in spite of the numerous benefits associated with this species (such as short generation times, genetic manipulability and ease of handling).
Here, we developed a D. melanogaster model of MSUD which facilitates the investigation of the changes to underlying mechanisms and molecular pathways in cases of MSUD, as well as the testing of novel therapeutics for use in treatment of disease. After generating various BCKDH enzyme and BCAA degradation pathway mutants using CRISPR-Cas9 techniques, we performed a variety of molecular, electrophysiological and behavioral tests in order to validate our model.
The protein sequences are highly conserved in human and Drosophila protein, their identity are 41%, 51%, 60%, 65%, 48% and 67% for BCAT1, BCAT2, BCKDHA, BCKDHB, DBT and DLD (Table S5) and also their domain have similar sequence (Table S6, Appendix i.), suggesting that the function of BCKDH in Drosophila is orthology of human. From mutant lines generated with our lab, CG1673, CG5599 and CG8199 mutant were used to establish a mimic MSUD model by validating elevated BCAAs level in hemolymph in Drosophila by Yu-Min Chen10. Further, phenotypes were similar to humans in Drosophila model such as decline of life span, reduction in
mobile ability and decreased body lengths and weights compared to wild type. Neuron defects were also first stated in Drosophila of MSUD. In order to create a more complete model, another BCAA catabolic enzyme (BCKDH complex) subunits, CG7430 (E3 subunit) and CG17691 (E1βsubunit) were also generated. Based on
previous research, we could go further study and make this model more trustworthy.
We found an accumulation of BCAAs in mutants and a change in lipid droplet (LD) size. We also identified changes to rhadomere morphology in two mutant lines (CG5599 and CG7430) when we used GMR Gal4 to drive RNAi for each of the five genes, suggesting these two genes may be the most important for neuronal development.
These two lines were also found to lack on- and off- transients and displayed defective depolarization when tested using a Drosophila electroretinogram (ERG) to examine mutant physiology, further suggestive of a neurodegenerative phenotype. Finally, administration of metformin rescued BCAA levels and locomotor activity in mutants.
In summary, two of the five mutant lines generated appear particularly promising for further research. Our results highlight the potential importance of dihydrolipoyl transacylase and dihydrolipoamide dehydrogenase in terms of the development of MSUD. Our newly established Drosophila model of MSUD could be applied to further research into these enzymes.
Overview of experiments in this study see Table S3.
Literature review
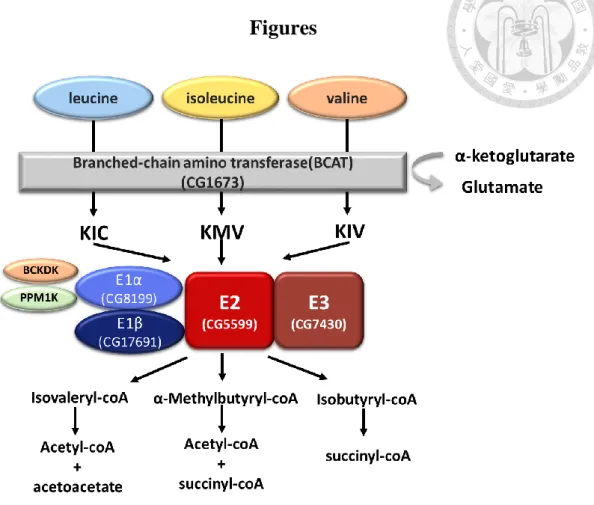
1. BCAA degradation pathway
BCAAs account for between 20% to 25% of most dietary proteins, with the major catalytic site for BCAAs being located in skeletal muscle11. BCAA catabolic
pathways begin with the transport of these amino acids into cells by the system L transporter located in the cytosolic membrane12. Inside the cell, the BCAA degradation pathway is comprised of two steps. The initial step is a reversible
transamination step to turn leucine, isoleucine and valine to BCKAs: α-ketoisocaproic acid (KIC), α-keto-3-methylvaleric acid (KMV) and α-ketoisovaleric acid (KIV)13. This step involves BCAA aminotransferase (BCAT), which have been linked with cytosolic and mitochondrial isoenzymes5.
The second step is an irreversible oxidative decarboxylation, catalyzed by the BCKD complex within the inner mitochondrial membrane. Acetyl-CoA (from isoleucine and leucine) and methylmalonyl-CoA (from valine and isoleucine) are yielded from a series of metabolic reactions. Methylmalonyl-CoA is then converted by methylmalonylCoA mutase (MUT) to succinyl-CoA, which can either be
incorporated into the tricarboxylic acid (TCA) cycle or enter complex II of the
into the TCA cycle14 (Figure 1).
The BCKD complex is a multi-enzyme macromolecule with four catalytic
components (E1α, E1β, E2 and E3). The E1 component is composed of two E1α and two E1β subunits forming a heterotetramer. The E1α subunit (BCKA decarboxylase
alpha) is encoded by BCKDHA whilst the E1β subunit (BCKA decarboxylase beta) is encoded by BCKDHB. The E2 (Dihydrolipoyl transacylase) component is encoded by DBT and E3 (Dihydrolipoamide dehydrogenase) is encoded by DLD15.
Studies have documented a strong correlation between the presence of mutant E2 proteins and a specific (thiamin-responsive) MSUD phenotype16. The normal E1 component should therefore possess residual decarboxylase activity, which is augmented by binding to a mutant E2 protein in the presence of the E1 cofactor thiamin diphosphate.
The catalytic components require the cofactors thiamin pyrophosphate (TPP) and flavin adenine dinucleotide (FAD) as well as the prosthetic group lipomide and two regulatory enzymes (a kinase and a phosphatase)16 .
2. Lipid metabolism
Lipids are responsible for providing cells with energy for daily usage.
Physiological metabolism must be precisely regulated to maintain proper homeostasis.
In most animals, lipids are stored mainly as triacylglycerol (TAG) in an organelle referred to as a lipid droplet17. The size of these lipid droplets depends on the balance
of lipogenesis and lipolysis. In nutrient-rich environments, excess fatty acids can be converted to TAG through lipogenesis and then stored in lipid droplets. Under some nutrient-limiting conditions such as starvation, lipids can be released from lipid droplets by lipolysis for usage.
BCAAs play a role in regulating lipogenesis and are associated with obesity and insulin-resistance, with circulating levels of BCAAs thought to be increased in people with obesity18. BCAAs and insulin are anabolic signals that alter the growth of
energy-consuming tissues, mediated in part through their ability to activate the mammalian target of rapamycin complex 1 (mTORC1) and then decrease protein breakdown. BCAAs may in fact cause obesity; excess dietary BCAAs increase plasma levels of leucine, which together with insulin activate mTORC1 and S6K1 (Appendix ii). Persistent activation leads to serine phosphorylation of IRS-1 and IRS- 2, which interferes with signalling and might target IRS1 for proteolysis via a
proteasomal pathway which leads to insulin resistance and Type 2 Diabetes18.
Furthermore, elevated BCAAs in plasma may play a pathogenic role in β-cell failure in obese individuals19. Experiments in mice also suggest that high levels of leucine may have detrimental effects on both β-cell function and insulin sensitivity, although the mechanism remains unclear20.
3. Monomethyl Branched-chain Fatty Acids (mmBCFAs)
Monomethyl branched-chain fatty acids (mmBCFAs) are fatty acids with a single methyl group whose position determines the molecule configuration. A methyl group located at the carbon next to the terminal carbon (ω-1) results in an ISO molecule, whilst a methyl group located at the second carbon to the terminal carbon (ω-2) results
in an anteISO molecule. mmBCFAs exist in many organisms from bacteria to mammals, where they can be found in hair, skin, brain, blood, and cancer cells21-22.
mmBCFA requires BCKDH activity for synthesis from BCAAs. The products produced from leucine, isoleucine and valine are C15ISO/C17ISO, C21anteISO and C16ISO respectively. C15ISO and C17ISO are the major mmBCFA species found in C.
elegans23. A recent study found that BCKDH gene mutations result in mmBCFA
deficiencies and a lack of mmBCFA causes larval arrested development at the first larval stage in C.elegans. However, the arrest was reversible and could be overcome by feeding the arrested animals mmBCFA supplements23. The study concluded that BCAA accumulation is not the sole cause of pathology and implicated mmBCFA deficiency as also playing a role.
4. mTOR and BCAA metabolic pathways
The phosphatidylinositol 3-kinase/protein kinase-B/mammalian target of rapamycin (PI3K/AKT/mTOR) is one of the intracellular signal pathways which can promote cellular proliferation in response to nutrients and growth factors24. In humans, TOR is a huge protein complex which includes both mTOR1 and mTOR2.
Moreover, TOR complex proteins are conserved in diverse eukaryotes including C.
elegans25, S. cerevisiae26, and Drosophila27.
The Drosophila homolog to mammalian TOR (dTOR) can combine with other proteins such as Raptor, Rictor, and PARS40 to downregulate p70 S6K
phosphorylation and 4e-bp de-phosphorylation28(Appendix ii.). In addition to rapamycin control, TOR protein activation, insulin, serum, nutritious and growth factors are upstream signals of the TOR protein29.
5. Neurodegeneration
Circulating leucine and KIC can cause severe damage to the brain by competing with precursors of amino acids neurotransmitters, potentially resulting in brain edema and encephalopathy30. Amino acid dysregulation results in aberrant neural networks with neurochemical deficiencies and correlates with neuropsychiatric morbidities31.
Hyperleucinemia inhibits the transport of tyrosine, tryptophan, and other essential amino acids across the blood-brain barrier and thereby limits substrate availability for cerebral catecholamine, serotonin, and protein synthesis32.
Accumulation of KIC favors synthesis of leucine, thus reversing the bidirectional transaminase reaction and depleting the brain of glutamate, an important metabolic currency used as both a neurotransmitter and a source of energy (Appendix iii).
Consistent with these mechanisms, reduced cerebral dopamine and glutamate levels have been reported in experimental MSUD animals and in postmortem brain tissue from a child who died of leucine intoxication31,33.
Overactivation of the mTOR signaling pathway is thought to be associated with many human diseases including obesity, cancer and Alzheimer’s disease34. However, the role of mTOR signaling pathways in these diseases have not been investigated in sufficient depth as to provide understanding as to the underlying mechanisms of disease.
6. Autophagy in Drosophila
Autophagy contributes to the turnover of proteins, lipids, nucleic acids, glycogen, and even whole organelles35. Recycling of these components helps to protect cells and organisms, with misregulation of autophagy likely involved in numerous human pathologies including aging, cancer, infections and neurodegeneration36.
The process of autophagy can be classified into three stages. First, in response to cellular stress, cytoplasm and protein aggregates and organelles are engulfed within double-membraned autophagosomes, which form through the growth of the cup- shaped phagophore membrane. Second, the autophagosome fuses with a lysosome to form an autolysosome. Third, contents delivered for degradation are broken down by acidic hydrolases within the autolysosome, and the resulting sequestered materials are recycled for biosynthesis and energy production in hydrolytic lysosomal
compartment. Each of these steps can be identified based on the substrate and pH profiles of their components37.
The first step of forming a phagophore is associated with Atg5, Atg12 and Atg16, which form a complex on the outer surface of the phagophore, and are removed upon its closure to form the autophagosome. One second step marker, Syntaxin 17, is recruited to the mature autophagosome and is released upon fusion with the lysosome. On the other hand, Atg8 is associated with the entire autophagic
pathway from phagophore to autolysosome. Autophagosome-lysosome fusion can be monitored through loss of GFP fluorescence from the GFP-mCherry-Atg8 marker.
Lamp1 and Lysotracker as general lysosomal markers can also be used to identify later autophagic vesicles (Appendix iv.). These responses are particularly apparent and well-studied in the Drosophila larval fat body, which uses autophagy to mobilize nutrients throughout the animal in response to starvation24.
.
7. Maple Syrup Urine Disease (MSUD)
Maple Syrup Urine Disease (MSUD) is a rare autosomal recessive inherited disease resulting from dysfunctions of the branched-chain α-keto-acid dehydrogenase (BCKDH) enzyme which results in insufficient breakdown of BCAAs. MSUD is named after the peculiar odor resembling that of maple syrup that comes from an intermediate metabolite of BCAAs, known as branched-chain alpha-keto acids, which along with excessive levels of BCAAs are toxic to brain cells and neuron cells9. Classic MSUD in newborns results in cerebral edema marked by lethargy, hypertonia, poor feeding, and severe neurological impairments. Rapid management is required to reduce the potential for permanent neural damage38.
The pathogenesis of MSUD commences in infants within the first week after birth and includes symptoms of poor appetite, weight loss and hypotonia39.
There are presently five known clinical phenotypes for MSUD, based on severity of the disease, response to thiamine therapy, residual BCKDH enzyme activity and the gene locus affected: classic, intermediate, intermittent, thiamine-responsive, and dihydrolipoamide dehydrogenase (E3)-deficient15. Classic MSUD cases demonstrate only 0-2% residual BCKDH activity, intermediate MSUD between 2-8% activity and intermittent between 8-15% activity. Classic and intermediate MSUD diagnoses account for the majority of cases worldwide (50-75% and 20% respectively)15.
Thiamine-responsive MSUD is a rare form of this disease with a phenotype very similar to that seen in intermediate MSUD. Enzyme activity can be restored by giving doses of thiamine hydrochloride, which is part of a cofactor needed for BCKDH to work. Individuals who have thiamin-responsive MSUD can also tolerate more protein in their diet as compared with other more severe types of the disease. Thiamine has improved the leucine tolerance in the few reported cases of this MSUD subtype, but some dietary branched-chain amino-acid (BCAA) restriction remains necessary40.
There are currently two therapeutic strategies for MSUD: dietary restriction and transplantation41. Of these options, a BCAA-free diet is the most common option
used, with patients avoiding consumption of food rich in BCAAs8. Dietary
compliance can be difficult however as it can lead to a deficiency in essential amino acids.
Liver transplantation is theoretically the preferred treatment as it leads to the cessation of all symptoms41. However, problems associated with this option include high financial costs, limited organ availability and risks associated with surgery. An alternative approach to treating MSUD would therefore be highly desirable. This would require an improved understanding of the fundamental mechanisms of the disease as well as a pipeline for testing novel therapeutics.
8. Animal models of MSUD
Several model organisms are currently used to investigate MSUD, including Hereford calves42, mice43 and zebrafish44. In 1986 for example, 2 day old Hereford calves were used to study central nervous system (CNS) disorders. Diffuse severe status
spongiosus of white matter in cerebellum was found by ultrastructural microscopic examination, suggestive of myelin oedema42.
More recently, in 2006, a murine model was generated by utilizing embryonic stem cell technologies to knock out a functional E2 subunit gene. This ‘classic’ MSUD model led to a 3-fold increase in circulating BCAAs. Intermediate models were also created used transgenic technology to express human E2 cDNA in the knockout background.
BCKDH activity reached at 5-6% and was sufficient to allow for survival, but was insufficient to normalize circulating BCAA levels43.
In 2012, a new zebrafish model of MSUD was established via mutation of the E2 component. Zebrafish mutant larvae showed abnormal swimming behavior,
reductions in the level of glutamate in the brain and developmental problems. As glutamate is a neurotransmitter precursor, reduced levels of glutamate likely contributed to aberrant CNS function and abnormal behavior45.
Although each of the existing animal models offers certain advantages, there are also significant associated limitations; these include relatively long generation times,
a paucity of genetic tools and animal maintenance expenses. The use of D.
melanogaster offers a potential solution to all of these limitations. However, this model system is not yet in widespread use for MSUD, despite its’ popularity in other
fields.
9. The biguanide antihyperglycemic agent metformin
Metformin, a biguanide antihyperglycemic agent, is widely used for type 2 diabetes treatment and its generic formulation is now available in several countries, despite the
molecular mechanism of metformin being incompletely understood. A 2016 study which screened drugs associated with AMPK, mTOR, fatty oxidation and oxidative
phosphorylation in patient derived fibroblasts found that Metformin showed potential for alleviating leucine levels and KIC46.Physiologically, Metformin can lower glucose levels by increasing glucose uptake in peripheral tissues such as the muscle and also reduce glucose production in the liver47.
Metformin has been shown to have at least two targets, activating AMPK and inhibiting complex I of the electron transport chain. Several potential mechanisms of
action have been put forward:
1. mitochondrial respiratory chain (complex I) inhibition 2. AMPK activation
3. Inhibition of cAMP which induces glucagon elevation 4.Inhibition of glycerophosphate dehydrogenase (GPDH) 5. Effect on gut microbiota48,49
Metformin ameliorates hyperglycemia through suppression of liver glucose production and increasesinsulin sensitivity. First, Metformin inhibits liver glucose
production by activation of AMPK, which is required for an increase in the expression of a small heterodimer partner (SHP), which inhibits the expression of the hepatic gluconeogenic genes phosphoenolpyruvate carboxykinase and glucose 6-phosphatase (G-6-P)50. AMPK is an enzyme that plays an important role in insulin signalling, whole body energy balance and the metabolism of glucose and fats. However, the mechanism by which biguanides increase the activity of AMPK remains uncertain51. Second, Metformin induces the phosphorylation of a GLUT4 enhancer factor,
enhances peripheral glucose uptake and decreases insulin-induced suppression of fatty acid oxidation52. This may be the result of improved insulin binding to insulin
receptors53.
Additionally, Metformin is also critical for lipid homeostasis. Its’ beneficial effects on circulating lipids have been linked to reductions in fat in the liver.
Metformin also decreases hepatic lipids in obese mice54. Metformin activates AMPK
in hepatocytes, resulting in a reduction in acetyl-CoA carboxylase (ACC) activity, inducement of fatty acid oxidation and suppression of lipogenic enzymes expression.
Activation of AMPK by metformin or an adenosine analogue suppresses expression of SREBP-1, a key lipogenic transcription factor, and therefore results in a reduction in triglyceride in plasma55,56.
Aims of thesis
It is clear from the above literature review that new therapeutics are required for MSUD, which in turn necessitates the development of novel models for the disease.
This project centered on developing and validating a model for MSUD using D.
melanogaster.
This required:
1. Generation of mutants for each potential stage of the BCAA degradation pathway
2. Validation of mutant phenotype (i.e. comparison of mutant phenotypes to supposed MSUD phenotypes in other models/ humans)
3. Testing of effect of novel therapeutic (Metformin)
Moreover, the validation process enabled us to explore the molecular and genetic pathways potentially involved in MSUD.
Materials and Methods 1.
Fly/larva stocks and geneticsAll Drosophila melanogaster lines were provided by the Bloomington Drosophila Stock Center or generated by our lab (see Table S1). Stocks were maintained on standard cornmeal food and housed at 25°C. W1118 was used as control in our experiments.
For the fly genetic background, CG1673 and CG5599 were both found to be located on the X chromosome; the X chromosome balancer, FM7a, was therefore used to establish the stock. The balancer Cyo was used for CG17691, which is on the second chromosome, whilst Tm3 was used for CG8199 and CG7430 with Tm6b, both of which are on the third chromosome.
In order to confirm larval genotypes, balancers were replaced by balancer-GFP fusion protein constructs. We could thus identify homozygous negative larvae as those lacking GFP fluorescence. For eye morphology experiments, GMR-gal4 was used for driving UAS RNAi line expression in retinas.
2.
Pupal eclosion rate analysisOne hundred homozygous negative pupae were collected from stock vials and transferred to vials containing fresh food. After approximately 48 hours, the number
of eclosed pupae were counted manually.
The eclosion rate was calculated as:
𝑛𝑢𝑚𝑏𝑒𝑟 𝑜𝑓 𝑎𝑑𝑢𝑙𝑡𝑠 𝑜𝑟𝑖𝑔𝑖𝑛𝑎𝑙 𝑛𝑢𝑚𝑏𝑒𝑟 𝑜𝑓 𝑝𝑢𝑝𝑎𝑒 Three biological repeats were conducted for each genotype.
3.
GC analysis of mmBCFA100 third instar (L3) wandering larvae from all genotypes were transferred into eppendorfs and frozen at -80℃ for at least 30 min. Frozen samples were mixed with 100μL methanol, 900μL hexane and 10mM Pentadecanoic acid as well as methyl
ester (C15 fatty acid) as an internal standard before being homogenized with an electric homogenizer and then vortexed for two minutes.
The samples were then centrifuged at 13,000 rpm for two minutes and the top hexane layer was collected in a glasses vial23. Samples were kept in a biosafety cabinet overnight for hexane volatizing. Samples were sent to the Academia Sinica, ABRC Metabolomics Core Laboratory for FAMES metabolite derivatization and then GC-Q-TOF. The area of each peak was calculated and then normalized with a control of Pentadecanoic acid and methyl ester.
4.
UPLC-MS-Multiple Reaction Monitoring (MRM) analysisUsing LC-based metabolomics, we measured levels of BCAAs in L3 wandering
larvae. 25 L3 wandering larvae were collected and frozen at -80℃ for at least 30
minutes. Samples were homogenized with 100μL 50 % methanol and 200ppm ribitol (an internal standard). 300μL chloroform was added and the samples were vortexed
for one minute.
After being stored at -80℃ overnight, samples were centrifuged at 10,000 for 15 minutes at 4℃. 75μL of the resulting aqueous layer was extracted and dried under
vacuum in a Concentrator plus (Eppendorf) for approximately 2 hours. Samples were then suspended in 20μL ddH2O and centrifuged in order to remove any residue prior
to LC-MS analysis.
5.
Fat body dissection and imagingL3 wandering larvae fat bodies were dissected in 1x phosphate buffered saline (PBS), with residual tissue such as gastric caeca and intestine being removed. Samples were fixed in 4% paraformaldehyde for 30 minutes. After fixation, samples were washed
three times with 1xPBS and then stained with BODIPY 493/503 (1 µg/ml) and DAPI for one hour. Samples were then washed three times with 1xPBS and mounted. Images were taken using LAS AF software on a Leica SP5 confocal microscope.
6.
Photoreceptor immunofluorescence (IF) and imagingEyes from 0, 7 and 14 day old fly (including all mutant and control genotypes) were dissected in 1x PBS. Using forceps to grasp the mouthparts, the head was detached from the body by severing the neck using a second set of forceps. A tear was created along the left/right midline of the head. The brain and fat body was then removed.
Ommatidia were fixed in 4% paraformaldehyde for 30 minutes at room temperature.
After fixation, the supernatant was removed and 0.5% PBST (1X PBS with 0.5%
Triton) was added. Samples were blocked with 2% BSA/ 0.5% PBST for one hour at
room temperature. The solution was then discarded and the samples washed three times with 0.5% PBST for 10 minutes each time.
Following addition of primary antibodies (1:200,anti-Na+/K+-ATPase
(Developmental Studies Hybridoma Bank, a5) ), samples were incubated overnight in 0.5% PBST at 4℃. After a further three washes using PBST, goat anti-mouse
secondary antibodies (Abcam) conjugated with Alexa 647 and phalloidin were added to the samples at 1:400. Samples were then incubated at 4℃ overnight.
Images were taken using LAS AF software on a Leica SP5 confocal microscope and the number of rhabdomeres was calculated per ommatidium.
7.
Lysotracker staining and imagingFat bodies from fed or starved 4 hr flies were dissected in 1x PBS and then incubated in 100µL Lysotracker red DND-99 and DAPI for 20 minutes. A longer fixation was not used as paraformaldehyde could compromise the Lysotracker staining. Samples were mounted and immediately imaged using LAS AF software on a Leica SP5
confocal microscope.
8.
Brain paraffin sectioningAt least ten flies aged 7 days old from each genotype were anesthetized using CO2, after which a pair of dissection scissors were used to remove the head of each fly. Samples were then transferred to eppendorfs. 1 mL bouin solution (Polysciences, Inc.) was added to the samples as a fix buffer, following which the samples were rotated on shaker at room temperature for five to seven days. Kim whips were torn into small pieces and put into the eppendorf to soak samples.
Following this, the bouin solution was replaced with a leaching buffer (1M Tris (pH8.0), NaCl, ddH2O) overnight. Samples were then transferred into 70% ethanol and sent to the core pathology lab at the NHRI for dehydration, clearing, infiltration, embedding, sectioning and H&E staining. Six to seven serial sections of each brain were taken. The number of vacuoles in each section were counted, with the median
number of lesions calculated per brain.
9.
Electroretinogram (ERG)0, 7 and 14 day old flies were fixed in one direction on glass slides using non-toxic glue.
2M NaCl (for use as a conductive medium) was used to fill both recording and reference
electrodes. A reference electrode was placed in the torso of each fly whist recording electrodes were put over the retina.
Electrode voltage was amplified using a Digidata 1440A, filtered through a Warner IE-210, and the Clampex 10.1 software (Axon Instruments) was used for all recordings.
A light stimulus was provided in 1 second pulses via a computer-controlled red LED system (Schott MC1500). All experiments were conducted in duplicate or triplicate with at least 10 recordings completed for each genotype and experimental condition.
10.
Larval locomotion analysisFor each experiment, six L3 wandering larvae were selected and placed upon a 200 mm x 115 mm x 30 mm agar plate using a paintbrush at 25℃. Each agar plate contained a
solution consisting of 1% agar, 0.1M sucrose and brilliant blue dye (in order to provide a dark background for contrast enhancement). The plate was then transferred to a light, temperature and humidity controlled incubator. A camera was placed onto a tripod and
focused on the plate. Larvae were then allowed to move for four minutes, all of which was filmed. All genotypes were recorded with approximately the same circadian time period (between six and nine hours after lights on), although no circadian locomotive rhythms have been reported in larvae57.
Following video collection, the middle 2 minutes of each video was analyzed using
the wrMTrck plug-in for ImageJ58. At least three biological repeats were used for each genotype.
11.
RNA extraction and real-time PCR (qPCR) 1. RNA extraction.RNA was extracted from five L3 larvae per genotype and then homogenized in 500μl TRI-reagent (Invitrogen). 100μl CHCl3 was added and the samples were vortexed for 15 seconds, after which they were allowed to stand for up to 15 minutes at room temperature. The samples were then centrifuged at 12,000g for 15 minutes at 4 ℃.
The aqueous phase of the samples was transferred to a fresh tube and 250μl of cool 15 isopropanol was added. The samples were then allowed to stand for 5-10 minutes on ice before being centrifuged at 12,000g for 10 minutes at 4℃.
The supernatant was removed and the RNA pellet was washed in 250μl of 75%
ethanol and then dried for at least five minutes. 30μl of RNA-free water solution was
added before total RNA was quantified using a NanoDrop ND-1000 (Thermo Fisher Scientific Inc).
2. Reverse transcription -PCR (RNA→cDNA).
Following RNA quantification samples were transferred to new PCR tubes, each of
which contained 3μg of total RNA and 8.8 μl dH2O. The samples were mixed with 1μl 10X buffer, 0.1μl RNase inhibitor and 0.1μl DNase I and then incubated at room temperature for 15 minutes before 1μl of EDTA was added to terminate the reaction.
Samples were incubated at 65℃ for 10 minutes to inactivate the DNase I before 11μl
of the RNA sample was mixed with 0.1 oligo (dT) 12-18 primer and 1.5μl 10mM dNTP.
The mixtures were then transferred to a PCR machine at 65℃ for 5 minutes and
then put on ice in order to denature the RNA. 6μl 5X First-Strand Buffer, 3μl 0.1M DTT, 0.1μl RNase inhibitor, 0.2μl SuperScriptTMIII RT and 8.1μl dH2O were then added to each sample. Samples were kept in a PCR machine at 50℃ for 60 minutes, then 70℃
for 15 minutes to inactive the enzymes. The resulting product was stored at -20℃.
3. SYBR-system.
Samples were mixed with 3μl 10ng/μl cDNA, 0.5μl 5μM 5'-primer, 0.5μl 5μM 3'primer, KAPA 2x SYBR®Green dye master mix (a fluorescent double-stranded DNA -binding
dye used to track the progress of DNA amplification in real-time PCR experiments) and ddH2O. Samples were then loaded into 384 wells and an AB ViiA-7 Real-Time PCR system was used to detect Ct values. Data was analyzed using QuantStudio software.
Primer sequences for individual genes are shown in (Table 1.).
12.
Statistical analysisStudents’ t tests, chi-squared tests and Mann-Whitney tests were used for statistical
analysis. In general, p values <0.05 were considered significant whilst p <0.1 were considered as indicating a trend.
For the eclosion rate experiments, post-hoc pairwise chi-squared tests utilizing a Bonferroni correction were computed in R to compare WT eclosion rates with individual mutant eclosion rates59. All tests used a (corrected) significance level of p < 0.01.
Results
1. Generation of BCKDH knock-out mutants via CRISPR-mediated
mutagenesis
We generated BCAA-associated gene knockout flies using the CRISPR/Cas9 system.
Each of the BCKDH complex genes was targeted via CRISPR/Cas9-mediated genome editing by homology-dependent repair (HDR), which used two guide RNAs and a dsDNA plasmid donor. The excision began near the start codon and the
majority of the coding sequence was deleted by knocking in a cassette containing attPX, two STOP codons and 3xP3-RFP. As a selection marker, 3xP3-RFP was used to facilitate genetic screening. Figure 2 shows excision sites and the insertion cassette. The balanced stocks were labelled CG1673△/FM7, CG8199△/TM3, CG17691△/Cyo, CG5599△/FM7 and CG7430△/TM6B.
2. Mutant larvae display elevated levels of BCAAs (valine, leucine and
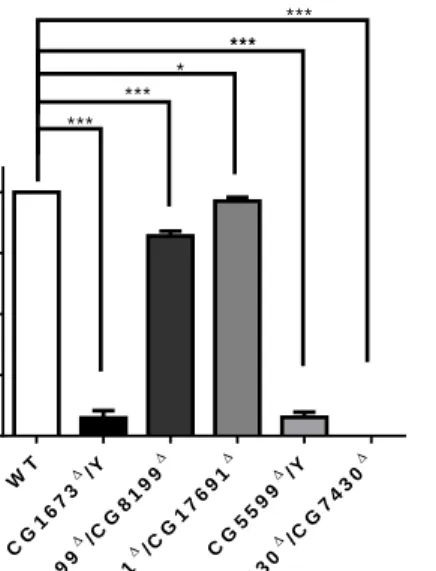
isoleucine).
To demonstrate that gene targeting was accurate, we investigated BCAAs level in mutant larvae. We found significant differences in the level of BCAAs in mutants as compared to wild-type controls (Figure 3). CG1673, CG5599, CG7430, CG8199 and CG17691 mutant lines showed 5.6~8.2 fold increases in leucine, 3~4.5 fold increases
in isoleucine and 2.3~4.3-fold increases in valine compared to wild-type (WT) flies.
These data validated BCAAs exactly accumulation in knock out larvae.
3. Loss of several BCKDH genes resulted in larval arrested development.
We found significant differences between controls and several mutant lines in terms of their eclosion rates (Figure 4). The majority of CG1673 and CG5599 mutant larvae could not eclose, with eclosion rates of just 7.3% and 7.6%. Those that did eclose died within a few hours. Furthermore, CG7430 mutant larvae were homozygous lethal, with no pupae found to have eclosed. However, CG8199 and CG17691 mutants were both found to be homozygous viable, with eclosion ratea of 82% and 96% respectively.
In total, eclosion rates were found to be significantly reduced in all mutant lines compared to WT (Figure 4). This implies that BCAA accumulation is toxic to immature forms of D. melanogaster, a similar phenotype to that observed in humans with the classic form of MSUD.
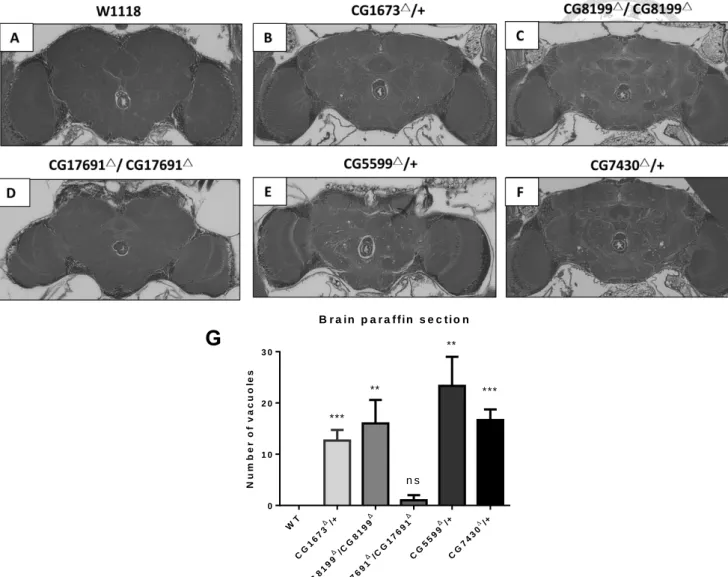
4. Vacuolar lesions increased in number in mutant brains.
As homozygous negative CG1673, CG5599 and CG7430 mutant lines were not viable to adulthood, we used heterozygous mutants for brain sectioning. We found that control
flies showed no lesions in any sample, whilst CG1673, CG8199, CG17691, CG5599, CG7430 mutants had median counts of 12, 16, 1, 23 and 16 respectively. We found a
significant difference in the number of lesions in CG1673, CG8199, CG5599 and CG7430 mutants as compared to WT (Student’s t test; p<0.05) (Figure 5.)
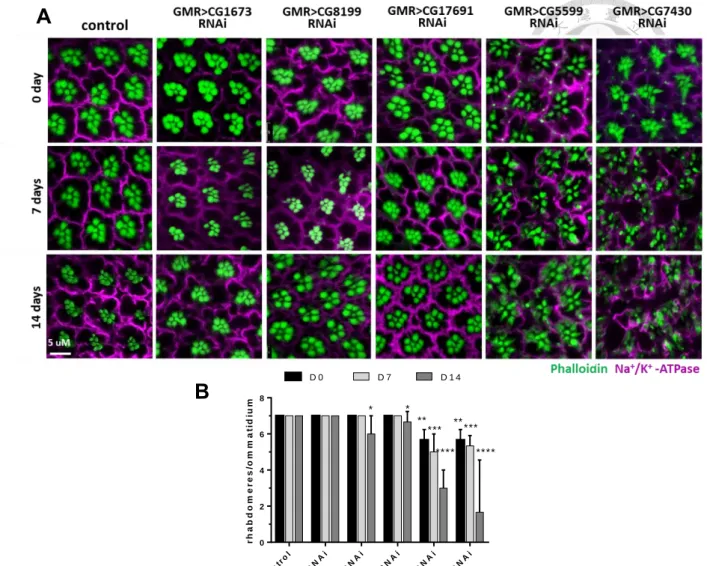
5.
Knockdown of BCKDH genes causes damage to neurons and results in neurodegeneration.Using the GMR-gal4 binary system to drive UAS-BCKDH RNAi expression we were able to observe morphological changes in photoreceptors. Since photoreceptors are light sensitive, we used a constant intensity of light to accelerate the neuronal damage process.
We found no significant morphological differences in the CG1673 knockdown line as compared to the control, whilst both CG8199 and CG17691 knockdowns showed slight decreases in the number of rhabdomeres after 14 days constant light exposure (6 and 6.6 rhabdomeres per ommatidium respectively) (Figure 6A). We also found a significant reduction for CG5599 and CG7430 knockdowns lines in the absence of light stimulation after both 7 and 14 days. Following 7 days of exposure, CG5599 and CG7430 knockdowns had 5 and 5.3 rhabdomeres per ommatidium. After exposure for 14 days, CG5599 and CG7430 knockdowns were left with only 3 and 1.6 rhabdomeres
per ommatidium.
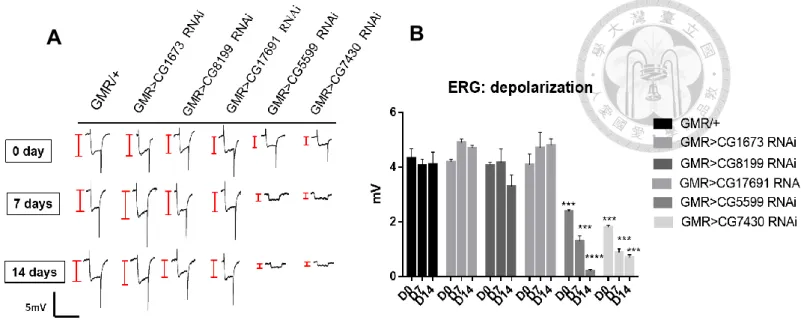
Likewise, we found a similar phenotype in the electroretinogram (ERG) data.
Results from both CG5599 and CG7430 knockdowns indicated a reduction in depolarization and a loss of on-transients, with the trend in agreement with their morphology. In CG5599 and CG7430 knockdowns, ERG showed significant decreases
in depolarization amplitude compared to WT (Student’s t test, p<0.001) after 7 and 14 days. These indicates that the knockdown of CG5599 and CG7430 genes results in age- dependent neurodegeneration. For all other knockdown lines and conditions, we found no significant differences from control results.
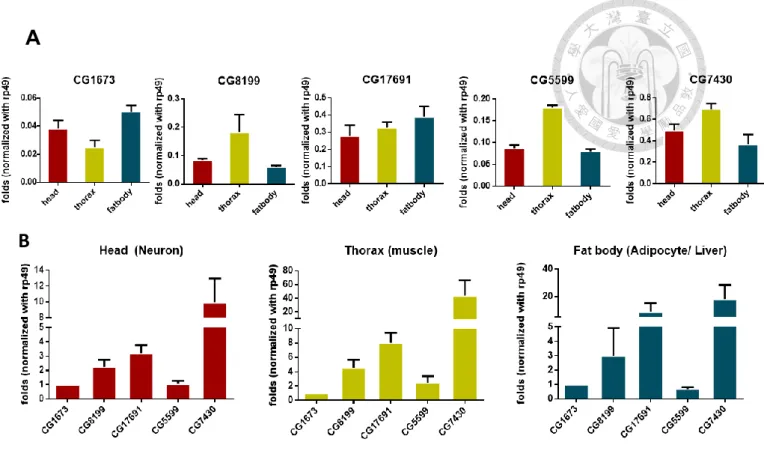
6. BCKDH complex expression site in WT flies.
We used qPCR to investigate the distribution of BCKDH genes in fly heads, thoraxes and fat bodies. Three of the five genes (CG8199, CG5599 and CG7430) were highly expressed in muscle tissue (0.182, 0.183 and 0.695 fold) whilst the remaining two, CG1673 and CG17691,were more highly expressed in the fat body (0.05 and 0.39 fold) (Figure 8A). Furthermore, we found that irrespective of the tissue type, CG7430 was more highly expressed than all other genes (Figure 8B).
7. Lipid droplets accumulate in larval fat bodies.
dynamics and lipid metabolism, we measured lipid droplet size in L3 wandering larvae fat bodies. We found that lipid droplet size was significantly enlarged in CG1673, CG5599 and CG7430 mutant flies compared to controls (Figure 9.). Average wild type
lipid droplet area was found to be 123 μm2, whilst CG1673, CG5599 and CG7430 mutants had averages of 158, 202 and 184 μm2 respectively (Mann-Whitney test, p <
0.001). Other mutants showed no significant difference from WT flies.
8. BCKDH mutant found increased lipid droplets size but not CG1673
mutant in high protein diet.
To test whether changes in lipid droplet size occurred in response to a high protein diet, we dissected L3 larvae which had been provided with high protein food. High protein food was comprised of excessive soy protein including most essential
proteins. To further investigate lipid droplet size under different nutrient regimes, we fed larvae high protein food for 5 days after egg laying. BODIPY was used for staining. Lipid droplets were found to be bigger in all lines (except CG1673 mutants) having fed on the high protein food as compared to normal food.
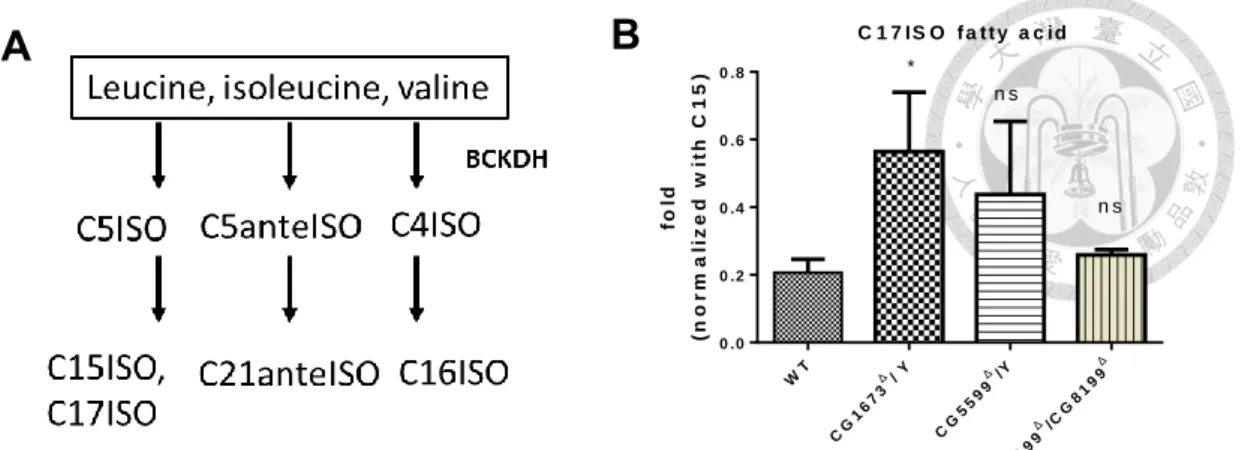
9. Production of mmBCFA increased in MSUD mutants.
mmBCFA is derived from BCAAs and is involved in BCKDH activity. In C. elegans,
the critical mmBCFAs are C15ISO and C17ISO (Figure 10A.). To determine whether the lack of BCKDH activity resulted in mmBCFA deficiency, we measured the level of mmBCFA in L3 larvae by GC-MS. In contrast to C.elegans we found that mmBCFA- C17ISO was significantly increased in CG1673 mutants (Mann-Whitney test, p <0.05) (Figure10), suggesting that there is another enzyme responsible for mmBCFA synthesis
in Drosophila. However, we found no significant differences between WT and CG5599 and CG8199 mutant larvae – in these cases we were unable to detect C15ISO in our samples.
10. Starvation-induced autophagy may be downregulated in the larval
fat body.
In addition to its role in nutrient storage (which includes proteins, lipids and carbohydrates), the larval fat body is also highly active in autophagy. In late larval stages, cytoplasmic organelles are enclosed by a double-membrane vesicle and are degraded by autophagy to provide nutrients for metamorphosis. Within three hours of the onset of starvation however, a striking increase in both size and number of
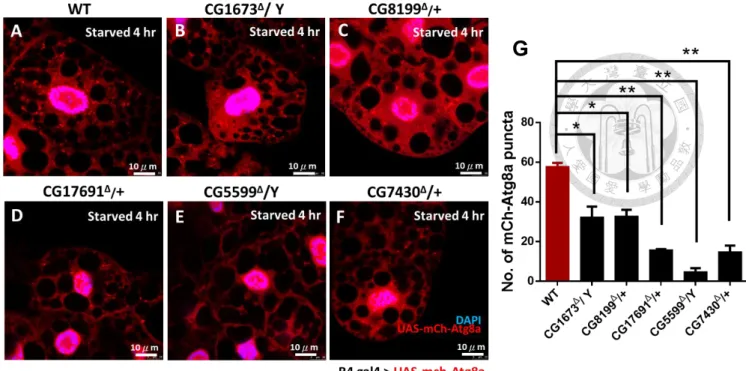
vesicles has been reported. We therefore hypothesized that elevation of the level of BCAAs should inhibit autophagy.
To determine this, UAS mch-Atg8a was driven in the fly fat body by r4 gal4 in the knockout background. After four hours of starvation, puncta could be seen and calculated in individual cells. Mutants showed reduced Atg8a signals (Figure11.) with this result support by Lysotracker data.
In the well-fed diet condition, we could not see abundant autolysosomes (Figure
12A.) We found clear autophagy puncta in well-fed WT larvae, whilst mutant larvae exhibited significantly fewer puncta (Mann-Whitney test; p<0.05).
As expression level was not sufficiently high in controls, we therefore
investigated starvation-induced autophagy. Abundant and intense puncta appeared in all cells of WT flies, from about 2.2 puncta per cell to 8 puncta per cell after
starvation (quantification data shown in Figure 12B.). In contrast, mutant fat bodies had fewer puncta compared to controls with a range of 0.5- 4.7 puncta per cell, significantly less than in WT (Mann-Whitney test.; p<0.001).
Our results imply that elevated BCAAs could modulate activity of autophagy.
11. Larval movement capability was significantly reduced in BCKDH
mutants.
Muscle is the major position to carry out BCAAs metabolism. In human, infants who suffered from MSUD also had obvious muscle atrophy symptoms. In addition to muscle problem, neuron defects would also affect locomotor behavior.
BCKDH mutant larvae showed significant reductions in travel length and average
speed (Student’s t test; p < 0.001.). We also observed differences in crawling patterns (Figure13A.) compared to WT larvae, with mutant larvae tending to have
uncoordinated paths and fewer turns.
12. Mitochondrial morphology changes in muscle and fat body
The BCAA degradation pathway eventually produces acetyl-coA, which is sent to mitochondria to produce energy. Dysfunction of mitochondria may cause many problems such as developmental arrest, muscle atrophy and neurodegenerative diseases. To understand mitochondrial distribution, shape, and dynamics in muscle and in fat bodies, we used immunostaining and examined mitochondrial patterns.
In muscle, mitochondrial shape obviously different in all BCKDH mutants except for the CG8199 and CG17691 line. Mitochondria became smaller than wildtype in CG1673 mutant and CG5599 mutant showed diffused, dispersed phenotype. Whereas, CG7430 mutant appeared cluster mitochondria (Figure14.).
Mitochondrial mass changes were observed in the fat body. All of mutants had lower mitochondria mass than WT (Mann-Whitney test; p<0.001) (Figure15.). In addition, we found that CG5599 and CG7430 mutants had particular clusters of mitochondria (arrow in Figure15 E,F.).
13. Metformin treatment reduces the accumulation of leucine and isoleucine in E2 mutant larvae.
To determine the efficiency of Metformin in Drosophila, we measured BCAA levels (using LC-MS) in L3 WT and CG5599 mutant larvae 24 hours after Metformin (10mM) treatment. Metformin effectively rescued accumulation levels of leucine and isoleucine in E2 mutants, with no significant differences being found between
controls and mutants that had been treated with Metformin (Mann-Whitney test;
p<0.001). On the other hand, valine levels were found to increase in mutants after Metformin treatment (Figure 16.).
14. Metformin treatment rescues locomotor behavior in mutant larvae.
Larval locomotor defects were previously found in most mutant larvae lines (Section 11). We thus tested if Metformin was also able to rescue this phenotype. After 3 days of 10mM Metformin treatment, CG5599 and CG7430 mutant larvae demonstrated
more straight locomotor behavior than untreated ones (Figure 17A.). Larval travelled length was also rescued in mutant lines treated with Metformin but not average larval speed, suggesting that Metformin could recover mutants discontinuous mobility.
(Student’s t test; p < 0.001.) (Figure 17B.).
Discussion
Here, we generated a novel model of MSUD in D. melanogaster, then validated it by comparing molecular, electrophysiological and behavioral phenotypes of mutant flies with other animal models/ humans.
Knock out flies for the CG1673 (BCAT), CG5599 (E2 component) and CG7430
(E3 component) genes showed severe developmental problems. Homozygous flies were unable to eclose from pupae in knockout lines under normal feeding conditions.
This is similar in some respects to the human form of MSUD, with consumption of protein potentially leading to lethality in infants11. On the other hand, CG8199 (E1α component) and CG17691 (E1β component) mutants were homozygous viable at all developmental stages. Both mutants were E1 component knockouts however; as both subunits serve the same function, it is potentially unsurprising that inactivation of only one of these genes has little or no effect on the biological phenotype.
MSUD causes not only metabolic problems but also central nervous system disorders. In humans, MSUD patients have been shown to suffer from both white matter and neuronal injuries in magnetic resonance imaging studies; this includes extensive brain edema and pathological changes in the basal ganglia60,61. Neuronal defects are therefore a significant risk for MSUD patients, with even the most effective available treatment (liver transplantation) only proven effective for preventing acute crises and
unable to reverse psychiatric disease62. Similar to this phenotype, we found the appearance of vacuoles in brain paraffin sections of mutant flies. Furthermore, eye morphology changes revealed severe injury traits in mutant larvae, particularly CG5599 and CG7430 mutants. This phenotype became more serious when intense light was supplied for 7 and 14 days, suggesting the absence of these two subunits led to
neurodegeneration.
Additionally, analysis of our ERG data on photoreceptor function lent credence to the morphological data. Reduction of ERG depolarization amplitudes and absence of on- and off- transients resulted in neuronal defects. These data indicate that CG5599 and CG7430 could play a critical role in the normal function of neurons.
High BCAA levels are often found in obese, insulin-resistant states and in Type 2 Diabetes patients35. BCAAs thus became as a biomarker to detect type 2 diabetes.
However, the mechanism of how BCAAs elevated in obesity individual is still unclear.
There is a hypothesis to explain increased plasma BCAA levels to insulin resistance linked to activation of mTORC1: excessive nutrients lead to increase plasma level of leucine which together with insulin activate mTORC1 and S6K1. Persistent activation leads to serine phosphorylation of IRS-1 and IRS-2, which interferes with signalling and might target IRS1 for proteolysis via a proteasomal pathway. The resulting insulin resistance increases demand on insulin to expenditure overmuch glucose. Long-term
demand for insulin secretion, ultimately fail to produce sufficient quantities of insulin and lead to the T2DM18(Appendix v.).
In adipose tissue, obesity was associated with declines in BCATm (homologous to CG1673) and BCKD E1α (homologous to CG8199) protein concentrations in
ob/ob mice and Zucker fatty rats37,63. However, we found BCAT (CG1673) and two
BCKDH (CG5599 and CG7430) mutants were associated with increasing lipid droplet size in fat body which is a sign od obesity. Due to the absence of CG1673, CG5599 and CG7430 in adipocytes, we hypothesized that elevated BCAAs activated dTOR, which
downregulated autophagy and protein/lipid synthesis. Therefore, decline of protein and lipid turnover rate made excessive lipid storage in fat body. We would also like to investigate whether elevated BCAAs regulate insulin resistant and hyperglycemia in the MSUD Drosophila model in the future.
In order to investigate autophagy in MSUD mutants, we used an Atg8a marker and Lysotracker to monitor the progress of autophagy and formation of lysosomes. Here, we found that Atg8a marker expression was markedly decreased in mutant lines, suggesting that autophagy was downregulated. We hypothesize that activation of dTOR resulted in an inhibition of autophagy. Misregulation of autophagy possibly represents protein degradation imbalances in the fat body and mitochondria homeostasis in muscle.
Alteration of muscular, neural, and olfactory systems should affect locomotor
behavior. Analysis of mutant mobility could therefore help to increase our understanding of the exact effects of MSUD. In human cases, twitching of limbs has been reported to happen in two month old infants64. In our model all mutants, apart from CG17691 mutants, demonstrated reduced mobility compared to controls. This includes not only the distance they were able to travel within a set time limit but also
their average speed. Their paths also tended to be highly irregularity, which is suggestive of damage to muscles and neurons.
It is increasingly clear that altered mitochondrial dynamics also underlie the pathology of many degenerative diseases65. Thus, understanding mitochondrial distribution, shape, and dynamics in all cell types should help to develop treatment regimens targeting different tissues66. We observed mitochondria in larval fat bodies and muscles and found that mitochondria in CG1673 mutant were smaller than in controls, whilst CG5599 and CG7430 mutant mitochondria were more dispersed.
Consistent with mitochondria in muscles, we found that the number of mitochondria decreased in mutant fat bodies and mitochondrial clusters appeared in CG5599 and CG7430 mutants, implying that this abnormal distribution contributed to functional
disorders. These results suggest that future research into mitochondrial function in mutants could be highly promising, particularly if done in conjunction with detection of TCA cycle substrate levels (which are linked to energy production).
Current treatments for MSUD are not satisfactory and require new approaches to combat this disease. A major hurdle in developing new treatments has been the lack of a suitable widespread animal model. Drosophila melanogaster is a well-studied, genetically pliable model organism that has proven useful for understanding molecular mechanisms of human diseases. Recently, scientists had been able to
establish live organism drug screening protocols based chemical screenings in Drosophila67. Though the value of flies in terms of human disease may be unclear, Drosophila and human cell had been found to often share the same molecular
mechanisms. This includes actin and microtubule poisons, inhibitors of DNA topoisomerases, kinases, and phosphatases, alkylating agents and modulators of membrane channels67. Thus small compounds identified for their activity in flies may show similar efficacy in mammals, whilst also being much quicker and easier to identify.
Our preliminary testing of the antidiabetic drug, Metformin (which has been previously used in mice models of MSUD) was able to rescued several mutant larvae phenotypes to the WT state46. In our findings, 10mM Metformin treatment was able to rescue BCAA levels and locomotor behavior, though mutant pupae from several lines were still unable to eclose. Testing of a range of concentrations is still required to
determine optimal dosage levels however, as well as determination of the most efficient treatment length.
Based on this newly established MUSD model, more metabolic processes and mechanism can be investigated and be understood. Taken together, our data illustrates how defects in BCAA metabolic processes can disrupt nervous system development
and function, and establishes D. melanogaster mutants as a model to better understand MSUD.
References
1. MartinLS. Maple Syrup Urine Disease. In: Encyclopedia of the Neurological Sciences. ; 2014. doi:10.1016/B978-0-12-385157-4.00076-2
2. ShimomuraY, MurakamiT, NakaiN, NagasakiM, HarrisR a. Exercise promotes branched-chain amino acids catabolism: Effects of branched-chain amino acids supplementation on skeletal muscle during exercise. J Nutr. 2004.
3. HermanMA, SheP, PeroniOD, LynchCJ, KahnBB. Adipose tissue Branched Chain Amino Acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem. 2010. doi:10.1074/jbc.M109.075184
4. ZhangS, ZengX, RenM, MaoX, QiaoS. Novel metabolic and physiological functions of branched chain amino acids: A review. J Anim Sci Biotechnol.
2017. doi:10.1186/s40104-016-0139-z
5. CharltonM. Branched-Chain Amino Acids: Metabolism, Physiological Function, and Application. J Nutr. 2006.
6. ShimomuraY, HondaT, ShirakiM, et al. Branched-chain amino acid catabolism in exercise and liver disease. J Nutr. 2006. doi:10.1093/jn/136.1.250S
7. NellisMM, DannerDJ. Gene Preference in Maple Syrup Urine Disease. Am J Hum Genet. 2002. doi:10.1086/316950
8. BlackburnPR, GassJM, Pinto e VairoF, et al. Maple syrup urine disease:
Mechanisms and management. Appl Clin Genet. 2017.
doi:10.2147/TACG.S125962
9. SkvorakKJ. Animal models of maple syrup urine disease. J Inherit Metab Dis.
2009. doi:10.1007/s10545-009-1086-z
10. GibsonKM, ElpelegON, MortonDH, WappnerRS. Disorders of Leucine Metabolism. In: Physician’s Guide to the Laboratory Diagnosis of Metabolic Diseases. ; 2011. doi:10.1007/978-3-642-55878-8_12
11. CharltonM. Branched-Chain Amino Acids: Metabolism, Physiological Function, and Application. J Nutr. 2006;136(3):295-298.
12. OzandP. Maple syrup urine disease (branched-chain oxoaciduria). In: Atlas of Metabolic Diseases Second Edition. ; 2013. doi:10.1201/b13565-27
13. FrazierDM, AllgeierC, HomerC, et al. Nutrition management guideline for maple syrup urine disease: An evidence- and consensus-based approach. Mol Genet Metab. 2014. doi:10.1016/j.ymgme.2014.05.006
14. LerinC, GoldfineAB, BoesT, et al. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol Metab. 2016.
doi:10.1016/j.molmet.2016.08.001
15. ChuangDT, ChuangJL, WynnRM. Branched-Chain Amino Acids :
Metabolism , Physiological Function , and Application Lessons from Genetic
Disorders of Branched-Chain Amino. Heal (San Fr. 2006.
16. ChuangJL, WynnRM, MossCC, et al. Structural and biochemical basis for novel mutations in homozygous Israeli maple syrup urine disease patients: A proposed mechanism for the thiamin-responsive phenotype. J Biol Chem. 2004.
doi:10.1074/jbc.M313879200
17. WangC, LiuZ, HuangX. Rab32 is important for autophagy and lipid storage in drosophila. PLoS One. 2012. doi:10.1371/journal.pone.0032086
18. AdamsSH. BCAA in metabolic signalling and IR. 2015;10(12):723-736.
doi:10.1038/nrendo.2014.171.Branched-chain
19. NewgardCB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012.
doi:10.1016/j.cmet.2012.01.024
20. LiuZ, JeppesenPB, GregersenS, LarsenLB, HermansenK. Chronic exposure to leucine in vitro induces β-cell dysfunction in INS-1E cells and mouse islets. J
Endocrinol. 2012. doi:10.1530/joe-12-0148
21. AungstBJ. Structure/Effect Studies of Fatty Acid Isomers as Skin Penetration Enhancers and Skin Irritants. Pharm Res An Off J Am Assoc Pharm Sci. 1989.
doi:10.1023/A:1015921702258
22. HradecJ, DufekP. Determination of cholesteryl 14-methylhexadecanoate in
blood serum by reversed-phase high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1994. doi:10.1016/0378-4347(94)00292-4
23. HanM, JiaF, CuiM, ThanMT. Developmental defects of Caenorhabditis elegans lacking branched-chain α-ketoacid dehydrogenase are mainly caused by
monomethyl branched-chain fatty acid deficiency. J Biol Chem.
2016;291(6):2967-2973. doi:10.1074/jbc.M115.676650
24. McPheeCK, BaehreckeEH. Autophagy in Drosophila melanogaster. Biochim Biophys Acta - Mol Cell Res. 2009. doi:10.1016/j.bbamcr.2009.02.009
25. VellaiT, Takacs-VellaiK, ZhangY, KovacsAL, OroszL, MüllerF. Influence of TOR kinase on lifespan in C. elegans. Nature. 2003. doi:10.1038/426620a 26. CrespoJL, HallMN. Elucidating TOR Signaling and Rapamycin Action:
Lessons from Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2003.
doi:10.1128/mmbr.66.4.579-591.2002
27. SarbassovDD, GuertinDA, AliSM, SabatiniDM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (80- ). 2005.
doi:10.1126/science.1106148
28. NojimaH, TokunagaC, EguchiS, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003.