Review
Roles of thioredoxin in nitric oxide-dependent preconditioning-induced tolerance against MPTP neurotoxin
Chuang C. Chiueh
a,b,*, Tsugunobu Andoh
c, P. Boon Chock
daSchool of Pharmacy, Taipei Medical University, Taipei 110, Taiwan, ROC
bLaboratory of Clinical Science, NIMH, NIH, Bethesda, MD 20892-1264, USA
cDepartment of Applied Pharmacology, Toyama Medical and Pharmaceutical University, Japan
dLaboratory of Biochemistry, NHLBI, NIH, Bethesda, MD 20892-8012, USA Received 11 July 2004; revised 2 March 2005; accepted 31 March 2005
Available online 7 July 2005
Abstract
Hormesis, a stress tolerance, can be induced by ischemic preconditioning stress. In addition to preconditioning, it may be induced by other means, such as gas anesthetics. Preconditioning mechanisms, which may be mediated by reprogramming survival genes and proteins, are obscure. A known neurotoxicant, 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), causes less neurotoxicity in the mice that are preconditioned. Pharmacological evidences suggest that the signaling pathway of S
NO – cGMP – PKG (protein kinase G) may mediate preconditioning phenomenon. We developed a human SH-SY5Y cell model for investigating S
NO-mediated signaling pathway, gene regulation, and protein expression following a sublethal preconditioning stress caused by a brief 2-h serum deprivation. Preconditioned human SH-SY5Y cells are more resistant against severe oxidative stress and apoptosis caused by lethal serum deprivation and 1-mehtyl-4- phenylpyridinium (MPP
+). Both sublethal and lethal oxidative stress caused by serum withdrawal increased neuronal nitric oxide synthase (nNOS/NOS1) expression and S
NO levels to a similar extent. In addition to free radical scavengers, inhibition of nNOS, guanylyl cyclase, and PKG blocks hormesis induced by preconditioning. S-nitrosothiols and 6-Br-cGMP produce a cytoprotection mimicking the action of preconditioning tolerance. There are two distinct cGMP-mediated survival pathways: (i) the up-regulation of a redox protein thioredoxin (Trx) for elevating mitochondrial levels of antioxidant protein Mn superoxide dismutase (MnSOD) and antiapoptotic protein Bcl-2, and (ii) the activation of mitochondrial ATP-sensitive potassium channels [K(ATP)]. Preconditioning induction of Trx increased tolerance against MPP
+, which was blocked by Trx mRNA antisense oligonucleotide and Trx reductase inhibitor. It is concluded that Trx plays a pivotal role in S
NO- dependent preconditioning hormesis against MPTP/MPP
+.
D 2005 Elsevier Inc. All rights reserved.
Keywords: Adaptation; Antiapoptotic protein Bcl-2; Cyclic GMP; Hormesis; Mitochondria; Mn superoxide dismutase; 1-Methyl-4-phenylpyridinium (MPP+);
ATP-sensitive potassium channel
Contents
Introduction . . . . S97 Selective and non-selective neurotoxicity caused by MPTP/MPP
+. . . . S97 Roles of free radicals in initiating and maintaining preconditioning adaptation . . . . S97 cGMP-dependent expression of thioredoxin mediates preconditioning adaptive neuroprotection . . . . S98 Neuroprotective mechanisms of thioredoxin . . . . S99 Preconditioning-induced thioredoxin mediates tolerance against MPTP/MPP
+. . . . S99 Concluding remarks . . . . S99 Drugs mimic cGMP-mediated preconditioning neuroprotection . . . . S99
0041-008X/$ - see front matterD 2005 Elsevier Inc. All rights reserved.
doi:10.1016/j.taap.2005.03.020
* Corresponding author. School of Pharmacy, Taipei Medical University, 250 Wu-Sing Street, Taipei 110, Taiwan, ROC.
E-mail address: chiueh@tmu.edu.tw (C.C. Chiueh).
www.elsevier.com/locate/ytaap
YTAAP-10269; No. of pages: 7; 4C:
Acknowledgments . . . . S100 References . . . . S100
Introduction
Selective and non-selective neurotoxicity caused by MPTP/MPP
+MPTP is a man-made neurotoxin which is converted to toxic metabolites such as 1-methyl-4-phenylpyridinium (MPP
+). MPP
+enters brain monoaminergic neurons and causes a selective destruction of the A9 nigrostriatal dopaminergic neurons in monkeys and humans at low milligram doses (Burns et al., 1983). At 1.5 mg/kg intra- venous (iv) dose of MPTP, MPP
+is taken up by brain dopaminergic neurons but preferentially it causes retrograde degeneration of the pigmented A9 nigral neurons only and spares non-pigmented brain dopamine neurons (A10, A12 and A 16) (Chiueh et al., 1985). It is worthy noting that the pigmented A9 nigral neurons contain relatively high levels of non-heme iron complexes (Chiueh, 2001). We proposed that sustained dopamine release caused by MPP
+could increase auto-oxidation of dopamine in the presence of iron com- plexes and oxygen (Chiueh et al., 1994). Dopamine oxidation generates hydroxyl radicals, lipid peroxidation, oxidative stress and retrograde degeneration predominantly in iron-rich A9 nigral neurons when the cellular antioxidative defense system is compromised by sustained high oxidant stress (Chiueh, 2001; Chiueh and Rauhala, 1998). In fact, ferrous citrate complexes are as toxic as MPP
+in causing nigral loss in vivo indicating that the mixture of iron, oxygen and dopamine is highly neurotoxic and selective to pigmented A9 nigral neurons (Sun et al., 1988; Sziraki et al., 1998).
Unexpectedly, the highest concentration of MPP
+is located in monkey’s noradrenergic cell bodies (locus caeruleus and adrenal medulla) where no significant neurotoxicity is found (Markey et al., 1984). When experimenting with high concentrations in non-dopaminergic cells, MPP
+causes oxidative damage and necrotic death in cells and neurons since it not only complexes with iron to generate cytotoxic- free radicals but also inhibits mitochondrial complex I and energy supply (Andoh et al., 2002a; Kotake and Ohta, 2003).
In the rodents MPTP (>100 mg/kg) causes a serotonin syndrome and a reversible acute dopamine depletion rather than chronic nigrostriatal neurodegeneration (Chiueh et al., 1984). The administration of low doses of MPTP in rhesus monkeys (<1.5 mg/kg, iv) creates an ideal primate model of parkinsonism reflected by a selective nigrostriatal degene- ration, dopamine depletion and nigral loss. This MPTP- induced primate model, but not rodent model, can be used for screening of new therapeutics, testing of brain dopamine imaging ligands, and transplantation efficacy using dopamine producing neurons and stem cells (Chiueh, 1988). However, unpublished information suggests that adaptive hormesis (e.g., concentrations of toxic substances below the amount
that cause toxicity will cause tolerance) may be developed in monkeys when much lower doses of MPTP are chronically administered (C. Freed, personal communications). This adaptation phenomenon could explain why only a few parkinsonian cases developed among approximately 200 MPTP abusers. For elucidating this hormesis mechanism, we employed a human neuroblastoma SH-SY5Y cells to examine preconditioning tolerance in preventing or decreas- ing MPP
+-induced neurotoxicity (Andoh et al., 2000). We also investigated preconditioning-induced survival proteins and to understand which gene or protein elicits precondition- ing-induced adaptive neuroprotection (Andoh et al., 2002a).
We focused on cGMP-dependent reprogramming of early genes in the nucleus and survival proteins in the mitochon- dria of preconditioned human neuroblastoma cells since free radicals often affect nuclear DNA and mitochondrial proteins (Andoh et al., 2002b, 2003).
Roles of free radicals in initiating and maintaining preconditioning adaptation
In the cardiovascular system, brief episodes of ischemia cause new protein synthesis and subsequently protect cells from prolonged period of lethal ischemia insult, which is now recognized as a new research field of preconditioning phenomenon. The exact preconditioning mechanism under- lying adaptive neuroprotection is largely unclear, which remains to be elucidated. Increasing evidence suggest that free radicals may be involved not only in the trigger of preconditioning phenomenon in the cell nucleus but also the maintenance of adaptive expression of survival proteins, especially in the mitochondria (Andoh et al., 2000; Nakano et al., 2000). Most preconditioning procedures are blocked by free radical scavengers, and some of delayed adaptive cytoprotections are prevented by inhibition of subtypes of nitric oxide synthase (NOS) depending on the types of cells studied (Andoh et al., 2000; Centeno et al., 1999; Gidday et al., 1999; Horimoto et al., 2000; Keynes and Garthwaite, 2004; Xi et al., 2002). Our pilot in vitro study indicates that serum withdrawal increases first the generation of hydroxyl radicals ( S
OH) and a delayed increase of nitric oxide ( S
NO) (Andoh et al., 2000).
In myocardial preparations, preconditioning activation of eNOS and associated S
NO – cGMP – PKG (protein kinase G) pathway in the cardiovascular system can phospho-activate ATP-sensitive potassium channels [K(ATP)] in the heart for cardioprotection (Garlid et al., 2003; Han et al., 2002;
Horimoto et al., 2000; Ockaili et al., 1999; Zaugg et al.,
2002). It is also known that a similar preconditioning
neuroprotective mechanism via activation of mitochondrial
K(ATP) channels can be observed in the central nervous
system (Kis et al., 2004). Ischemic preconditioning-induced
Hsp70 and Bcl-2 are also mitochondrial proteins, which may or may not be mediated by the proposed S
NO – cGMP – PKG signaling pathway (Ciani et al., 2002; Suzuki et al., 2003) A recent report suggests that S
NO induces Hsp70 in rat brain for cytoprotection (Suzuki et al., 2002). Ischemic precondition- ing increases both S
NO and Hsp70 while lipopolysaccharide (LPS) elevates S
NO and ceramide but not Hsp70 indicating different preconditioning signaling pathways may be acti- vated by S
NO (Furuya et al., 2001; Zimmermann et al., 2001).
Based on results obtained from preconditioned human SH-SY5Y cells following a 2-h serum withdrawal, we proposed that reactive oxygen species activate redox factor Ref-1 and activator protein AP-1 in cell nucleus via yet to be defined signaling pathway (Andoh et al., 2000). In other cell types, preconditioning activates either HIF-1a or TNFa (Digicaylioglu and Lipton, 2001; He et al., 2001; Ruscher et al., 2002). Elevated AP-1 levels leads to a delayed synthesis of nNOS for the activation of the signaling pathway of S
NO – cGMP – PKG for reprogramming expression of survival genes and proteins in adaptive neuroprotection. Recently, we discovered that cGMP-mediated induction of the redox protein thioredoxin (Trx) and mitochondrial antioxidant pro- tein Mn superoxide dismutase (MnSOD) and antiapoptotic protein Bcl-2, all of which may participate in precondition- ing-induced adaptation for neuroprotection (Andoh et al., 2000, 2002b, 2003; Ciani et al., 2002). Furthermore, we are currently investigating an unexpected observation of a pos- sible role of suppression of the pro-oxidative adaptor protein p66
shcfor suppressing oxidative stress in preconditioned human neurotrophic cells (Andoh et al., 2000). In this mini- review, we focus on cGMP-dependent induction of mito- chondrial survival proteins 24 h post-preconditioning stress.
cGMP-dependent expression of thioredoxin mediates preconditioning adaptive neuroprotection
In the resting stage, human neuroblastoma SH-SY5Y cells contain relatively low levels of nNOS and Trx. Both nNOS and Trx are significantly elevated 24 h following a brief non- lethal serum deprivation for 2 h, and these preconditioned human neurotrophic cells develop tolerance 12 to 24 h thereafter against lethal oxidative stress evoked by a prolonged and severe serum deprivation. Inhibition of nNOS/cGMP/PKG bocks preconditioning-induced Trx expression and associated adaptive neuroprotection indicat- ing cGMP-dependent Trx expression may mediate mecha- nism underlying preconditioning tolerance (see Fig. 1, Andoh et al., 2002a, 2003). Cell permeable 6Br-cGMP mimics preconditioning adaptation of Trx expression and cytopro- tection. PKG enhances the expression of Trx through its c- Myc, AP-1 and PEA-3 nuclear binding motifs since PKG phospho-activates the transcription factor c-Myc (Andoh et al., 2003). Furthermore, exogenously administered Trx activates cAMP-response element binding protein (CREB) in cell nucleus and induces in mitochondria not only Bcl-2 for suppressing caspase-mediated apoptosis but also MnSOD for
inhibiting generation of superoxide anion radicals, lipid peroxidation and oxidative damage (Andoh et al., 2002a, 2003). Moreover, overexpression of either Bcl-2 or MnSOD also enhances cellular tolerance against severe oxidative stress (Chiueh et al., 2003; Klivenyi et al., 1998;
Offen et al., 1998). Furthermore, the participation of Trx in preconditioning-induced hormesis is supported by the finding that inhibition of Trx synthesis in these human SH-SY5Y cells by Trx mRNA antisense but not sense
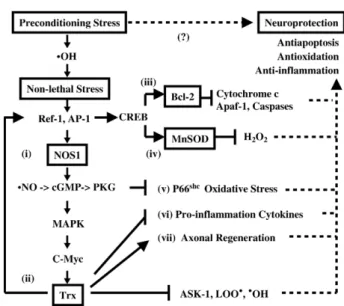
Fig. 1. Schematic diagram of nitric oxide-dependent and Trx-mediated preconditioning hormesis against MPP+. Severe oxidative stress causes sustained generation of reactive oxygen species, lipid peroxidation, oxidative damage and apoptotic cell death due to the activation of mitochondria-mediated apoptotic pathways. The binding of Apaf-1 to the release cytochrome c catalyzes procaspase 9 to activate caspase 9-mediated apoptotic pathway. A brief preconditioning episode causes hormetic effects, which protects human neuroblastoma cells from apoptosis via the release of reactive oxygen species and activation of a preconditioning cascade: (i) The induction of NOS1 mRNA and synthesis of nNOS, (ii) the activation of cGMP-PKG signaling pathway for elevating Trx expression via activation of c-Myc. In addition to suppress generation of hydroxyl radicals, lipid peroxidation and ASK-1, Trx also activate CREB for delayed expression of mitochondrial survival proteins including (iii) antiapoptotic Bcl-2 and (iv) antioxidative MnSOD. (v) Furthermore, cGMP down regulates pro- oxidative protein p66shcthereby inhibiting oxidative stress. (vi) It has an unique anti-inflammatory property since Trx overexpression in mice results in tolerance against acute lung distress syndrome. (vii) Finally, Trx may participate in sprouting and regeneration of nerve terminals. This multi- faceted neuroprotective mechanism may increase tolerance of this human neurotrophic cell line against MPP+. In conclusion, free radicals may mediate not only triggering but also maintaining of preconditioning cascade. cGMP-mediated expression of Trx and delayed expression of Bcl-2 and MnSOD could play a pivotal role in preconditioning-induced cardio- and neuroprotection induced by preconditioning procedures. Drugs mimic preconditioning induction of Trx and related survival proteins may provide new insights of developing neuroprotective strategy and therapeu- tics. Abbreviations:
S
OH, hydroxyl radical; Ref-1, redox factor 1; AP-1, activator protein 1; PKG, protein kinase G; MAPK, mitogen-associated protein kinase; LOO, lipid peroxyl radical; ASK-1, apoptosis signal- regulating kinase 1; c-Myc, transcriptor factor; CREB, cAMP-response element binding protein; Bcl-2, antiapoptotic protein; MnSOD, Mn superoxide dismutase; Apaf-1, apoptotic protease activating factor-1.
phosphothionate oligonucleotides reduces the hormesis effects (Andoh et al., 2002a).
Neuroprotective mechanisms of thioredoxin
Previous studies indicate that knock out of either cytosolic Trx1 or mitochondrial Trx2 is detrimental to cell viability and early embryonic lethality, whereas up-regulation of Trx would decrease oxidative stress-induced apoptosis (Kahlos et al., 2001; Mitsui et al., 2002; Nonn et al., 2003; Tanaka et al., 2002; Ueda et al., 2002; Yamamoto et al., 2003). Trx1 contains a conserved active site which consists of two redox active cysteine residues (Tyr-Cys-Gly-Pro-Cys-Lys), Trx2 has two additional zinc binding motifs of Cys-X-X-Cys in the N-terminal. The reduced form of Trx-(SH)
2suppresses free radical formation, lipid peroxidation and caspase-mediated apoptosis since reduced Trx is oxidized to form disulfide bonds (Trx-S
2). This is catalyzed by Trx reductase with NADPH as the cofactor and Trx-S
2is reduced back to its active form Trx-(SH)
2. The redox of Trx would help to reactivate functional thiol groups of enzymes and tran- scription factors for maintaining vital cellular functions. Trx increases DNA binding to AP-1 transcription factor in conjunction with the nuclear Ref-1 by specifically reducing a cysteine residue of the DNA binding domain of Jun and Fos dimmer, those make a composition of functional AP-1 transcription factor. Trx is capable of removing H
2O
2and thus suppressing generation of reactive oxygen species and lipid peroxyl radicals. Reduced Trx may complex with apoptosis signaling-regulating kinase 1 (ASK-1) and thus reduce ASK-1 activity resulting in protection of cells from apoptosis (Saitoh et al., 1998; Zhang et al., 2004). Further- more, Trx activates CREB and enhances the biosynthesis of antiapoptotic protein Bcl-2 to prevent cytochrome c release from mitochondria and antioxidative protein MnSOD to reduce mitochondrial levels of superoxide anion radicals (Andoh et al., 2003). Bcl-2 could prevent apoptosis through the activation of the pathway of caspase-9 and apoptotic protease activating factor-1 (Apaf-1) by inhibiting the release of cytochrome c. These multifaceted direct and/or indirect neuroprotective properties of Trx may contribute to adaptive neuroprotection evoked by preconditioning phenomenon in human neurotrophic cells, SH-SY5Y.
Preconditioning-induced thioredoxin mediates tolerance against MPTP/MPP
+Both serum deprivation and MPP
+generate cytotoxic reactive oxygen species and 4-hydroxy-1,2-nonenol, which in turn lead to oxidative stress-induced apoptosis due to the release of cytochrome c and the activation of proapoptotic cascade of caspase 9 and 3 (Andoh et al., 2002a). Moreover, MPP
+-induced hydroxyl radical generation and brain lipid peroxidation have been demonstrated in vivo (Chiueh and Rauhala, 1998; Chiueh et al., 1994). We recently showed that exogenously administered Trx (1 AM) protects SH-SY5Y
neurotrophic cells from apoptosis caused by MPP
+support- ing a notion that free radical-induced oxidative stress may cause neurodegeneration (Andoh et al., 2002a). Precondi- tioning-induced cross tolerance against MPP
+-induced neu- rotoxicity is also mediated by Trx since this adaptive tolerance can be prevented by pretreatment of cells with either Trx reductase inhibitor or using antisense oligonucleo- tides. Moreover, reduced Trx also induces the synthesis of mitochondrial protein Bcl-2 and MnSOD, all of which could suppress oxidative stress/caspase-mediated apoptosis pro- duced by a parkinsonism-producing neurotoxin, MPP
+(Andoh et al., 2003). Interestingly, the concentration – response curve of MPP
+in human neurotrophic cells is right shifted in the preconditioning hormesis group. The LD
50for MPP
+-induced apoptotic cell death increased by approx- imately 30-fold shifting from 0.1 mM for the control cell group to 3 mM obtained from the preconditioned cell group.
Using serum deprivation and MPP
+as model of oxidative stress, our study of neuroprotective mechanism by which Trx prevents oxidative stress-induced apoptosis reveals that Trx biosynthesis is required for hormesis induced by precondi- tioning in either human neurotrophic cells. Acting via a common adaptive mechanism of Trx serum deprivation- induced preconditioned cells develops a cross tolerance against oxidative stress produced by the parkinsonism- producing neurotoxin MPP
+(Andoh et al., 2002b). A recent report suggests that elevated Trx levels are observed following ischemic preconditioning in myocardial prepara- tions (Das and Maulik, 2003). The protective effects of Trx against MPP
+may be derived in part from its ability to active CREB in cell nucleus for elevating mitochondrial levels of Bcl-2 and MnSOD. Furthermore, overexpression of Trx, MnSOD or Bcl-2 in mice leads to tolerance against MPTP- induced dopamine depletion (Klivenyi et al., 1998; Kojima et al., 1999; Mitsui et al., 2002; Offen et al., 1998). Lastly, we are wondering whether cancer cells might up-regulate Trx for developing drug resistance for survival during chemotherapy since relatively high Trx levels are often observed in malignant tumors (Kahlos et al., 2001; Powis et al., 2000).
Concluding remarks
Drugs mimic cGMP-mediated preconditioning neuroprotection
Preconditioning adaptive cytoprotection is undergoing clinical trials for its potential clinical benefits in cell survival and vitality of donor liver following organ transplantation (Berrevoet et al., 2003; Carini and Albano, 2003; Fung, 2001;
Klein et al., 1999; Koti et al., 2003). With the development of
neuroprotective strategy and therapeutic agents in mind, it is
now recognized that preconditioning-induced adaptive neu-
roprotective mechanisms become a very attractive means for
identifying lead neuroprotective compounds. Preclinical in-
vestigation of cardioprotective role of activation of mito-
chondrial K(ATP) channels in preconditioned myocardial preparations leads to the development of potassium channel openers and/or protonophores (i.e., pinacidil, diazoxide) for restoring functions in ischemic hearts (Han et al., 2002;
Holmuhamedov et al., 2004; Sebille et al., 2004; Zaugg et al., 2002). Some of potassium channel openers may enter blood – brain barrier and produce assumed neuroprotective effects in helping neurodegenerative brain disorders and stroke (Kornhuber et al., 1999). The proposed S
NO – cGMP – PKG signaling pathway that induces Trx and subsequently Bcl-2 and MnSOD for supporting preconditioning-induced adap- tive neuroprotection could be used to design lead compounds targeted for neuroprotection and/or cardioprotection. Ebselen can mimic selenium-containing Trx reductase for elevating levels of reduced Trx, and it is now entering clinical trials for its potential applications in enhancing neuroprotection or functional recovery in ischemic stroke patients (Zhao and Holmgren, 2004). Other drugs that activate the S
NO – cGMP – PKG pathway for enhancing Trx biosynthesis could be screened and developed for their potential in providing neuroprotection against oxidative stress-induced neurode- generation such as Parkinson’s disease and stroke.
One of the lead neuroprotective compounds is 17h- estradiol, which activates nuclear estrogen receptors for the induction of nNOS, Trx, MnSOD and associated neuro- protection (Lee et al., 2003). It remains to be determined whether estrogen analogues in nanomolar concentrations act like a chemical preconditioning-producing agent. Gas anesthetics (i.e., desflurane, isoflurane, sevoflurane) are known for their ability in producing chemical precondition- ing for either neuroprotection or cardioprotection against ischemia/reperfusion injury (Zaugg et al., 2002). S-nitro- sothiols and cell membrane-permeable cGMP are used to activate the signaling pathway S
NO – cGMP – PKG for inducing beneficial survival genes and their proteins for cardio- and/or neuroprotection. For example, both GSNO and SNAP have been shown to protect nigrostriatal dopaminergic neurons against iron-catalyzed oxidative damage and neurodegeneration in vivo (Rauhala and Chiueh, 2000; Rauhala and Chiueh, this Proceedings; Rauhala et al., 1998). A new lead compound YC-1 [3-(5V-hydroxymethyl- 2V-furyl)-1-benzyl Indazole] discovered by Taiwanese researchers could be evaluated for its unique property in elevating cGMP levels and associated beneficial biological actions (Pan et al., 2004; Teng et al., 1997). It is concluded that potential therapeutics derived from current investiga- tions of roles of Trx in cGMP-dependent adaptive mecha- nisms underlying preconditioning-induced cardio- and/or neuroprotection may become a new research direction for both biotechnological and pharmaceutical developments.
Acknowledgments
Supported, in part, by NSC (C.C.C.), JSPS (T.A.) and NIH IRP (P.B.C.) grants.
References
Andoh, T., Lee, S.Y., Chiueh, C.C., 2000. Preconditioning regulation of bcl-2 and p66shc by human NOS1 enhances tolerance to oxidative stress. FASEB J. 14, 2144 – 2146.
Andoh, T., Chock, P.B., Chiueh, C.C., 2002a. The roles of thioredoxin in protection against oxidative stress-induced apoptosis in SH-SY5Y cells.
J. Biol. Chem. 277, 9655 – 9660.
Andoh, T., Chock, P.B., Chiueh, C.C., 2002b. Preconditioning-mediated neuroprotection: role of nitric oxide, cGMP, and new protein expres- sion. Ann. N. Y. Acad. Sci. 962, 1 – 7.
Andoh, T., Chiueh, C.C., Chock, P.B., 2003. Cyclic GMP-dependent protein kinase regulates the expression of thioredoxin and thioredoxin peroxidase-1 during hormesis in response to oxidative stress-induced apoptosis. J. Biol. Chem. 278, 885 – 890.
Berrevoet, F., Schafer, T., Vollmar, B., Menger, M.D., 2003. Ischemic preconditioning: enough evidence to support clinical application in liver surgery and transplantation? Acta Chir. Belg. 103, 485 – 489.
Burns, R.S., Chiueh, C.C., Markey, S.P., Ebert, M.H., Jacobowitz, D.M., Kopin, I.J., 1983. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
Proc. Natl. Acad. Sci. U.S.A. 80, 4546 – 4550.
Carini, R., Albano, E., 2003. Recent insights on the mechanisms of liver preconditioning. Gastroenterology 125, 1480 – 1491.
Centeno, J.M., Orti, M., Salom, J.B., Sick, T.J., Perez-Pinzon, M.A., 1999.
Nitric oxide is involved in anoxic preconditioning neuroprotection in rat hippocampal slices. Brain Res. 836, 62 – 69.
Chiueh, C.C., 1988. Dopamine in the extrapyramidal motor function. A study based upon the MPTP-induced primate model of parkinsonism.
Ann. N. Y. Acad. Sci. 515, 226 – 238.
Chiueh, C.C., 2001. Iron overload, oxidative stress, and axonal dystrophy in brain disorders. Pediatr. Neurol. 25, 138 – 147.
Chiueh, C.C., Rauhala, P., 1998. Free radicals and MPTP-induced selective destruction of substantia nigra compacta neurons. Adv. Pharmacol. 42, 796 – 800.
Chiueh, C.C., Markey, S.P., Burns, R.S., Johannessen, J.N., Jacobowitz, D.M., Kopin, I.J., 1984. Neurochemical and behavioral effects of 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in rat, guinea pig, and monkey. Psychopharmacol. Bull. 20, 548 – 553.
Chiueh, C.C., Burns, R.S., Markey, S.P., Jacobowitz, D.M., Kopin, I.J., 1985. Primate model of parkinsonism: selective lesion of nigrostriatal neurons by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine produces an extrapyramidal syndrome in rhesus monkeys. Life Sci. 36, 213 – 218.
Chiueh, C.C., Wu, R.M., Mohanakumar, K.P., Sternberger, L.M., Krishna, G., Obata, T., Murphy, D.L., 1994. In vivo generation of hydroxyl radicals and MPTP-induced dopaminergic toxicity in the basal ganglia.
Ann. N. Y. Acad. Sci. 738, 25 – 36.
Chiueh, C., Lee, S., Andoh, T., Murphy, D., 2003. Induction of antioxidative and antiapoptotic thioredoxin supports neuroprotective hypothesis of estrogen. Endocrine 21, 27 – 31.
Ciani, E., Guidi, S., Bartesaghi, R., Contestabile, A., 2002. Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: implication for a survival role of nitric oxide. J. Neurochem. 82, 1282 – 1289.
Das, D.K., Maulik, N., 2003. Preconditioning potentiates redox signaling and converts death signal into survival signal. Arch. Biochem. Biophys.
420, 305 – 311.
Digicaylioglu, M., Lipton, S.A., 2001. Erythropoietin-mediated neuro- protection involves cross-talk between Jak2 and NF-nB signalling cascades. Nature 412, 641 – 647.
Fung, J.J., 2001. Ischemic preconditioning: application in clinical liver transplantation. Liver Transplant. 7, 300 – 301.
Furuya, K., Ginis, I., Takeda, H., Chen, Y., Hallenbeck, J.M., 2001. Cell permeable exogenous ceramide reduces infarct size in spontaneously hypertensive rats supporting in vitro studies that have implicated
ceramide in induction of tolerance to ischemia. J. Cereb. Blood Flow Metab. 21, 226 – 232.
Garlid, K.D., Dos Santos, P., Xie, Z.J., Costa, A.D., Paucek, P., 2003.
Mitochondrial potassium transport: the role of the mitochondrial ATP- sensitive K+channel in cardiac function and cardioprotection. Biochem.
Biophys. Acta 1606, 1 – 21.
Gidday, J.M., Shah, A.R., Maceren, R.G., Wang, Q., Pelligrino, D.A., Holtzman, D.M., Park, T.S., 1999. Nitric oxide mediates cerebral ischemic tolerance in a neonatal rat model of hypoxic preconditioning.
J. Cereb. Blood Flow Metab. 19, 331 – 340.
Han, J., Kim, N., Joo, H., Kim, E., Earm, Y.E., 2002. ATP-sensitive K+ channel activation by nitric oxide and protein kinase G in rabbit ventricular myocytes. Am. J. Physiol.: Heart Circ. Physiol. 283, H1545 – H1554.
He, S.Y., Deng, H.W., Li, Y.J., 2001. Monophosphoryl lipid A-induced delayed preconditioning is mediated by calcitonin gene-related peptide.
Eur. J. Pharmacol. 420, 143 – 149.
Holmuhamedov, E.L., Jahangir, A., Oberlin, A., Komarov, A., Colombini, M., Terzic, A., 2004. Potassium channel openers are uncoupling proto- nophores: implication in cardioprotection. FEBS Lett. 568, 167 – 170.
Horimoto, H., Gaudette, G.R., Saltman, A.E., Krukenkamp, I.B., 2000. The role of nitric oxide, K+(ATP) channels, and cGMP in the precondition- ing response of the rabbit. J. Surg. Res. 92, 56 – 63.
Kahlos, K., Soini, Y., Saily, M., Koistinen, P., Kakko, S., Paakko, P., Holmgren, A., Kinnula, V.L., 2001. Up-regulation of thioredoxin and thioredoxin reductase in human malignant pleural mesothelioma. Int. J.
Cancer 95, 198 – 204.
Keynes, R.G., Garthwaite, J., 2004. Nitric oxide and its role in ischaemic brain injury. Curr. Mol. Med. 4, 179 – 191.
Kis, B., Nagy, K., Snipes, J.A., Rajapakse, N.C., Horiguchi, T., Grover, G.J., Busija, D.W., 2004. The mitochondrial K(ATP) channel opener BMS-191095 induces neuronal preconditioning. NeuroReport 15, 345 – 349.
Klein, M., Geoghegan, J., Wangemann, R., Bockler, D., Schmidt, K., Scheele, J., 1999. Preconditioning of donor livers with prostaglandin I2
before retrieval decreases hepatocellular ischemia – reperfusion injury.
Transplantation 67, 1128 – 1132.
Klivenyi, P., St Clair, D., Wermer, M., Yen, H.C., Oberley, T., Yang, L., Flint Beal, M., 1998. Manganese superoxide dismutase overexpression attenuates MPTP toxicity. Neurobiol. Dis. 5, 253 – 258.
Kojima, S., Matsuki, O., Nomura, T., Yamaoka, K., Takahashi, M., Niki, E., 1999. Elevation of antioxidant potency in the brain of mice by low-dose gamma-ray irradiation and its effect on 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP)-induced brain damage. Free Radical Biol.
Med. 26, 388 – 395.
Kornhuber, J., Maler, M., Wiltfang, J., Bleich, S., Degner, D., Ruther, E., 1999. Neuronal potassium channel opening with flupirtine. Fortschr.
Neurol. Psychiatr. 67, 466 – 475.
Kotake, Y., Ohta, S., 2003. MPP+ analogs acting on mitochondria and inducing neuro-degeneration. Curr. Med. Chem. 10, 2507 – 2516.
Koti, R.S., Seifalian, A.M., Davidson, B.R., 2003. Protection of the liver by ischemic preconditioning: a review of mechanisms and clinical applications. Dig. Surg. 20, 383 – 396.
Lee, S.Y., Andoh, T., Murphy, D.L., Chiueh, C.C., 2003. 17h-estradiol activates ICI 182,780-sensitive estrogen receptors and cyclic GMP- dependent thioredoxin expression for neuroprotection. FASEB J. 17, 947 – 948.
Markey, S.P., Johannessen, J.N., Chiueh, C.C., Burns, R.S., Herkenham, M.A., 1984. Intraneuronal generation of a pyridinium metabolite may cause drug-induced parkinsonism. Nature 311, 464 – 467.
Mitsui, A., Hamuro, J., Nakamura, H., Kondo, N., Hirabayashi, Y., Ishizaki-Koizumi, S., Hirakawa, T., Inoue, T., Yodoi, J., 2002.
Overexpression of human thioredoxin in transgenic mice controls oxidative stress and life span. Antioxid. Redox Signal 4, 693 – 696.
Nakano, A., Liu, G.S., Heusch, G., Downey, J.M., Cohen, M.V., 2000.
Exogenous nitric oxide can trigger a preconditioned state through a free radical mechanism, but endogenous nitric oxide is not a trigger of classical ischemic preconditioning. J. Mol. Cell. Cardiol. 32, 1159 – 1167.
Nonn, L., Williams, R.R., Erickson, R.P., Powis, G., 2003. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 23, 916 – 922.
Ockaili, R., Emani, V.R., Okubo, S., Brown, M., Krottapalli, K., Kukreja, R.C., 1999. Opening of mitochondrial K(ATP) channel induces early and delayed cardioprotective effect: role of nitric oxide. Am. J. Physiol.
277, H2425 – H2434.
Offen, D., Beart, P.M., Cheung, N.S., Pascoe, C.J., Hochman, A., Gorodin, S., Melamed, E., Bernard, R., Bernard, O., 1998. Transgenic mice expressing human Bcl-2 in their neurons are resistant to 6-hydroxydop- amine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity.
Proc. Natl. Acad. Sci. U.S.A. 95, 5789 – 5794.
Pan, S.L., Guh, J.H., Chang, Y.L., Kuo, S.C., Lee, F.Y., Teng, C.M., 2004.
YC-1 prevents sodium nitroprusside-mediated apoptosis in vascular smooth muscle cells. Cardiovasc. Res. 61, 152 – 158.
Powis, G., Mustacich, D., Coon, A., 2000. The role of the redox protein thioredoxin in cell growth and cancer. Free Radical Biol. Med. 29, 312 – 322.
Rauhala, P., Lin, A.M., Chiueh, C.C., 1998. Neuroprotection by S- nitrosoglutathione of brain dopamine neurons from oxidative stress.
FASEB J. 12, 165 – 173.
Ruscher, K., Freyer, D., Karsch, M., Isaev, N., Megow, D., Sawitzki, B., Priller, J., Dirnagl, U., Meisel, A., 2002. Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J. Neurosci. 22, 10291 – 10301.
Saitoh, M., Nishitoh, H., Fujii, M., Takeda, K., Tobiume, K., Sawada, Y., Kawabata, M., Miyazono, K., Ichijo, H., 1998. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1.
EMBO J. 17, 2596 – 2606.
Sebille, S., De Tullio, P., Boverie, S., Antoine, M.H., Lebrun, P., Pirotte, B., 2004. Recent developments in the chemistry of potassium channel activators: the cromakalim analogs. Curr. Med. Chem. 11, 1213 – 1222.
Sun, C.J., Johannessen, J.N., Gessner, W., Namura, I., Singhaniyom, W., Brossi, A., Chiueh, C.C., 1988. Neurotoxic damage to the nigrostriatal system in rats following intranigral administration of MPDP+ and MPP+. J. Neural Transm. 74, 75 – 86.
Suzuki, T., Kumamoto, H., Ooya, K., Motegi, K., 2002. Expression of inducible nitric oxide synthase and heat shock proteins in periapical inflammatory lesions. J. Oral Pathol. Med. 31, 488 – 493.
Suzuki, E., Kageyama, H., Nakaki, T., Kanba, S., Inoue, S., Miyaoka, H., 2003. Nitric oxide induced heat shock protein 70 mRNA in rat hypothalamus during acute restraint stress under sucrose diet. Cell.
Mol. Neurobiol. 23, 907 – 915.
Sziraki, I., Mohanakumar, K.P., Rauhala, P., Kim, H.G., Yeh, K.J., Chiueh, C.C., 1998. Manganese: a transition metal protects nigrostriatal neurons from oxidative stress in the iron-induced animal model of parkinsonism.
Neuroscience 85, 1101 – 1111.
Tanaka, T., Hosoi, F., Yamaguchi-Iwai, Y., Nakamura, H., Masutani, H., Ueda, S., Nishiyama, A., Takeda, S., Wada, H., Spyrou, G., Yodoi, J., 2002. Thioredoxin-2 (TRX-2) is an essential gene regulating mito- chondria-dependent apoptosis. EMBO J. 21, 1695 – 1703.
Teng, C.M., Wu, C.C., Ko, F.N., Lee, F.Y., Kuo, S.C., 1997. YC-1, a nitric oxide-independent activator of soluble guanylate cyclase, inhibits platelet-rich thrombosis in mice. Eur. J. Pharmacol. 320, 161 – 166.
Ueda, S., Masutani, H., Nakamura, H., Tanaka, T., Ueno, M., Yodoi, J., 2002. Redox control of cell death. Antioxid. Redox Signal. 4, 405 – 414.
Xi, L., Tekin, D., Gursoy, E., Salloum, F., Levasseur, J.E., Kukreja, R.C., 2002. Evidence that NOS2 acts as a trigger and mediator of late preconditioning induced by acute systemic hypoxia. Am. J. Physiol.:
Heart Circ. Physiol. 283, H5 – H12.
Yamamoto, M., Yang, G., Hong, C., Liu, J., Holle, E., Yu, X., Wagner, T., Vatner, S.F., Sadoshima, J., 2003. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J. Clin.
Invest. 112, 1395 – 1406.
Zaugg, M., Lucchinetti, E., Spahn, D.R., Pasch, T., Schaub, M.C., 2002.
Volatile anesthetics mimic cardiac preconditioning by priming the
activation of mitochondrial K(ATP) channels via multiple signaling pathways. Anesthesiology 97, 4 – 14.
Zhang, R., Al-Lamki, R., Bai, L., Streb, J.W., Miano, J.M., Bradley, J., Min, W., 2004. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ. Res. 94, 1483 – 1491.
Zhao, R., Holmgren, A., 2004. Ebselen is a dehydroascorbate reductase
mimic, facilitating the recycling of ascorbate via mammalian thiore- doxin systems. Antioxid. Redox Signal. 6, 99 – 104.
Zimmermann, C., Ginis, I., Furuya, K., Klimanis, D., Ruetzler, C., Spatz, M., Hallenbeck, J.M., 2001. Lipopolysaccharide-induced ischemic tolerance is associated with increased levels of ceramide in brain and in plasma. Brain Res. 895, 59 – 65.