The low-lying states of the scandium dimera)
Cristopher Camacho, Henryk A. Witek, and Renzo Cimiraglia
Citation: The Journal of Chemical Physics 132, 244306 (2010); doi: 10.1063/1.3442374
View online: http://dx.doi.org/10.1063/1.3442374
View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/132/24?ver=pdfcov Published by the AIP Publishing
Articles you may be interested in
Critical appraisal of excited state nonadiabatic dynamics simulations of 9H-adeninea) J. Chem. Phys. 137, 22A503 (2012); 10.1063/1.4731649
Sensitivity of transport and stability to the current profile in steady-state scenario plasmas in DIII-Da) Phys. Plasmas 19, 122506 (2012); 10.1063/1.4772765
Relation of astrophysical turbulence and magnetic reconnectiona) Phys. Plasmas 19, 012105 (2012); 10.1063/1.3672516
Intermolecular potential energy surface and second virial coefficients for the water–CO2 dimera) J. Chem. Phys. 134, 134309 (2011); 10.1063/1.3574345
( H Cl ) 2 and ( H F ) 2 in small helium clusters: Quantum solvation of hydrogen-bonded dimersa) J. Chem. Phys. 123, 224313 (2005); 10.1063/1.2136358
The low-lying states of the scandium dimer
a… Cristopher Camacho,1Henryk A. Witek,1,b兲and Renzo Cimiraglia2 1Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung University, 1001 Ta-Hsueh Road, Hsinchu 30010, Taiwan
2
Dipartimento di Chimica, Università di Ferrara, Via Borsari 46, Ferrara I-44100, Italy 共Received 30 March 2010; accepted 11 May 2010; published online 25 June 2010兲
A systematic investigation of low-lying states of Sc2 using multireference perturbation theory 共NEVPT2 and NEVPT3兲 indicates that the ground state of this system is 5⌺
u
− with r
e= 2.611 Å,
e= 241.8 cm−1, and De= 1.78 eV. This state is closely followed by other low-lying states of Sc2: 3⌺
u
−, 5⌬
u, 3⌸g, 1⌸g, and 1⌺u−. Our energy ordering of the 5⌺u− and3⌺u− states confirms the recent MRCI results of Kalemos et al.关J. Chem. Phys. 132, 024309 共2010兲兴 and is at variance with the earlier diffusion Monte Carlo predictions of Matxain et al.关J. Chem. Phys. 128, 194315 共2008兲兴. An excellent agreement between the second- and third-order NEVPT results and between the computed and experimental values ofe共241.8 versus 238.9 cm−1兲 for the 5⌺u−state suggests high accuracy of our predictions. © 2010 American Institute of Physics.关doi:10.1063/1.3442374兴
I. INTRODUCTION
The electronic structure of bulk metals is quite well understood,1,2 but an accurate description of small metal clusters is still far from satisfactory and constitutes a field of vigorous scientific investigations.3–22 An example of a sys-tem that received considerable attention—both in theory and in experiment—is the scandium dimer. Even though the Sc2
molecule is seemingly among the simplest transition metal dimers, the description of its potential energy curve 共PEC兲 along the whole range of internuclear distances still raises a challenge to nowadays’ most refined quantum chemical methods. Whereas at large internuclear distances the ground state of Sc2is described by the two atoms in their2Dgground state, at short distances, due to the relatively low energy required to promote an atom from 2Dg 共4s23d1兲 to 4Fg 共4s13d2兲, the nature of the bond in the ground state is
domi-nated by a 4s24s1interaction. For a long time, there seemed to be a consensus with respect to the nature of the ground state of Sc2. However, a recent quantum diffusion Monte
Carlo 共DMC兲 study of Matxain et al.8 suggested that the ground state should be classified as 3⌺u
−
rather than 5⌺u
−
, as was previously widely accepted. We feel that an extensive study of the scandium dimer aiming at the definite character-ization of the ground and low-lying excited states and their properties is timely, also in regard of the recent success of encapsulating the Sc2molecules inside fullerene cages.23The
metal-fullerene interaction was found to significantly stabi-lize the nonisolated-pentagon fullerene isomers of C64, C66,
C72, C74, and C82.24–30 It appears obvious that in-depth
un-derstanding of the stabilization mechanism must be preceded by a thorough elucidation of the electronic structure of the isolated dimer.
An excellent complete review of experimental and
theo-retical results on Sc2 has been given recently by Kalemos et al.31 Here, we give only an overview of the most impor-tant results that are relevant in the context of the present work. The equilibrium distance in Sc2 is not well known;
crude estimations32based on empirical rules locate it around 2.20–2.48 Å. In contrast, the vibrational constants of the ground state 共e= 238.91 cm−1 and exe= 0.93 cm−1兲 are determined33very accurately from resonance Raman experi-ments in an argon matrix. The corresponding gas-phase har-monic frequency is expected34to differ only slightly共within a few wave numbers兲 from its matrix value. The experimen-tal value of the dissociation energy of the scandium dimer is a subject of controversy. It was estimated in 1964 as
1.12⫾0.22 eV by mass-spectroscopic measurements
fol-lowed by the third-law thermodynamic method calculations
by Drowart and co-workers.35 In a review published two
years later, Drowart36 already quoted a different value of
1.65⫾0.22 eV, which apparently was obtained from the
same data. Further details concerning the corrected value have been given recently by Kalemos et al.31A detailed mul-tireference theoretical study performed by Åkeby and co-workers37,38suggested that the presence of previously un-known low-lying electronic states of Sc2contributing to the
molecular partition function should lower De to
1.05⫾0.2 eV. In the context of this discussion, it is appro-priate to mention here another attempt39 to estimate De of Sc2 using the LeRoy–Bernstein analysis40,41 of the
vibra-tional energy levels of the scandium dimer, which yielded a lower limit of the dissociation energy of 0.79 eV. It is impor-tant to stress that this value should just be viewed as a lower limit for the dissociation energy since the only five observed transitions in the resonance Raman progression33are not suf-ficient to show an evidence of theeyeanharmonicity in the Dunham fit, which is required for the LeRoy–Bernstein analysis to yield reasonable results.39
The analysis42 of the hyperfine splitting of two ESR bands of Sc2 trapped in solid neon matrix showed that the
ground state is a quintet共S=2兲. It was further demonstrated
a兲
This paper is dedicated to the memory of Professor Björn O. Roos 共1937–2010兲.
b兲Electronic mail: [email protected].
0021-9606/2010/132共24兲/244306/9/$30.00 132, 244306-1 © 2010 American Institute of Physics
that the observed pattern is consistent with a simulated elec-tron spin resonance 共ESR兲 spectrum of a5⌺ state. It is im-portant to stress here that the positions of the two observed perpendicular fine-structure lines in the ESR spectrum could not be reproduced for S = 1 and S = 3, which effectively ex-cludes the possibility that the ground state of Sc2 is either a
triplet or a septet. An UV-vis spectrum of scandium atoms trapped in an argon matrix shows evidence of three absorp-tions at 15 100, 21 050, and 29 850 cm−1, which were
as-signed to three low-lying excited states of Sc2.43
Early semiempirical and HF calculations designated the
1⌺
g
+ and 5⌬
g electronic states as candidates for the ground state.43–46 The first multireference 共MCSCF/MRCI兲 study47 of Sc2 showed that the 5⌺u
−
state with a minimum at 2.6 Å lies considerably lower than the previously investigated states. The first global PEC for the5⌺u
−
state was constructed
by Tatewaki and co-workers48 using an 共22o, 6e兲 active
space built from the 3d, 4s, and 4d atomic orbitals; state averaging was needed to assure the connected and smooth character of the curve. The electronic nature of the5⌺u−state was investigated by a number of authors.8,31,49–51As already pointed out, this state dissociates formally to an excited manifold of separated atoms. The electronic states arising from the lowest2Dg+2Dgdissociation channel were found to be very closely spaced and only weakly bound.31,52,53 Their PECs have been constructed recently by Kalemos et al.31An extensive study of low-lying states of Sc2 was given by
Åkeby and co-workers.38 A number of DFT studies on the
low-lying states of Sc2 appeared,51,54–60 which were mainly
focused on the accurate determination of the ground state using various exchange-correlation functionals and a com-parison between the lowest state in each of the spin mani-folds. A compilation of spectroscopic parameters obtained with various correlated methods for covalently bound low-lying states of Sc2is given in TableI.
This well-established theoretical picture has been re-cently upset by the quantum DMC study of Sc2performed by
Matxain et al.8This study reports single point DMC energies computed at the DFT/B3LYP equilibrium geometries for ten low-lying states of Sc2. The symmetry of the ground state is found to be3⌺u
−
instead of the previously believed5⌺u
−
.共Note that no details on the +/− symmetry of the ⌺ states are provided in Ref.8.兲 Both states are found to be separated by
only 0.17 eV 共DMC兲. Their spectroscopic parameters are
also similar. The ground character of3⌺u−is further corrobo-rated by MRMP calculations,61–63 which give almost
identi-cal energy spacing 共0.16 eV兲 as DMC. It is important to
stress here that these MRMP results are affected by con-spicuous procedural faults, as have been shown in a recently published erratum.64 It was demonstrated recently65 by the present authors that MRMP and CASPT2 calculations using
the chemically correct 共12o, 6e兲 complete active space
共CAS兲 are plagued by numerous intruder states. Intruder state removal techniques 共i.e., shift techniques兲 produce smooth and continuous curves but simultaneously introduce unpleasant artifacts in the energy ordering of the3⌺u−and5⌺u− states. The energy ordering depends on the magnitude of the shift parameter; 3⌺u− is the ground state for small values of the shift, and5⌺u
−
for large values. This unexpected behavior
casts many a doubt on the effectiveness of MRMP and CASPT2 as an appropriate tool for the reliable investigation of the low-lying states of Sc2. Note that the3⌺u−state was not studied using multireference methods prior to the calcula-tions of Matxain et al.; previous DFT calculacalcula-tions8,51,56 pre-dicted it higher than the5⌺u−state by 0.07–0.22 eV. Also the recent MRCI calculations of Kalemos et al.31 predict it higher by 0.04 eV.
The present study has two goals. First of all, we want to reaffirm that the ground state of Sc2is5⌺u
−
and that3⌺u
−
state is one of a few low-lying excited states of this molecule. The
present results are obtained using NEVPT266,67 and
NEVPT3,10,68 recently developed variants of multireference perturbation theory. The particular features of NEVPT, viz., the absence of intruder states and the property of size con-sistence, are particularly useful in the study of a molecule such as Sc2, where both statical and dynamical correlation
energies need to be adequately accounted for. An excellent agreement between the results from the second- and third-order multireference perturbation theory combined with a large atomic basis set and including the relativistic and semi-core correlation effects suggests high accuracy of our predic-tions. The correctness of this assignment is further corrobo-rated by an excellent agreement between the computed and experimental values ofefor the5⌺u−state. The second pur-pose of the present study is to give a systematic account of the low-lying states of Sc2 in order to characterize all the
potential candidates for the ground state. To this end, the PEC for the ground state in each irreducible representation of the D⬁hpoint group is computed for the singlet, triplet,
quin-tet, and septet spin manifolds 共S=0,1,2,3兲 using CASSCF
with the 共18o, 6e兲 full valence active space followed by NEVPT2 corrections for the low-lying curves. We believe that the presented results are a significant contribution to the quest for finding the ground state of Sc2.
II. COMPUTATIONAL DETAILS
The all-electron relativistic 共21s15p10d6f4g2h兲/
关10s9p8d5f4g2h兴 atomic natural orbital 共ANO兲 type basis set of Roos et al.69has been adopted for this work. The PECs of Sc2have been computed with the n-electron valence state perturbation theory共NEVPT兲 method.66,68 Scalar relativistic effects have been accounted for by using the second-order Douglas–Kroll–Hess Hamiltonian.70Spin-orbit共SO兲 calcula-tions have been performed using the full Breit–Pauli SO op-erator. The diagonal elements of the SO matrix have been substituted with the state-specific 共18o, 6e兲 NEVPT2 ener-gies; the off-diagonal elements have been calculated using the state-averaged CASSCF orbitals averaged over the six lowest-lying states共5⌺u−,3⌺u−,3⌬u,3⌸g,1⌸g, and1⌺u−兲 of Sc2.
We used the full valence active space 共18o, 6e兲 com-posed of the 4s, 3d, and 4p atomic orbitals 共AOs兲 for this work. Two other active spaces have been tested in our cal-culations, the reduced valence space共12o, 6e兲 composed of the 4s and 3d AOs of each scandium atom and the aug-mented reduced valence space共14o, 6e兲 composed of the 4s, 3d, and 4pzAOs. Dynamical correlation has been taken into account by means of the second- and third-order10 NEVPT
244306-2 Camacho, Witek, and Cimiraglia J. Chem. Phys. 132, 244306共2010兲
method. The 3s and 3p electrons have been correlated at the perturbation level in all calculations. The second-order cal-culations have been performed using the partially contracted formalism共PC-NEVPT2兲, while the third-order calculations employed both the strongly共SC-NEVPT3兲 and partially
共PC-NEVPT3兲 contracted approach. It is noteworthy that this is the first time that 18 active orbitals have been used in junction with the NEVPT method and that the partially con-tracted variant of NEVPT3 has been used in quantum chemi-cal chemi-calculations. All the chemi-calculations have been carried out
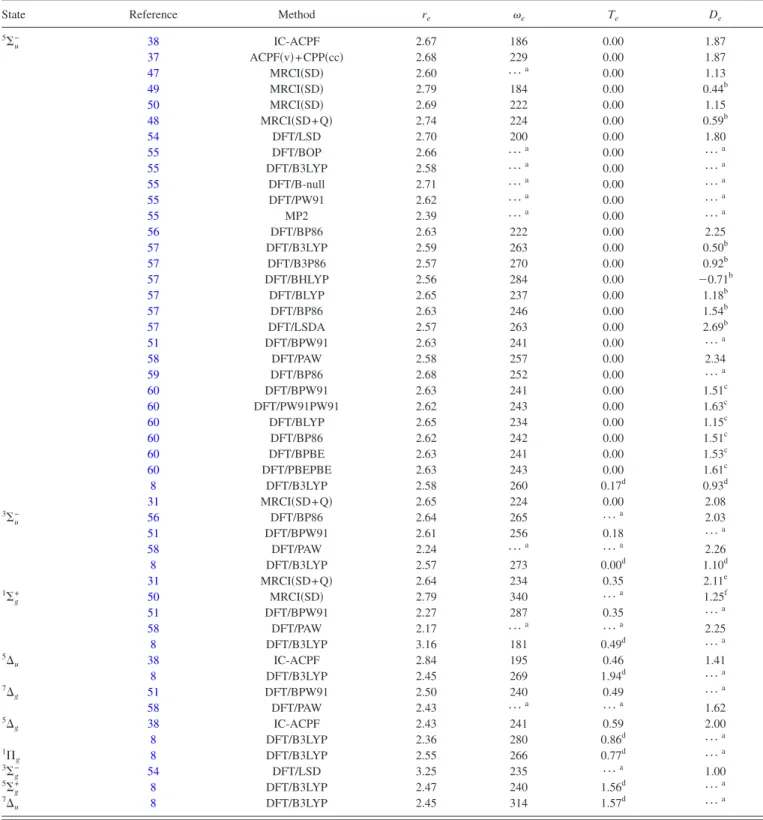
TABLE I. Compilation of spectroscopic parameters computed with a variety of quantum mechanical methods for covalently bonded low-lying states of Sc2.
Bond lengths re are given in angstroms, harmonic frequencies ein cm−1, adiabatic excitation energies Tein eV, and dissociation energies Dein eV.
Dissociation energies are determined with respect to the2D
g+4Fg共if otherwise, see footnote兲.
State Reference Method re e Te De
5⌺ u − 38 IC-ACPF 2.67 186 0.00 1.87 37 ACPF共v兲+CPP共cc兲 2.68 229 0.00 1.87 47 MRCI共SD兲 2.60 ¯a 0.00 1.13 49 MRCI共SD兲 2.79 184 0.00 0.44b 50 MRCI共SD兲 2.69 222 0.00 1.15 48 MRCI共SD+Q兲 2.74 224 0.00 0.59b 54 DFT/LSD 2.70 200 0.00 1.80 55 DFT/BOP 2.66 ¯a 0.00 ¯a 55 DFT/B3LYP 2.58 ¯a 0.00 ¯a 55 DFT/B-null 2.71 ¯a 0.00 ¯a 55 DFT/PW91 2.62 ¯a 0.00 ¯a 55 MP2 2.39 ¯a 0.00 ¯a 56 DFT/BP86 2.63 222 0.00 2.25 57 DFT/B3LYP 2.59 263 0.00 0.50b 57 DFT/B3P86 2.57 270 0.00 0.92b 57 DFT/BHLYP 2.56 284 0.00 ⫺0.71b 57 DFT/BLYP 2.65 237 0.00 1.18b 57 DFT/BP86 2.63 246 0.00 1.54b 57 DFT/LSDA 2.57 263 0.00 2.69b 51 DFT/BPW91 2.63 241 0.00 ¯a 58 DFT/PAW 2.58 257 0.00 2.34 59 DFT/BP86 2.68 252 0.00 ¯a 60 DFT/BPW91 2.63 241 0.00 1.51c 60 DFT/PW91PW91 2.62 243 0.00 1.63c 60 DFT/BLYP 2.65 234 0.00 1.15c 60 DFT/BP86 2.62 242 0.00 1.51c 60 DFT/BPBE 2.63 241 0.00 1.53c 60 DFT/PBEPBE 2.63 243 0.00 1.61c 8 DFT/B3LYP 2.58 260 0.17d 0.93d 31 MRCI共SD+Q兲 2.65 224 0.00 2.08 3⌺ u − 56 DFT/BP86 2.64 265 ¯a 2.03 51 DFT/BPW91 2.61 256 0.18 ¯a 58 DFT/PAW 2.24 ¯a ¯a 2.26 8 DFT/B3LYP 2.57 273 0.00d 1.10d 31 MRCI共SD+Q兲 2.64 234 0.35 2.11e 1⌺ g + 50 MRCI共SD兲 2.79 340 ¯a 1.25f 51 DFT/BPW91 2.27 287 0.35 ¯a 58 DFT/PAW 2.17 ¯a ¯a 2.25 8 DFT/B3LYP 3.16 181 0.49d ¯a 5⌬ u 38 IC-ACPF 2.84 195 0.46 1.41 8 DFT/B3LYP 2.45 269 1.94d ¯a 7⌬ g 51 DFT/BPW91 2.50 240 0.49 ¯a 58 DFT/PAW 2.43 ¯a ¯a 1.62 5⌬ g 38 IC-ACPF 2.43 241 0.59 2.00 8 DFT/B3LYP 2.36 280 0.86d ¯a 1⌸ g 8 DFT/B3LYP 2.55 266 0.77 d ¯a 3⌺ g − 54 DFT/LSD 3.25 235 ¯a 1.00 5⌺ g + 8 DFT/B3LYP 2.47 240 1.56d ¯a 7⌬ u 8 DFT/B3LYP 2.45 314 1.57d ¯a
aData not available.
bDetermined with respect to the2D
g+2Dgasymptote. cThe reported value is D
0.
dThe reported value is given at the DMC//DFT/B3LYP level of theory. eUncorrected value共see Ref.31兲.
fDetermined with respect to the4F
g+4Fgasymptote.
with theMOLPRO共Ref.71兲 computational package in the D2h
point group symmetry. The proper⌳ quantum number for all the studied states has been ensured by means of theLQUANT
option implemented inMOLPRO.
III. RESULTS AND DISCUSSION A. Atomic calculations
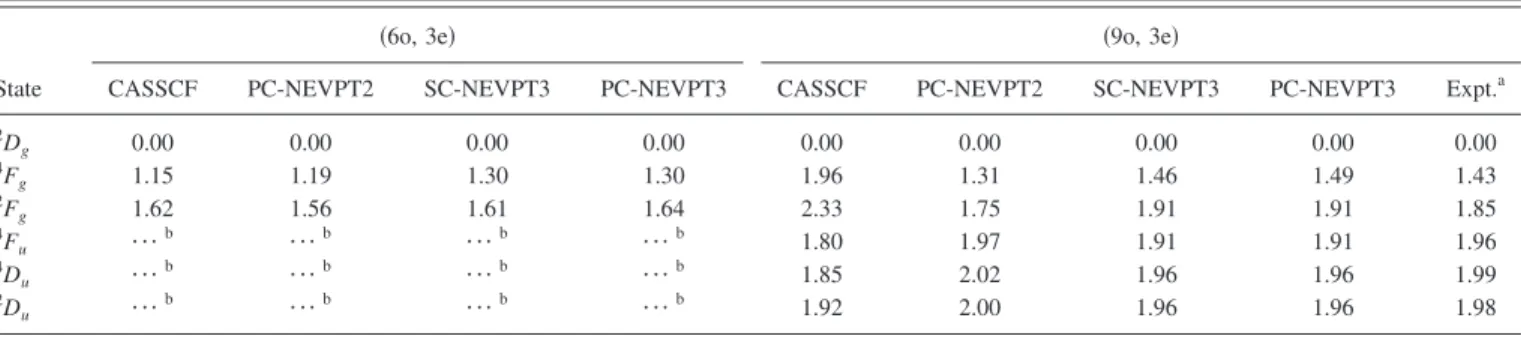
Before proceeding to the molecular calculations, we briefly inspect here the low-lying atomic terms of the scan-dium atom. These results help us elucidate the energy order-ing of various dissociation asymptotes formally available for the molecular states. It should be clear that such information is quite crucial in obtaining accurate dissociation energies of the molecular states. Atomic calculations for the low-lying states of scandium 共2Dg, 2Du,2Fg, 4Du, 4Fg, and 4Fu兲 have been performed using the full and reduced valence active spaces 共9o, 3e兲 and 共6o, 3e兲. Note that the reduced valence CAS is capable of describing only the atomic terms of even parity:2Dg,4Fg, and2Fg. The calculations have been carried out in D2hpoint group symmetry including all the
appropri-ate components共five and seven, respectively兲 of the D and F terms. State-specific full-valence CASSCF calculations for the six lowest atomic terms of Sc reveal serious discrepan-cies with the experimental energy ordering72共for details, see Table II兲. Accounting for dynamical correlation yields the
correct ordering of the states and considerably improves the energetics. The results from NEVPT3 are slightly more ac-curate than those from NEVPT2, especially for the terms corresponding to the 4s23d and 4s3d2 configurations. There
is only a very small deviation between the strongly and par-tially contracted versions of NEVPT3. Employing the full valence active space is quite crucial for obtaining accurate energetics. The mean absolute deviation for the NEVPT2 results obtained with the共9o, 3e兲 CAS is only 0.06 eV, while for the共6o, 3e兲 it is approximately five times as large 共0.26 eV兲. A similar trend is also observed for the PC-NEVPT3 共0.04 versus 0.17 eV兲 and SC-NEVPT3 共0.04 versus 0.18 eV兲 approaches.
Special attention is given here to the2Dg,4Du,4Fu, and
4F
g atomic terms, since the ground state 共5⌺u
−兲 of Sc 2 may
formally originate from the 2Dg+4Fg, 2Dg+4Du, or 2Dg +4Fu dissociation channels. An inspection of the 5⌺u− wave function around its minimum shows almost negligible con-tributions from the atomic 4p orbitals, suggesting that the
correct dissociation asymptote for 5⌺u−is2Dg+4Fg.
Unfortu-nately, the CASSCF method erroneously places the 2Dg
+4Fu and 2Dg+4Du lower than 2Dg+4Fg by 0.16 and 0.12 eV, respectively. This fact has serious consequences in the estimation of the dissociation energy. If the dissociation en-ergy for the 5⌺u−ground state is computed directly from the PEC, rather than from the separated atoms limit, a correction equivalent to the 4Fu−4Fg energy spacing must be intro-duced in the final result to compensate for the automatic choice of the wrong asymptote.
B. Low-lying states of Sc2
The lowest PECs of Sc2 for each of the
1,3,5,7关⌺+,⌺−,⌸,⌬,⌽,⌫兴
g,u manifolds have been computed
using the state-specific CASSCF method with the 共18o, 6e兲 full valence active space. The curves, shown in Fig.1, have been computed between 1.95 and 3.55 Å in order to desig-nate viable candidates for the low-lying covalently bound states of the scandium dimer. As earlier anticipated,31 the resulting curves are quite closely spaced. The dynamical cor-relation contribution has been estimated for each of the curve around its minimum using the NEVPT2 method. The singlet and triplet states of purely van der Waals character that were studied previously by Kalemos et al.31 were omitted in this procedure. Out of the 48 studied states, the lowest six共5⌺u−,
3⌺
u
−,5⌬
u,3⌸g,1⌸g, and1⌺u−兲 have been further considered as possible candidates for the ground state of Sc2.
Figure2 shows the NEVPT2 curves for the six lowest-lying states of Sc2. It is found that the ground state is 5⌺u−. This state is closely followed by 3⌺u− 共+0.10 eV兲, 5⌬u 共+0.25 eV兲, the two almost degenerate 3⌸
g and1⌸g states
共+0.26 and +0.27 eV, respectively兲, and the 1⌺
u
− state
共+0.41 eV兲. The next low-lying states of Sc2 are 1⌺g+, 3⌸u,
5⌬
g,3⌬u, and 1⌬u, all with minima between 0.4 and 0.5 eV higher than that of5⌺u−. Our energy ordering of the5⌺u−and
3⌺
u
− states confirms the recent MRCI results of Kalemos et al.31and contradicts the earlier DMC predictions of Matxain
et al.8
Spectroscopic parameters computed for the six NEVPT2 curves presented in Fig. 2are shown in Table III. Note that the spectroscopic parameters for all the states 共except 5⌬u兲 are quite similar. It is important to emphasize here the excel-lent agreement between the computed and experimental har-monic vibrational frequency of 5⌺u
−
, which is the only
spec-TABLE II. Excitation energies for the low-lying atomic terms of scandium in eV calculated at the CASSCF, NEVPT2, and NEVPT3 level using the共9o, 3e兲 full valence and共6o, 3e兲 reduce valence active space.
State
共6o, 3e兲 共9o, 3e兲
CASSCF PC-NEVPT2 SC-NEVPT3 PC-NEVPT3 CASSCF PC-NEVPT2 SC-NEVPT3 PC-NEVPT3 Expt.a
2D g 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 4F g 1.15 1.19 1.30 1.30 1.96 1.31 1.46 1.49 1.43 2F g 1.62 1.56 1.61 1.64 2.33 1.75 1.91 1.91 1.85 4F u ¯b ¯b ¯b ¯b 1.80 1.97 1.91 1.91 1.96 4D u ¯b ¯b ¯b ¯b 1.85 2.02 1.96 1.96 1.99 2D u ¯b ¯b ¯b ¯b 1.92 2.00 1.96 1.96 1.98 aReference73.
bThe共6o, 3e兲 CAS does not permit the description of the given atomic term.
244306-4 Camacho, Witek, and Cimiraglia J. Chem. Phys. 132, 244306共2010兲
troscopic parameter known with certainty from experiment. Such a good concordance suggests that also the other spec-troscopic parameters may be in good agreement with the corresponding共as yet unknown兲 experimental values. One of the most serious controversies concerns the dissociation
en-ergy of Sc2. The dissociation energy computed with
NEVPT2 is 1.78 eV with respect to the separated atoms 共2D
g+4Fg兲 and 0.51 eV with respect to the ground state of the atoms共2Dg+2Dg兲. The analogous MRCI results recently given by Kalemos et al.31are 2.08 and 0.35 eV, respectively.
We recall that NEVPT is a size consistent theory and the quoted results are not affected by misbehaviors at large in-ternuclear distances 共as for example in MRCI兲. It is impor-tant to stress that the size consistency of NEVPT follows only if the underlying CASSCF calculations are size consis-tent. In the case of a CASSCF wave function not providing a well balanced mixture of all the components corresponding to the atomic states at the dissociation limit, the lack of size consistency of the zeroth-order wave function breaks the size -1526.60 -1526.50
Energy
[h
artree]
Singlets Triplets 2.1 2.4 2.7 3.0 3.3 -1526.60 -1526.50 Quintets 5∆ u 2.1 2.4 2.7 3.0 3.3r
Sc-Sc[Å]
Septets 5Σ -u 3Σ -u 3Π g 1Π g 1Σ-u FIG. 1. The low-lying states of Sc2determined by the
共18o, 6e兲 CASSCF calculations. The lowest energy state in each irreducible representation共⌺, ⌸, ⌬, ⌽, and ⌫兲 of the D⬁hpoint group with S = 0 , 1 , 2 , 3 is presented.
The viable candidates for the ground state are depicted in color. -1526.60 -1526.55
Energy
[hartree]
CASSCF (18o,6e) 1Π
g 1Σ
-u 3Π
g 3Σ
-u 5Σ
-u 5∆
u 2.1 2.4 2.7 3.0 3.3r
Sc-Sc[Å]
-1527.30 -1527.25 NEVPT2 (18o,6e)FIG. 2. A comparison of the共18o, 6e兲 CASSCF 共upper panel兲 and NEVPT2 共lower panel兲 PECs for the six low-lying states of Sc2.
TABLE III. Spectroscopic parameters for the five low-lying states of Sc2
calculated at the NEVPT2 level using the共18o, 6e兲, 共14o, 6e兲, and 共12o, 6e兲 CAS. Bond lengths reare given in angstrom, harmonic frequencieseand
first anharmonicitiesexein cm−1, and dissociation energies Dein eV.
Dis-sociation energies are determined with respect to the2D
g+4Fgasymptote.
State re e exe De
NEVPT共18o, 6e兲
5⌺ u − 2.611 241.8 0.70 1.78 238.9a 0.93a 3⌺ u − 2.620 254.0 0.71 1.69 5⌬ u 2.792 212.0 0.53 1.53 3⌸ g 2.644 221.3 0.74 1.52 1⌸ g 2.645 226.9 0.71 1.52 1⌺ u − 2.673 235.7 0.76 1.37
NEVPT共14o, 6e兲
5⌺ u − 2.588 253.9 0.83 1.72 238.9a 0.93a 3⌺ u − 2.595 265.6 0.78 1.60 5⌬ u 2.775 218.0 0.76 1.42 3⌸ g 2.592 234.7 1.26 1.56 1⌸ g 2.584 238.1 1.03 1.53 1⌺ u − 2.644 244.7 0.72 1.28
NEVPT共12o, 6e兲
5⌺ u − 2.580 257.5 0.87 1.74 238.9a 0.93a 3⌺ u − 2.595 260.1 0.76 1.65 5⌬ g ¯b ¯b ¯b ¯b 3⌸ g ¯b ¯b ¯b ¯b 1⌸ g ¯ b ¯b ¯b ¯b 1⌺ u − 2.637 247.0 0.78 1.34
aExperimental value, Ref.33.
bThe共12o, 6e兲 CAS does not permit a proper description of the electronic
structure of Sc2for the given state.
consistency property of NEVPT. We found that the size con-sistency error estimated with respect to the 2Dg+4Fg disso-ciation asymptote at 20.00 a.u. for the 5⌺u− in the reduced valence space is as small as 0.028 eV.
Among the six lowest-lying states of Sc2, only1⌸g,3⌸g, and 5⌬u interact via the SO Hamiltonian. The energy sepa-ration between these three states is very small and therefore it is important to examine the SO splitting effects for them. Estimation of the SO coupling for 3⌸g and1⌸g shows that the interaction is indeed very weak. The computed splittings at 2.59 Å with respect to the zeroth-order spin-free3⌸gstate are −36 cm−1共⍀=0+and 0−兲, −21 cm−1 共⍀=1兲, +64 cm−1
共⍀=1兲, and +36 cm−1共⍀=2兲. For the5⌬
ustate, the interac-tion is noticeably larger and causes a splitting of 206 cm−1
between the outermost ⍀=0 and ⍀=4 SO components of
this state. As expected, none of these splittings modifies se-riously the energy landscape for the low-lying states of the scandium dimer.
For some of the states, namely for those that formally dissociate to the ground state of the separated atoms, the lowest PECs consist of two crossing segments. The long-distance segment corresponds to the interaction of two 4s23d1scandium atoms, while the short-distance segment to the interaction of the 4s23d1 and 4s13d2 scandium atoms. Their analytical structure and consequently also the physical nature are very different. The best evidence of the dissimi-larity follows from the fact that the two CASSCF solutions can be analytically continued also to metastable regions yielding the crossed, two-segment patterns observed in Fig.
1. Clearly, such a situation is not desirable for physical so-lutions, which should vary smoothly while passing from one region to the other. Usually, an appropriately averaged CASSCF procedure can resolve this situation. Unfortunately, it has not proved possible to achieve this result for the Sc2
molecule. The two-segment curves presented in Fig.1can be thus regarded as diabatic PECs that should be coupled via an appropriate adiabatic coupling matrix element. Note that such an interaction is expected to modify the shape of these curves only in the vicinity of the crossing point owing to their very different physical nature. Actually, for our pur-pose, it is more convenient to analyze the diabatic curves,
since the crossing point is usually shifted after accounting for the dynamical correlation. In case of adiabatic states, one would need to employ a multistate perturbation theory for-malism, such as the quasidegenerate NEVPT2,73 to recover the smooth character of PECs at the crossing region. Fortu-nately, for the five states analyzed here, the crossing points occur far away from their minima, thereby not influencing the final conclusions drawn from our work.
C. NEVPT3 results for the 5⌺ u
−and3⌺ u −states
The results presented in Sec. III B have been obtained using the second-order NEVPT method. In this section we extend the correlation treatment to the third order using the 共12o, 6e兲 reduced valence space. In addition to its intrinsic value, the third-order approach is a good test to assess the efficiency and stability of the perturbation series. Actually, a large discord between the second- and third-order results would be a clear indication of the inadequacy of the zero-order wave function. So far, third-zero-order NEVPT in its strongly contracted variant共SC-NEVPT3兲 has been only ap-plied to the study of transition metal dimers for the group VI elements6,10共Cr2, Mo2, W2, and CrMo兲 and for Mn2.11In the
present work, besides the usual strongly contracted variant, we also made use of the partially contracted NEVPT3 ap-proach 共PC-NEVPT3兲. We recall that in the partially con-tracted variant of NEVPT the whole dimensionality of the internally contracted first-order interacting space 共IC-FOIS兲 is taken into account, whereas in the strongly contracted ap-proach only a subspace of IC-FOIS is considered. The PC-NEVPT3 calculations are computationally more demanding than the SC-NEVPT3 ones and thus they have been per-formed only for the 5⌺u−state, with the purpose to assess the magnitude of the differences between the two variants of
NEVPT3. The resulting PECs are reported in Fig. 3 along
with second-order PC-NEVPT2 ones. As can be seen, the difference between SC- and PC-NEVPT3 is very small and there is hardly any advantage in choosing the more laborious partially contracted variant. The third-order PECs for the5⌺u− and3⌺u−states run parallel to the corresponding second-order curves and are lower in energy with respect to the
PC-2.1 2.4 2.7 3.0
r
Sc-Sc[Å]
-1527.30 -1527.25Energy
[hartree]
5Σ -u PC-NEVPT2 3Σ -u PC-NEVPT2 5Σ -u SC-NEVPT3 3Σ -u SC-NEVPT3 5Σ-u PC-NEVPT3 FIG. 3. A comparison of the partially contracted共PC兲
second-order 共in blue兲 and strongly contracted 共SC兲 third-order共in red兲 NEVPT PECs for the two lowest-lying states of the scandium dimer, 5⌺
u − and 3⌺
u −. In
addition, the PC third order共in black兲 is given for the
5⌺ u −state.
244306-6 Camacho, Witek, and Cimiraglia J. Chem. Phys. 132, 244306共2010兲
NEVPT2 PECs, showing that the perturbation series does not suffer from oscillations. The difference in energy between the two states turns out to be slightly increased in the SC-NEVPT3 treatment with respect to PC-NEVPT2; the minimum-to-minimum energy difference changes from
0.12 eV共SC-NEVPT3兲 to 0.08 eV 共PC-NEVPT2兲. These
es-timations using the 共12o, 6e兲 active space embrace the
0.10 eV 共PC-NEVPT2兲 energy spacing obtained with the
共18o, 6e兲 CAS. The corresponding spectroscopic constants are reported in Table IV. A comparison of the 共12o, 6e兲 NEVPT2 and NEVPT3 values shows that the harmonic vi-brational frequencies are almost identical, while one can ob-serve a small elongation in the equilibrium distance of both states, in agreement with the NEVPT2 results obtained with the full valence space共18o, 6e兲 and shown in TableIII.
D. A comment on the applicability of reduced valence active spaces
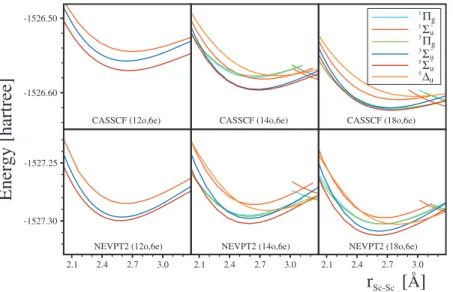
Full-valence active space calculations for systems with a large number of configuration state functions are prohibi-tively expensive. Unfortunately, this is the situation for most transition metal dimers. In this respect Sc2 is an exception; the full-valence active space calculations are not only pos-sible, but quite robust. Therefore, Sc2 constitutes a perfect benchmark system for verifying the applicability of reduced valence spaces in electronic structure calculations. In our
previous studies12 of Mn2 we investigated the use of the
so-called nonclosed active spaces. As stressed by Buch-achenko et al.,74the nonclosed active spaces do not possess a complete set of AOs representing a particular atomic shell, and might not be appropriate for this type of calculations. For Mn2, where the bond is of van der Waals character, a
large variance in the spectroscopic parameters of the ground state was found.12,74 It would be very interesting to investi-gate the applicability of reduced valence active spaces for covalently bonded transition metal dimers.
In order to evaluate the appropriateness of this particular approach, the PECs of Sc2 shown in Fig.4 have been
com-puted with the traditional choice of reduced 共12o, 6e兲 and full共18o, 6e兲 valence spaces along with the nonclosed 共14o, 6e兲 active space. Spectroscopic parameters computed for the NEVPT2 PECs shown in TableIIIdisplay rather small varia-tions with the change in the active space. For instance, the largest difference ine—between the共18o, 6e兲 and 共12o, 6e兲
CAS for the 5⌺u− state—is only 16 cm−1 共6.5%兲. Bond
lengths present even smaller fluctuations. The largest differ-ence is 0.06 Å共2.3%兲 and corresponds to the deviation be-tween the共18o, 6e兲 and 共14o, 6e兲 CAS for the 1⌸gstate.
The differences observed foreand reof Sc2are rather
small, but the most serious criticism against the nonclosed active space approach concerned the computation of the dis-sociation energy De. Since the use of nonclosed active spaces leads to an artificial symmetry breaking at the atomic level, it is expected that large discrepancies between the nonclosed and traditional active spaces can be observed. This intuitive expectation is not fully confirmed for Sc2. It is true that the mean absolute deviation between the closed active spaces 共12 orbital CAS versus 18 orbital CAS兲 is twice as small as the nonclosed case共14 orbital CAS versus 18 orbital CAS兲, but the absolute magnitude of the error is rather small in both cases: 0.04 and 0.07 eV, respectively. The largest difference detected for Deis of the order of 0.11 eV共7.2%兲; it is found between the共18o, 6e兲 and 共14o, 6e兲 active spaces for the5⌬u state. It has to be noted here that the traditional choice of active space for transition metal dimers 共12o, 6e兲 causes much more serious problems since it does not permit a proper description of the3⌸g,1⌸g, and5⌬ustates due to the
TABLE IV. Spectroscopic parameters for the two low-lying states of Sc2
calculated at the NEVPT2 and NEVPT3 levels using the reduced valence 共12o, 6e兲 active space. Bond lengths reare given in angstrom, harmonic
frequencieseand first anharmonicitiesexein cm−1, and dissociation
en-ergies Dein eV. Dissociation energies are determined with respect to the 2D g+4Fgasymptote. State Method re e exe De 5⌺ u − PC-NEVPT2 2.580 257.5 0.87 1.74 SC-NEVPT3 2.607 257.4 0.87 1.56 PC-NEVPT3 2.605 260.3 0.87 1.61 238.9a 0.93a 3⌺ u − PC-NEVPT2 2.595 260.1 0.76 1.65 SC-NEVPT3 2.615 261.5 0.75 1.44
aExperimental value, Ref.33.
-1526.60 -1526.50
Energy
[h
artree]
CASSCF (12o,6e) CASSCF (14o,6e) CASSCF (18o,6e)1Π g 1Σ -u 3Π g 3Σ -u 5Σ -u 5∆ u 2.1 2.4 2.7 3.0 -1527.30 -1527.25 NEVPT2 (12o,6e) 2.1 2.4 2.7 3.0 NEVPT2 (14o,6e) 2.1 2.4 2.7 3.0
r
Sc-Sc[Å]
NEVPT2 (18o,6e)FIG. 4. PECs of the six low-lying states of the scan-dium dimer computed using CASSCF and NEVPT2 with the full valence共18o, 6e兲 CAS 共right panel兲, aug-mented reduced valence共14o, 6e兲 CAS 共middle panel兲, and the reduced valence 共12o, 6e兲 CAS 共left panel兲. Note that three states—3⌸
g,1⌸g, and5⌬u—cannot be
described in the reduced valence space.
lack of the indispensable 4p AOs in the active space. Note also that in the case of other spectroscopic parameters, reand
e, the deviation between the共12o, 6e兲 and 共18o, 6e兲 is ac-tually sizably larger than between共14o, 6e兲 and 共18o, 6e兲.
IV. CONCLUSION
The PECs of low-lying states of the scandium dimer have been studied with multireference perturbation theory 共NEVPT2 and NEVPT3兲 using the full valence active space 共18o, 6e兲 and a large 共21s15p10d6f4g2h兲/关10s9p8d5f4g2h兴 ANO-type basis set. We found that the ground state of Sc2is
5⌺
u
−. This state is closely followed by other low-lying states: 3⌺
u
−, 5⌬
u, 3⌸g, 1⌸g, and 1⌺u
− with energy separations of
+0.10, +0.25, +0.26, +0.27, and +0.41 eV, respectively. A systematic examination of the lowest energy state in each of the 1,3,5,7关⌺+,⌺−,⌸,⌬,⌽,⌫兴
g,u irreducible representation of the D⬁hpoint group suggests that no other state of Sc2can be regarded as a viable candidate for the ground state of this system. Our energy ordering of the5⌺u−and3⌺u−states bears out the recent MRCI results of Kalemos et al.31 and is in contrast with the earlier DMC predictions of Matxain et al.8 An excellent agreement between the second- and third-order NEVPT results and between the computed and experimental values ofefor the 5⌺u−state indicates high accuracy of our predictions.
Another significant aspect of our work concerns the in-vestigation of the possible reduction of the size of the active space. We found that for the low-lying states of Sc2the
spec-troscopic parameters depend only weakly on the choice of the active space with deviations of a few percent. As ex-pected, the most accurate results are obtained with the full valence active space共18o, 6e兲. The traditional choice of the 共12o, 6e兲 CAS does not permit a proper description of the electronic structure of Sc2in the3⌸g,1⌸g, and5⌬ustates. No particular problems are detected for the nonclosed共14o, 6e兲 active space. These observations suggest that a strong depen-dence of the computed molecular parameters and potential energy surfaces on the choice of the active space may be limited to noncovalently bound species,12,74 where the ad-equate description of the electronic structure requires a prop-erly balanced composition of the active space. It can be hoped that the results shown in the present study, along with the recent surge of interest in the theoretical description of the scandium dimer, will prompt experimental groups to re-investigate this intriguing molecule so as to remedy the cur-rent paucity of reliable experimental data on Sc2.
ACKNOWLEDGMENTS
H.A.W. thanks the National Center for
High-Performance Computing of Taiwan for computer facilities. Financial support from the National Science Council of
Taiwan 共Grant No. NSC96-2113-M-009-022兲 and the ATU
project of the Ministry of Education, Taiwan is acknowl-edged. R.C. wishes to express his indebtedness to the
CINECA of Bologna 共Italy兲 for a generous amount of
com-puter time and to the University of Ferrara 共Italy兲 for par-tially financing this research through its FAR funds.
1C. Kittel, Introduction to Solid State Physics共Wiley, New York, 2005兲. 2N. W. Ashcroft and N. D. Mermin, Solid State Physics共Brooks-Cole,
Belmont, MA, 1976兲.
3A. D. McLean,J. Chem. Phys. 79, 3392共1983兲.
4D. Tzeli, U. Miranda, I. G. Kaplan, and A. Mavridis,J. Chem. Phys.129,
154310共2008兲.
5S. F. Li, Z. Shao, S. Han, X. Xue, F. Wang, Q. Sun, Y. Jia, and Z. X. Guo,
J. Chem. Phys. 131, 184301共2009兲.
6C. Angeli, A. Cavallini, and R. Cimiraglia,J. Chem. Phys. 127, 074306
共2007兲.
7A. C. Borin, J. P. Gobbo, and B. O. Roos,Chem. Phys. 343, 210共2008兲. 8J. M. Matxain, E. Rezabal, X. Lopez, J. M. Ugalde, and L. Gagliardi,J.
Chem. Phys. 128, 194315共2008兲.
9K. Ellingsen, T. Saue, C. Pouchan, and O. Gropen,Chem. Phys. 311, 35
共2005兲.
10C. Angeli, B. Bories, A. Cavallini, and R. Cimiraglia,J. Chem. Phys.
124, 054108共2006兲.
11C. Angeli, A. Cavallini, and R. Cimiraglia,J. Chem. Phys. 128, 244317
共2008兲.
12C. Camacho, S. Yamamoto, and H. A. Witek,Phys. Chem. Chem. Phys.
10, 5128共2008兲.
13C. Camacho, H. A. Witek, and S. Yamamoto,J. Comput. Chem. 30, 468
共2009兲.
14B. Minaev,Spectrochim. Acta, Part A 62, 790共2005兲.
15Z. Wu, B. Han, Z. Dai, and P. Jin,Chem. Phys. Lett. 403, 367共2005兲. 16S. Castillo, A. Cruz, V. Bertin, E. Poulain, J. S. Arellano, and G. D.
Angel,Int. J. Quantum Chem. 62, 29共1997兲.
17G. Bravo-Pérez, I. L. Garzón, and O. Novaro, J. Mol. Struct.:
THEOCHEM 493, 225共1999兲.
18R. Beuc, M. Movre, V. Horvatic, C. Vadla, O. Dulieu, and M. Aymar,
Phys. Rev. A 75, 032512共2007兲.
19H.-K. Chung, K. Kirby, and J. F. Babb,Phys. Rev. A63, 032516共2001兲. 20D. Dai, S. Roszak, and K. Balasubramanian,Chem. Phys. Lett. 308, 495
共1999兲.
21L. A. Kaledin, A. L. Kaledin, M. C. Heaven, and V. E. Bondybey,J. Mol.
Struct.: THEOCHEM 461–462, 177共1999兲.
22E. Czuchaj, M. Krośnicki, and H. Stoll,Chem. Phys. Lett. 371, 401
共2003兲.
23C.-R. Wang, T. Kai, T. Tomiyama, T. Yoshida, Y. Kobayashi, E.
Nishi-bori, M. Takata, M. Sakata, and H. Shinohara,Nature共London兲 408, 426
共2000兲.
24K. Kobayashi and S. Nagase,Chem. Phys. Lett. 362, 373共2002兲. 25M. Takata, E. Nishibori, M. Sakata, C.-R. Wang, and H. Shinohara,
Chem. Phys. Lett. 372, 512共2003兲.
26Z. Slanina, Z. Chen, P. v. R. Schleyer, F. Uhlík, X. Lu, and S. Nagase,J.
Phys. Chem. A 110, 2231共2006兲.
27Y. Ito, W. Fujita, T. Okazaki, T. Sugai, K. Awaga, E. Nishibori, M.
Takata, M. Sakata, and H. Shinohara,ChemPhysChem 8, 1019共2007兲.
28A. R. Khamatgalimov and V. I. Kovalenko, Russ. J. Phys. Chem. 82,
1164共2008兲.
29D. Liu and F. Hagelberg,Int. J. Quantum Chem. 107, 2253共2007兲. 30Y.-H. Cui, W. Q. Tian, J.-K. Feng, and D.-L. Chen,J. Comput. Chem.29,
2623共2008兲.
31A. Kalemos, I. G. Kaplan, and A. Mavridis,J. Chem. Phys. 132, 024309
共2010兲.
32J. L. Jules and J. R. Lombardi,J. Phys. Chem. A 107, 1268共2003兲. 33M. Moskovits, D. P. DiLella, and W. Limm,J. Chem. Phys. 80, 626
共1984兲.
34M. E. Jacox, J. Phys. Chem. Ref. Data Monogr. 3, 461共1994兲. 35G. Verhaegen, S. Smoes, and J. Drowart,J. Chem. Phys. 40, 239共1964兲. 36J. Drowart, in Phase Stability in Metals and Alloys, edited by P. S.
Rud-man, J. Stringer, and R. I. Jafee共McGraw-Hill, New York, 1967兲, pp. 305–307.
37H. Åkeby, L. G. M. Pettersson, and P. E. M. Siegbahn,J. Chem. Phys.
97, 1850共1992兲.
38H. Åkeby and L. G. M. Pettersson,J. Mol. Spectrosc. 159, 17共1993兲. 39T. L. Haslett, M. Moskovits, and A. L. Weitzman,J. Mol. Spectrosc.135,
259共1989兲.
40R. J. LeRoy and R. B. Bernstein,J. Chem. Phys. 52, 3869共1970兲. 41R. J. Le Roy,J. Chem. Phys. 73, 6003共1980兲.
42L. B. Knight, Jr., R. J. V. Zee, and W. Weltner, Jr.,Chem. Phys. Lett.94,
296共1983兲.
43
R. Busby, W. Klotzbücher, and G. A. Ozin,J. Am. Chem. Soc. 98, 4013
共1976兲.
244306-8 Camacho, Witek, and Cimiraglia J. Chem. Phys. 132, 244306共2010兲
44W. F. Cooper, G. A. Clarke, and C. R. Hare,J. Phys. Chem. 76, 2268
共1972兲.
45V. D. Fursova, A. P. Klyagina, A. A. Levin, and G. L. Gutsev,Chem.
Phys. Lett. 116, 317共1985兲.
46A. Wolf and H.-H. Schmidtke,Int. J. Quantum Chem. 18, 1187共1980兲. 47C. Wood, M. Doran, I. H. Hillier, and M. F. Guest,Faraday Symp. Chem.
Soc. 14, 159共1980兲.
48Y. Suzuki, S. Asai, K. Kobayashi, T. Noro, F. Sasaki, and H. Tatewaki,
Chem. Phys. Lett. 268, 213共1997兲.
49S. P. Walch and C. W. Bauschlicher, Jr.,J. Chem. Phys. 79, 3590共1983兲. 50G. H. Jeung,Chem. Phys. Lett. 125, 407共1986兲.
51G. L. Gutsev, P. Jena, B. K. Rao, and S. N. Khanna,J. Chem. Phys. 114,
10738共2001兲.
52G. Das,Chem. Phys. Lett. 86, 482共1982兲.
53S. P. Walch and C. W. Bauschlicher, Jr., Chem. Phys. Lett. 94, 290
共1983兲.
54J. Harris and R. O. Jones,J. Chem. Phys. 70, 830共1979兲.
55S. Yanagisawa, T. Tsuneda, and K. Hirao, J. Chem. Phys. 112, 545
共2000兲.
56I. Pápai and M. Castro,Chem. Phys. Lett. 267, 551共1997兲.
57C. J. Barden, J. C. Rienstra-Kiracofe, and H. F. Schaefer III,J. Chem.
Phys. 113, 690共2000兲.
58M. Valiev, E. J. Bylaska, and J. H. Weare,J. Chem. Phys. 119, 5955
共2003兲.
59A. Bérces,Spectrochim. Acta, Part A 53, 1257共1997兲.
60G. L. Gutsev and C. W. Bauschlicher, Jr.,J. Phys. Chem. A 107, 4755
共2003兲.
61K. Hirao,Chem. Phys. Lett. 196, 397共1992兲. 62H. Nakano,J. Chem. Phys. 99, 7983共1993兲.
63H. A. Witek, Y.-K. Choe, J. P. Finley, and K. Hirao,J. Comput. Chem.
23, 957共2002兲.
64J. M. Matxain, E. Rezabal, X. Lopez, J. M. Ugalde, and L. Gagliardi,J.
Chem. Phys. 132, 139901共2010兲.
65C. Camacho, R. Cimiraglia, and H. A. Witek,Phys. Chem. Chem. Phys.
12, 5058共2010兲.
66C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger, and J.-P. Malrieu,
J. Chem. Phys. 114, 10252共2001兲.
67C. Angeli, R. Cimiraglia, and J. P. Malrieu,J. Chem. Phys. 117, 9138
共2002兲.
68C. Angeli, M. Pastore, and R. Cimiraglia,Theor. Chem. Acc. 117, 743
共2007兲.
69B. O. Roos, R. Lindh, P. Å. Malmqvist, V. Veryazov, and P.-O. Widmark,
J. Phys. Chem. A 109, 6575共2005兲.
70B. A. Hess,Phys. Rev. A 33, 3742共1986兲. 71
MOLPRO, a package of ab initio programs designed by H.-J. Werner and P. J. Knowles, Version 2009.1, R. Lindh, F. R. Manby, M. Schütz et al.
72C. E. Moore, Atomic Energy Levels, Circular of the National Bureau of
Standards 共U.S. Government Printing Office, Washington, DC, 1971兲, Vol. I.
73C. Angeli, S. Borini, M. Cestari, and R. Cimiraglia,J. Chem. Phys.121,
4043共2004兲.
74A. A. Buchachenko, G. Chałasiński, and M. M. Szcześniak,J. Chem.
Phys. 132, 024312共2010兲.