鉭基氮化物擴散阻礙層在砷化鎵與銅金屬間阻礙機制之研究
79
0
0
全文
(2) 謝 誌 本論文得以順利完成,首應感謝指導教授謝光宇博士、黃升龍博 士的悉心教誨指導。黃老師的泱泱氣度、謝老師的助人危難,學生獲 益良多、銘感五內,在此深致謝意。 感謝工研院電子所石儲榮先生的寶貴意見,陸軍官校黃興祿博士 的熱心協助。感謝中山大學貴儀中心技術員李秀月小姐、陳貴香小 姐、江宏達先生和成功大學貴儀中心李瑞欽先生在儀器分析上面給予 的幫助。感謝學長姊、同學、學弟妹的協助陪伴,使本論文得以完成。 最後要感謝家人給予我的關愛和支持,及女友紅菊的鼓勵陪伴, 使我能安心於學問研究並順利完成學業,僅此論文與你們分享。. 2.
(3) 中文摘要 本研究主要探討鉭基氮化物擴散阻礙層在不同溫度下對銅金屬 和砷化鎵間擴散阻礙行為與失效的機制。實驗以反應式磁控法在砷化 鎵基板上以不同的 Ar/N2 分壓比例濺鍍鉭靶或氮化鉭靶成長鉭基氮 化物阻礙層,銅金屬以相同方式濺鍍其上,隨後進行真空封管並在不 同回火氣氛下進行高溫退火。藉由 X 光繞射的結構觀察以及 X 光光 電子能譜能量的定量分析,探討鉭基氮化物阻礙層的阻礙失效行為。 實驗結果顯示鉭基氮化物可以在真空環境 575℃下一小時內阻礙 銅金屬和砷化鎵間之擴散效應,而在鎵或砷的氣氛下進行退火,並不 能有效提升耐熱溫度和壓抑銅金屬和砷化鎵間之擴散問題。其失效機 制可能源自於當外界溫度高於 580℃時,砷化鎵基底開始自身解離,高 氣壓砷原子外溢,加速降低鉭基氮化物阻礙銅與砷化鎵相互間擴散的 能力。. 3.
(4) Abstract The behaviors of the TaNx barrier layer that placed between the Cu metal and GaAs have been studied by using X-ray diffraction, X-ray photoelectron spectroscopy and scanning electron microscopy. The TaNx and Cu films were deposited on GaAs sequentially with RF magnetron sputter. With a 250 nm thick TaNx barrier layer, the Cu metal can be impeded from reacting with GaAs substrate at 575℃ annealed for one hour. Within an As or Ga overpressure environment condition, the failure temperature still occurred below 600℃. The failure of TaNx diffusion barrier layer for preventing the reaction of the Cu and GaAs was originated for the dissociation of the GaAs itself at 580℃. The outgoing As atoms increased the deterioration speed of the TaNx film and reduced its blocking ability.. 4.
(5) 目錄 目錄.......................................................................................................I 圖目錄 ................................................................................................ Ⅳ 表目錄 ................................................................................................ Ⅶ 第一章 導論......................................................................................... 1 1.1 砷化鎵 MOS 場效電晶體的發展............................................... 1 1.2 金屬鋁的瓶頸與銅金屬時代的來臨.......................................... 2 1.2.1 金屬鋁的瓶頸.................................................................. 2 1.2.2 銅金屬的發展.................................................................. 3 1.3 擴散阻礙層 ............................................................................... 3 1.4 研究緣起 ................................................................................... 5 第二章 理論......................................................................................... 6 2.1 砷化鎵基板材料特性 ................................................................ 6 2.2 鉭金屬和鉭基氮化物的結構與特性.......................................... 7 2.2.1 鉭金屬和鉭基氮化物的結構與性質................................ 7 2.2.2 鉭金屬和鉭基氮化物的電阻率........................................ 7 2.3 文獻回顧 ................................................................................... 8 2.3.1 鉭基氮化物在矽基板和銅金屬間之擴散阻礙行為機制... 8. 5.
(6) 2.3.2 砷化鎵基板上擴散阻礙層的研究.................................... 8 2.4 反應性射頻磁控濺鍍原理......................................................... 9 2.4.1 濺鍍原理 ......................................................................... 9 2.4.2 射頻磁控原理................................................................ 10 2.5 X 光光電子電子能譜(XPS)原理.......................................... 10 第三章 實驗步驟與分析方法............................................................. 13 3.1 實驗設備介紹.......................................................................... 13 3.2 實驗流程 ................................................................................. 13 3.2.1 基板清潔 ....................................................................... 14 3.2.2 鉭基氮化物薄膜和銅膜的鍍製...................................... 15 3.2.3 封管............................................................................... 16 3.2.4 退火(annealing)......................................................... 17 3.3 薄膜的分析 ............................................................................. 17 3.3.1 X 光繞射儀分析............................................................. 17 3.3.2 X 光光電子電子能譜分析(XPS)................................ 18 3.3.3 掃描式電子顯微鏡分析................................................. 18 3.3.4 電子微探針分析儀(EPMA) ...................................... 18 3.3.5 鉭基氮化物電性分析 .................................................... 19 3.3.6 表面測量儀(Surface profile)...................................... 20. 6.
(7) 第四章 實驗結果與討論.................................................................... 21 4.1 X 光光電子能譜儀定量分析結果............................................. 21 4.1.1 鉭基氮化物定量分析結果............................................. 21 4.1.2 鉭基氮化物束縛能偏移的探討...................................... 21 4.2 鉭基氮化物的 X 光繞射儀分析............................................... 22 4.3 鉭基氮化物擴散阻礙層在真空環境下熱處理後之分析 .......... 23 4.3.1 有無鉭基氮化物擴散阻礙層的 XRD 分析比較............. 23 4.3.2 鉭基氮化物擴散阻礙層在砷化鎵基板上耐火程度之探 之分析 .................................................................................... 23 4.3.3 鉭基氮化物擴散阻礙層在玻璃基板上耐火程度之探討 分析........................................................................................ 24 4.4 不同氣氛下退火之 XRD 分析................................................. 25 4.4.1 鎵氣氛環境退火之 XRD 分析....................................... 25 4.4.2 砷氣氛環境退火之 XRD 分析....................................... 27 4.5 掃瞄式電子顯微鏡下退火後晶粒大小的探討......................... 27 4.6 電子微探針分析儀(EPMA)定量分析結果.......................... 29 4.7 電性量測 ................................................................................. 29 第五章 結論....................................................................................... 30 參考文獻 ............................................................................................ 31. 7.
(8) 圖目錄 圖 2-1 f.c.c 晶體結構........................................................................... 36 圖 2-2 砷化鎵晶體結構(閃鋅礦結構,zincblende)........................ 36 圖 2-3 砷化鎵之電子漂移速度圖....................................................... 37 圖 2-4 砷化鎵之能隙圖...................................................................... 37 圖 3-1 濺鍍實驗系統示意圖.............................................................. 38 圖 3-2 實驗流程架構示意圖.............................................................. 39 圖 3-3 真空封管系統架構圖.............................................................. 40 圖 3-4 真空封管過程示意圖.............................................................. 40 圖 3-5 置放雜質源的封管示意圖....................................................... 41 圖 3-6 出爐時冷卻方法意圖.............................................................. 41 圖 4-1 鉭基氮化物(Ta、Ta2N、TaN )外層電子 4f7/2 和 4f5/2 軌域 的束縛能 ................................................................................ 42 圖 4-2 鉭基氮化物外層電子 4f7/2 和 4f5/2 軌域的束縛能( a)150W Si (b)150W Glass(c)200W Si(d)200W Glass.................. 43 圖 4-3 表 4-2 中 M、N、O、P 組的 X 光繞射圖譜(a)M (200W, 9.5/0.5) (b)N (150W, 9.5/0.5) (c)O (150W, 13/0.5) (d)P (150W, 18/0.5)..................................................................................... 44 圖 4-4 表 4-2 中Ⅰ、Ⅱ、Ⅲ、Ⅳ組的 X 光繞射圖譜(a)Ⅰ(50W, 5/0) 8.
(9) (b)Ⅱ(50W, 5/0.5)(c)Ⅲ(100W, 7.5/0)(d)Ⅳ(100W, 7.5/1) ............................................................................................... 45 圖 4-5 表 4-2 中Ⅴ、Ⅵ、Ⅶ、Ⅷ組的 X 光繞射圖譜(a)Ⅴ(150W, 7.5/1) (b)Ⅵ(175W, 6.5/1) (c)Ⅶ(175W, 8.5/1) (d)Ⅷ(200W, 9.5/0.5) ............................................................................................... 46 圖 4-6 真空氣氛、不退火情況下的 X 光繞射圖譜(a)無鉭基氮化物 阻礙層(Cu/GaAs) (b)有鉭基氮化物阻礙層(Cu/TaNx/GaAs) ............................................................................................... 47 圖 4-7 真空氣氛、不同熱處理一小時下,鉭基氮化物在砷化鎵基板 和銅膜間〔Cu(324nm) / TaNx (141nm) / GaAs〕的 X 光繞射圖 譜(a)未退火(b)500℃(c)600℃ .................................. 48 圖 4-8 鉭 基 氮 化 物 在 砷 化 鎵 基 板 和 銅 膜 間 〔 Cu(284nm)/TaNx (212nm)/GaAs〕退火一小時後的 X 光繞射圖譜( a)575℃(b) 625℃ ...................................................................................... 49 圖 4-9 鉭基氮化物在玻璃基板上退火一小時後的 X 光繞射圖譜(a) 未退火(b)400℃(c)500℃(d)600℃ ............................ 50 圖 4-10 鎵氣氛環境、不同基板〔Cu (284nm)/ TaNx (212nm)/ Sub.)、 不同溫度下退火一小時後的 X 光繞射圖譜(a)600℃玻璃基 板(b)600℃砷化鎵基板(c)650℃砷化鎵基板................. 51 圖 4-11 砷氣氛環境、不同基板〔Cu (284nm)/ TaNx (212nm)/ Sub.〕、 9.
(10) 不同溫度下退火一小時後的 X 光繞射圖譜(a)575℃砷化鎵 基板(a1)575℃玻璃基板(b)600℃砷化鎵基板(b1)600 ℃玻璃基板(c)650℃砷化鎵基板........................................ 52 圖 4-12 真空氣氛下(a)不退火(b)575℃(c)650℃(d)700℃退 火一小時後,鉭基氮化物在矽基板上(TaNx/Si)經酸蝕刻後 表面之 SEM 圖....................................................................... 53 圖 4-13 真空氣氛下(a)575℃(b)700℃退火一小時後,銅膜在鉭 基氮化物上(Cu/TaNx/Si)經酸蝕刻後表面之 SEM 圖 ........ 55. 10.
(11) 表目錄. 表 2-1 砷化鎵在室溫時(300 K)之特性...................................... 56 表 2-2 鉭金屬和鉭基氮化物之結構特性........................................ 57 表 2-3 砷化鎵基板上擴散阻礙層之相關文獻整理......................... 58 表 3-1 銅、銅鎵、銅砷化合物之 X 光繞射資料............................ 59 表 3-2 鉭、鉭基氮化物、鉭鎵、鉭砷化合物之 X 光繞射資料 ..... 62 表 4-1 XPS 數據整理表格............................................................... 65 表 4-2 成長鉭基氮化物實驗之變動參數及數值 ............................ 66 表 4-3 不同氣氛、溫度退火實驗整理 ........................................... 67 表 4-4 EPMA 實驗結果................................................................... 68. 11.
(12) 第一章 導論 近年來微波無線通訊已成了日常生活中重要的應用科技,舉凡行 動電話、衛星節目、無線區域網路迴路、無線電纜電視及其他的射頻 /中頻(RF/IF)積體電路技術應用等,均為此一科技之應用實例。早年 受限於砷化鎵晶體本身性質的影響,使得砷化鎵晶體在金屬-半導體 場效電晶體(MESFET)的應用發展上較受重視:例如砷化鎵高功率 場效電晶體,可在 2GHz 操作下,輸出 1.6W 的功率,且場效電晶體 的閘極長度(gate length)也從早先發展的 100 ìm,在數年間即縮減 為數μm 左右程度,此後,低雜音化、高功率化的研究發展不斷向前, 廣泛地使用砷化鎵場效電晶體的時代終於來臨〔1〕。. 1.1 砷化鎵 MOS 場效電晶體的發展 在半絕緣性的砷化鎵基板表面上,利用離子佈植或是分子束磊晶 等方法,形成 n 型之砷化鎵通道層(channel),通常使用四族的矽做 為摻雜三五族砷化鎵的 n 型雜質。一般而言,砷化鎵與二氧化矽等的 氧化膜之界面狀態品質很差,表面能態密度很高,所以電力線到達界 面即終止;對砷化鎵而言,利用類似於矽 MOS 電晶體那種金屬-氧化 膜-半導體構造來控制通道層中之電子數的方法很困難,因此使用蕭 特基接合面所產生的空乏區,來控制從源極(source)到汲極(drain) 間之通道層的電子,也因此 GaAs FET 被稱為 MESFET ( metal semiconductor FET) 。然而近幾年來,由於磊晶技術的成熟,已可用 電子束蒸鍍法在砷化鎵基板上成長單晶的氧化鎵 Ga2O3 等氧化層不 再是難事〔2〕,所以砷化鎵 MOS 電晶體的發展備受矚目。. 12.
(13) 1.2 金屬鋁的瓶頸與銅金屬時代的來臨 1.2.1 金屬鋁的瓶頸 鋁的電阻率頗低(2.74 ìÙ -cm),且對二氧化矽層的附著情形良 好,金屬鋁已普遍使用為元件的主要導電材料,以降低 RC 時間延遲 (time delay),並提升元件的開關(switching)頻率;雖然鋁與二氧 化矽的介面化性與物性十分的理想,但是金屬鋁和矽間的界面就沒有 那樣幸運了:當製程有歷經溫度約 400℃以上的步驟時,因矽、鋁間 本身固態溶解度( solid solubility)的關係,使得矽藉由擴散效應進入 鋁,且鋁也會回填矽擴散後所遺留下的空隙,而在鋁和矽接觸的部 分,造成尖峰(spike)的情形,當尖峰的長度超過 MOS 的接面深度 (junction depth) ,元件會因迴路斷路而失效;防止尖峰現象的發生, 可在沈積金屬鋁時,加入相當於 1﹪的矽,如此鋁矽金屬合金與矽基 材接觸時,便不易產生尖峰現象〔3〕。 金屬鋁另一個最大的缺點就是其抗電致遷移(electromigration) 能力差:當製程線寬進入更小尺寸,且鋁導線處於高電場、傳導電流 作用狀態下時,鋁因抗電致遷移能力差,致使在高電場附近作用的鋁 金屬離子藉電子碰撞的動量轉移,往電場的負極移動,這會造成連線 的局部地方產生斷路或不連續的現象,影響元件的可靠性甚大;一般 可藉由加入約 0.5-4﹪含量的銅來解決此電致遷移現象〔3〕。 在金屬鋁內摻雜適量的矽、銅,可以預防上述尖峰和電致遷移這 兩種最常見於鋁金屬連線的失效方式( failure modes) ,並延長其失效 平均時間(mean time between failure) ,但是摻雜不同元素卻會使電阻 率上升,也增加蝕刻時的困難度〔3〕。 13.
(14) 1.2.2 銅金屬的發展 銅金屬材料在抗電致遷移的能力表現上比鋁來的優異,且銅金屬 的電阻率(1.67µΩ-cm)也不比鋁金屬材料來的差,在半導體元件的 線寬進入更小尺寸時,電阻率的些微差距將使 RC 時間延遲更形明 顯,且元件的設計積集度(integration)越來越高時,銅製程可以有 效減少相同電路設計的層數,降低成本〔4〕;考量元件的可靠性、 操作速度和成本等諸因素,銅製程皆可是取代鋁金屬連線的下一代製 程技術〔5〕。 當然銅製程也有其缺點:首先是在活性離子蝕刻(RIE)時,因 CuF x、CuClx 等反應產物在 200℃以下是固體而不會氣化,故反應進 行時需提高溫度,然此光阻卻無法承受此溫度,導致銅線圖案化 ( pattern ) 無 法 進 行 。 其 次 在 化 學 機 械 研 磨 平 坦 化 (chemical mechanical planarization, CMP)時,因對銅和阻礙層金屬的研磨速率 不同,將使 CMP 平坦化效果不佳,且增加尺寸控制上的困難度;此 外包括 CMP 的研磨液、清洗液等耗材研發的不夠成熟,皆造成銅製 程實用化的困難,所幸這些問題已經由工程師不斷的研發改進和業界 的共同努力而逐一克服〔6〕。另外銅原子若擴散進入矽與二氧化矽 中,並在主動區形成載子捕獲中心,將降低載子的活動能力,導致漏 電流增加,嚴重者甚或改變破壞元件特性;其他諸如銅金屬本身穩定 度不佳,極易和環境中的氧分子結合氧化,且銅金屬和大部分介電層 或其他金屬之附著力不佳等因素影響,也都是全面進入銅製程的瓶 頸,然這些問題都已逐漸克服並進入量產階段〔5〕。. 1.3 擴散阻礙層 14.
(15) 兩種不同材料彼此間接觸,就會因元素濃度不同開始彼此擴散, 而產生新的界面相,此時如果在這兩層不同材料間加入第三種材料以 防止彼此間的擴散或阻礙其發生化學反應,此第三種材料就是擴散阻 礙層(diffusion barrier layer)。擴散阻礙層是避免防止在接觸的兩種 材料間,因彼此產生化學反應形成新的界面相〔7〕。 在半導體製程中,因為要完成元件電路設計目的,增加金屬導線 的層數是必須的,而連接不同金屬層所使用的鎢插塞(plug),因和 其他材質的附著能力不太理想,我們必須在鋁、鎢金屬與其他材料接 觸面之間,加入一層阻礙層以提升附著能力,也可避免鋁和矽接觸面 的尖峰現象〔3〕。在積體電路進入銅製程後,為防止銅金屬擴散進 入矽基材,適當的阻礙層是必須的,目前已被研究選用為阻障銅金屬 的阻礙層,包括有 W〔8〕、W-N、Mo〔8-9〕、Mo-N〔9〕、Ti〔10〕、 Ti-N〔10-11〕、Ti-W〔12〕、TaSi〔13〕、Ta〔8、13-15〕、Ta-N 〔16-24〕、Ta-Si-N〔25-27〕等,這些材料都是耐火的過渡金屬或是 其氮化物,本身材質堅硬,具有高熔點、高導電性、強電子鍵結(strong electronic-bonds)和低溫超導〔21〕等的特性;而在擴散阻礙層加入 氮或矽元素,雖然會提高電阻率,但是阻礙能力卻可增強。一般擴散 阻礙層的厚度要求約為 100 埃( Å ),才能降低電阻率(electrical resistivity),為減少擴散阻礙層的厚度,在重金屬內加入氮元素,使 更具阻礙特性。事實上,在現行研究發現的擴散阻礙層材料,並非完 美到可以無限延伸元件的壽命,而只能延長元件壽命到某種一定的程 度。一般擴散阻礙層可歸結具備以下的特性〔7〕: 1.. 擴散阻礙層要能穩定的存在於上下兩種不同材料間。. 2. 不會擴散進入上下兩層材料裡或是擴散進入的量要很低,不至於破. 15.
(16) 壞元件結構。 3. 擴散阻礙層和上下兩種材料間,要有良好的黏附能力。 4. 擴散阻礙層和上下兩種材料維持低接觸電阻及好的導電能力。 5. 擴散阻礙層可以抗熱和抗機械應力。. 1.4 研究緣起 基於上述三節的討論,我們希冀對 GaAs 的 MOS 元件,將來在 進入銅製程後,可能會遭遇到銅製程在矽基板上相同的問題,先行做 研究探討,對鉭基氮化物擴散阻礙層在砷化鎵和銅金屬間的阻礙機 制,和高溫熱處理產生的界面相和失效機制進行瞭解。. 本文第二章將介紹理論部分和相關文獻回顧﹔第三章介紹實驗 步驟流程及實驗分析儀器﹔第四章探討實驗的結果﹔第五章則是歸 納總結。. 16.
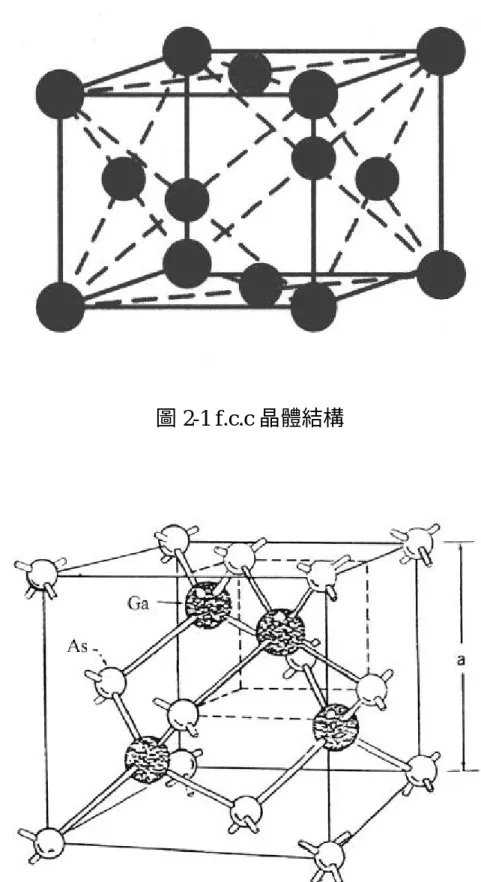
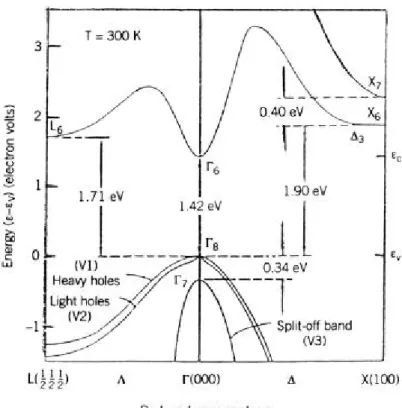
(17) 第二章 理論 2.1 砷化鎵基板材料特性 砷化鎵晶體的電子遷移率高,高飽和遷移速率使得砷化鎵材料比 矽材料更適用於高頻微波電路元件,所以被廣泛應用在通訊工業上。 其晶格為兩個面心立方結構(f.c.c),如圖 2-1,沿對角線四分之一處 重疊,如圖 2-2,為閃鋅晶格結構(zincblende)。 砷化鎵為Ⅲ-Ⅴ族合成半導體材料,其在室溫時特性如表 2-1 所 示〔1〕 。砷化鎵載子之遷移率和漂移速度高,在相同外加電場下,載 子很快的獲得一個相當的速度,大約 10-12 秒數量級,在低電場區其 速度與電場為線性,如圖 2-3 所示,關係式如(2-1)所示,其斜率 為載子之遷移率(mobility):. (2-1). v=μE. v 為電子速度(cm/sec),E 為電場強度,μ為遷移率(cm2/V-sec)。 載子的速度源自電場加速電子之力,以及電子在晶格中散射兩種機制 的總和,其最大速度為飽和漂移速度。 砷化鎵材料為直接能隙( direct bandgap) ,能階圖如圖 2-4 所示, 其導電帶(conduction band)的最低點直接位於價電帶( valance band) 最高點的正上方。能帶間的電子轉移只需要藉由吸收或放出一個光 子,便可以完成。砷化鎵能隙與溫度的關係如(2-2)式所示〔28〕:. E(T ) =. 1.519 − 5.405 ×10 −4 T 2 T + 204. 17. eV. (2-2).
(18) 2.2 鉭金屬和鉭基氮化物的結構與特性 2.2.1 鉭金屬和鉭基氮化物的結構與性質 鉭金屬的熔點高達 3293 K,具有良好的熱穩定度,在熱動力學 上和銅金屬間是穩定的。相關研究指出在含有 SiO2 介電層(熱氧化 法成長)的矽晶圓上,或在玻璃(Corning 7059)、氧化鈹(BeO)、 藍寶石(sapphire)、銅(111)金屬上濺鍍沈積鉭金屬,會形成正方 晶系的β型鉭金屬,而在鋁金屬或鉻金屬上沈積鉭金屬,則會形成體 心立方的α型鉭金屬〔14〕。α型鉭金屬較穩定,而β型鉭金屬則屬 於暫態不穩定型,但β型可經由 700℃的高溫退火過程,轉變成α型 (110)面的鉭金屬結構;當然諸如鉭金屬厚度、基板成長時溫度等, 皆會影響鉭金屬的結構形式〔14〕。 鉭基氮化物有多種不同的相,最穩定的結構不是立方晶格結構, 而是六角最密堆積結構;表 2-2 為鉭金屬和鉭基氮化物的晶格結構、 晶格常數〔29〕。其中 Ta2N 屬六角最密堆積結構,晶格常數為 as = 5.283Å,cs =4.928Å;另一種是結晶態的 TaN,晶格常數 a=2.936Å, c=2.885Å〔29〕。. 2.2.2 鉭金屬和鉭基氮化物的電阻率 相關研究指出α型體心立方結構的鉭金屬電阻率約 為 40μΩ -cm,β型暫態鉭金屬電阻率約為 150μΩ-cm;而在成長鉭基氮化物 薄膜條件時,略為加入極少量氮氣,電阻值會下降至 80μΩ-cm,推 論電阻值下降應是β型暫態鉭金屬元素發生相轉變成α型體心立方 晶格結構的緣故;不同的鉭基氮化物因氮含量不同而有所差異,當氮 18.
(19) 氣比例含量增加時,電阻率會逐步上升至 220~260μΩ-cm,估計 TaN 電阻率約為 250~330μΩ-cm,Ta2N 約為 240μΩ-cm〔17-18〕。. 2.3 文獻回顧 2.3.1 鉭基氮化物在矽基板和銅金屬間之擴散阻礙行為機制 先前的研究指出,介於矽基板與銅金屬之間的非晶質鉭基氮化 物,會因高溫誘發的結晶機制,晶粒開始成長,進而產生局部缺陷或 晶界裂縫,銅膜在此局部位置產生破裂並快速擴散進入矽基材,當溫 度提高時,銅原子會貫穿擴散阻礙層與矽基材反應生成銅矽化合物 (Cu3Si) ,而在更高溫度時,鉭基氮化物自身會裂解並與矽基材生成 鉭矽化合物(TaSi2)〔30〕。 相關文獻指出可藉由改變濺鍍功率、氮氣氬氣比例、腔室壓力來 尋找鉭基氮化物薄膜之最佳成長條件:在鉭基氮化物薄膜的成長過程 中,若提高氮氣流量,則定量分析發現薄膜中的氮含量成分比例會增 加,且阻障擴散效果愈好,能承受較高的熱處理溫度,但電阻率會提 高,當然厚度越厚,阻障效果也越佳;值得考量的是當元件進入深次 微米時,薄膜的厚度和電阻值是要和阻礙效能取得一個適當的平衡點 〔30〕。. 2.3.2 砷化鎵基板上擴散阻礙層的研究 在很多相關研究報告中都有指出,大部分的金屬在高溫下皆會因 為擴散機制,而在和砷化鎵接觸的介面上產生一些介面化合物 〔31-33〕。例如砷化鎵和金、銀、銅等貴金屬形成接觸面時,三價. 19.
(20) 的鎵離子會受具有強負電性的金屬電子吸引,而發生擴散遷移進入這 些金屬層;而諸如 Cr、Mo、W、V、Nb、Ta、Ti、Zr、Hf 等過渡金 屬和砷化鎵形成介面時,因其電負性不若貴金屬,故和砷化鎵接觸介 面會相對較穩定,但在高溫時則會產生介面化合物〔31〕。 砷化鎵承受高溫的熱穩定度不佳,相關文獻曾對鈦金屬(Ti)和 砷化鎵接觸面系統進行不同條件的退火實驗,證實約在 400℃左右, 鈦金屬和砷化鎵的接觸面開始產生反應,形成 Ti/TixGa1-x/TiAs/GaAs 的微細結構,並且進一步發現在較低溫退火後,產生 Ti2Ga3 和 TiAs 的化合物,而在較高溫度的退火下,則生成 Ti5Ga4 和 Ti5As3 的化合 物結構〔31-32〕。另外在砷化鎵基板上,也有選用 TiN、ZrB2、W2B 等擴散阻礙層對金做擴散阻礙能力研究,利用拉塞福回向散射儀 (Rutherford back-scattering spectroscopy, RBS)量測單位面積溢出的 砷原子數目來評定比較阻礙性能優劣,結果顯示硼化物的擴散阻礙層 比氮化物擴散阻礙層更能抑制砷元素的溢出,但是氮化物的電導率比 硼化物好〔33〕。表 2-3 為在砷化鎵基板上擴散阻礙層的相關文獻整 理結果〔31-34〕。. 2.4 反應性射頻磁控濺鍍原理 2.4.1 濺鍍(sputtering)原理 濺鍍製程是指利用電漿(plasma)對一塊靶材(target)進行離子 轟擊(ion bombardment),藉由離子轟擊的動量轉移(momentum transfer),將靶材表面的原子撞擊出來。原子以氣體分子的形式發射 出來,藉擴散效應以原子或分子的形態到達所要沈積的基材表面上, 再經過附著、吸附(adsorption)、表面遷徙(surface migration)、 20.
(21) 成核(nucleation)等過程後,在基板上成長形成薄膜的一種製程 〔38〕;其在基板上的活動力則受到原子與基材間的鍵結能、基板溫 度及離子撞擊程度等因素影響。. 2.4.2 射頻磁控原理 射頻(radio frequency, RF)是指頻率 13.56 MHz 的交變電源信 號,在射頻電源的電場作用下,真空腔體中的惰性氣體可從高能電子 的碰撞中得到能量並離子化成電漿態,這些電漿態的正離子受負偏壓 的靶材端電場作用後加速撞擊靶面,轟擊出靶面原子並產生濺射效 應。靶面的實際電位會隨射頻電源的週期而變化,為一固定負偏壓和 射頻弦波脈動的疊加和電位,當和電位為正時,靶面收集電漿中的電 子,和電位為負時,則收集離子,此交互收集電子和離子的結果,恰 可維持靶面上淨電流為零,此優點使濺鍍製程可採用導電度不佳的靶 材,這也正是採用射頻的最大好處和目的〔39-40〕。 在濺射系統中加裝磁控裝置,可藉磁場將電子限制在靶材附近作 螺旋狀運動,增加與惰性氣體分子碰撞離子化的機會,提升濺鍍的速 率。當工作氣體解離率增加時,產生更多的離子撞擊靶材,並濺射出 更多的靶材粒子沈積在基板上,因此磁控裝置可有效地提升濺鍍的速 率〔39-40〕。. 2.5 X 光光電子電子能譜(XPS)原理 本實驗在製鍍鉭基氮化物初期,沒有適當的參考經驗值,因此必 須藉助 XPS,對試片進行定量分析,視其結果來作為判斷調整實驗的. 21.
(22) 射頻功率、氬氣和氮氣流量比等參數,所以對於 XPS 的工作原理, 我們有必要做進一步瞭解。 X 光光電子能譜儀是藉著量測 X 光光電子能譜來分析材料表面 各種元素的化學狀態,因此化學分析功能,也稱為電子能譜化學分析 儀(electron spectroscopy for chemical analyzer, ESCA );其特色是可 鑑定材料表面上各種元素的化學鍵能,適合分析材料表面的化學性 質。 當 X 光由材料表面入射進入材料內部時,X 光會將原子內的核 心電子(core electron)或鏈結電子(valence electron)激發至高能態 (excited state),靠近材料表面的高能態電子,有機會逃逸出材料表 面進入真空,這些逃逸出材料表面的電子稱為 X 光光電子。X 光光 電子能譜包括各種核心電子、鍵結電子、歐傑電子( Auger electron), 以及非彈性碰撞電子( inelastic electron) ;其中核心電子、鏈結電子、 歐傑電子的能量,都會因化學元素不同而不同,也會因化學元素的鏈 結狀態不同而不同。此三種電子常被用來分析材料表面元素的化學狀 態,但因核心電子在 X 光波段的訊號最強,能量解析度最佳,X 光 光電子能譜儀都採用核心電子能譜來鑑定材料表面原子的化學狀 態。核心 X 光光電子的訊號是由材料表面附近的數層原子表面貢獻 的,核心 X 光光電子的表面靈敏度(surface sensitivity)是由核心 X 光光電子的動能大小決定。傳統的 X 光光源有鋁 Ká 或鎂 KáX 譜線, 其能量分別為 1486.6 及 1253.6 電子伏特(eV),鎂 Ká 的光子通量 (photon flux)較鋁 Ká 高,習慣以鎂 Ká 當光源來取數據。核心 X 光光電子的動能(K.E.)大小,等於鎂 Ká X 光光子能量(hν)減去 該核心電子的束縛能(binding energy, B.E.)再減去能譜分析器的工 作函數(work function, Φ),其關係式如下(2-3)所示,因此一般. 22.
(23) 的動能值約在 20-1200 電子伏特之間。 K .E. = hν − B.E. − Φ. (2-3). X 光光電子能譜也常用來鑑定材料表面的化學反應是否發生,以 及反應生成物的種類;當有化學變化時,原子間的鍵結電荷密度 (valence charge density)會重新分佈,促使原子內的各個核心電子的 能量產生位移,此能量位移現象稱為化學位移( chemical shift) 。各個 核心電子的化學位移可由 X 光光電子能譜直接測出,藉此可判斷反 應生成物的種類。 X 光光電子能譜儀的裝置可分為真空系統、光源、試片操縱裝 置、電子能量分析器,以及數據擷取等五大部分。其中真空系統是由 渦輪幫浦(turbo pump)和離子幫浦組成,利用超高真空技術將系統 抽至 1×10-10 torr;X 光光源是由雙陽極 X 光產生器(dual anode X- ray source)提供鋁 Ká 以及鎂 Ká X 光;試片操縱裝置包括傳送,以 及 XYZ 定位;較常用的電子能量分析器是半球型電子能量分析器 (hemi-spherical electron energy analyzer),因其能量解析度好;X 光 光電子能譜是由能譜擷取軟體取得,並與資料庫的數據比較,確定所 測物的化學態。 不同型式的 X 光光電子能譜儀,其濃度靈敏度、空間解析度、 能量解析度差異很大。一般的 X 光光電子能譜儀具有 0.3-1.0%的濃 度靈敏度、200μm 的空間解析度,以及 0.8-1.0 電子伏特的能量解 析度。若要研究化學位移,單頻 X 光光電子能譜儀是較好的選擇, 因其能量解析度較佳,約 0.5 電子伏特。若要有較佳的空間解析度, 須選擇小面積 X 光光電子能譜儀,其空間解析度可好到 5μm〔41〕。. 23.
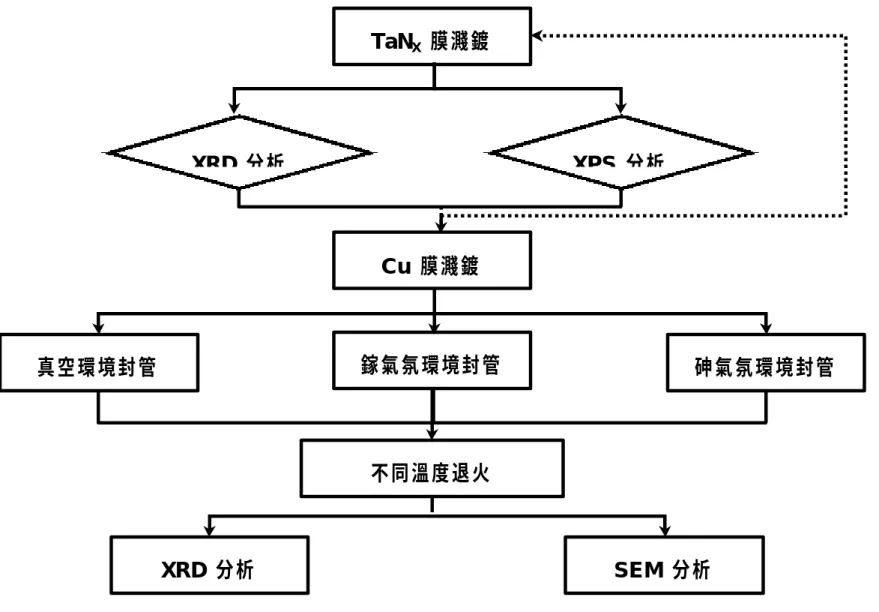
(24) 第三章 實驗步驟與分析方法 3.1 實驗設備介紹 本實驗採用反應性射頻磁控濺鍍法成長鉭基氮化物薄膜,實驗設 備如圖 3-1 所示。可概分為真空系統、濺鍍腔室、電漿磁控電源系統、 作用氣體系統、冷卻系統等五大部分,分述如下: 1. 真空系統:採用機械式幫浦( mechanical pump)和渦輪幫浦( turbo -6. pump)可將濺鍍腔室內壓力抽至 5×10. torr 以下。量測真空系統 -3. 的壓力計,可分低真空計和高真空計,分別為量測範圍界於 1~10. torr 間的熱偶真空計( thermocouple gauge),和量測範圍界於 -4. -6. 10 ~10 torr 間的離子真空計(ion gauge)。 2. 濺鍍腔室:用以腔體內含有機板乘載台(substrate holder) 、濺鍍擋 板(shutter)、陰極磁控盤、石英震盪片。 3. 電漿磁控電源系統:採用射頻頻率 13.56 MHz 的電源供應器,最 大輸出功率 350W,利用同軸電纜匹配傳入真空室內的陰極靶材 端,藉以將惰性氣體離子化形成電漿態。 4. 作用氣體系統:包括控制氣體流量的質流控制器( mass flow controller)、管路控制閥和傳送氣體管路等,可控制作用氣體導入 濺鍍腔室。 5. 冷卻系統:包括熱交換機、冷卻水循環系統,用以冷卻靶材和磁 控盤,避免高溫退磁現象發生。. 3.2 實驗流程 24.
(25) 本實驗的流程架構如圖 3-2 所示,其實驗步驟詳述如下:. 3.2.1 基板清潔 在此實驗中,我們選用三種不材質基板進行濺鍍薄膜和比較: GaAs、Si 與 Soda-lime 玻璃。不同基板的清潔處理方式亦不同,其清 潔處理步驟如下: A. 砷化鎵基板處理步驟: 1. 將 GaAs 晶片切成適當大小 2. 在丙酮〔Acetone, (CH3)2CO〕中以超音波震 10 分鐘,以除去 油漬。 3. 在異丙醇〔2-Propanol, (CH3)2CHOH〕中以超音波震 10 分鐘, 以除去丙酮殘留。 4. 在去離子水(D.I. Water)中以超音波震 10 分鐘,以除去異丙 醇殘留。 5. 以 H2SO4:H2O2:H2O = 5:1:1 溶液浸蝕 15 秒,以去除氧 化層。 6. 置於 HCl 煮 3 分鐘使表面具疏水性。 7. 試片以 DI water 沖洗乾淨。 8. 以氮氣吹乾。 B. 矽基板處理步驟: 1. 將 Si 晶片切成適當大小。 2. 在丙酮中以超音波震 10 分鐘,以除去油漬。 3. 在異丙醇中以超音波震 10 分鐘,以除去丙酮殘留。 4. 在去離子水中以超音波震 10 分鐘,除去異丙醇殘留。 5. 試片以 DI water 沖洗乾淨。. 25.
(26) 6. 以氮氣吹乾。 7. 將試片以 120℃高溫烘烤 1 小時,以除去水氣。 C. 玻璃基板處理步驟: 1. 將玻璃切成適當大小。 2. 在丙酮中以超音波震 20 分鐘,以除去油漬。 3. 在異丙醇中以超音波震 20 分鐘,以除去丙酮殘留。 4. 在去離子水中以超音波震 20 分鐘,除去異丙醇殘留。 5. 以氮氣吹乾。 6. 將試片以 120℃高溫烘烤 1 小時,以除去水氣。. 3.2.2 鉭基氮化物薄膜和銅膜的鍍製流程 本實驗以反應性射頻磁控濺鍍法成長鉭基氮化物和銅的薄膜。基 板採用玻璃、矽或砷化鎵基板,以不同的濺鍍功率和不同的氣氛比例 濺鍍成長鉭基氮化物薄膜(如表 4-1) ,所採用的濺鍍氣氛為氬氣和氮 氣。實驗所用的靶材選用純度 99.95%的 Ta 靶及純度 99.997%的 Cu 靶,並配合高純度的氮氣(N2)和氬氣(Ar)作為反應氣體。濺鍍實 驗流程概分為以下八大部分: 1. 濺鍍室腔體真空度的粗抽與細抽:將清洗乾淨、烘烤完成的試片 置於基板座(holder)上,在陰極靶材基座塗上導熱銀膠,並放上 欲濺鍍的靶材,關閉腔體上孔蓋,用機械幫浦( mechanical pump) -2. 粗抽至 5×10. torr 以下,再利用渦輪幫浦(turbo pump)細抽至. -6. 5×10 torr 以下,完成濺鍍前置準備工作。當腔室內真空度的壓力 至 5×10-6 torr 以下時,則可開始進行下一步驟。 2. 預濺鍍靶材(presputtering):經由氣體流量控制儀,調整成膜需 26.
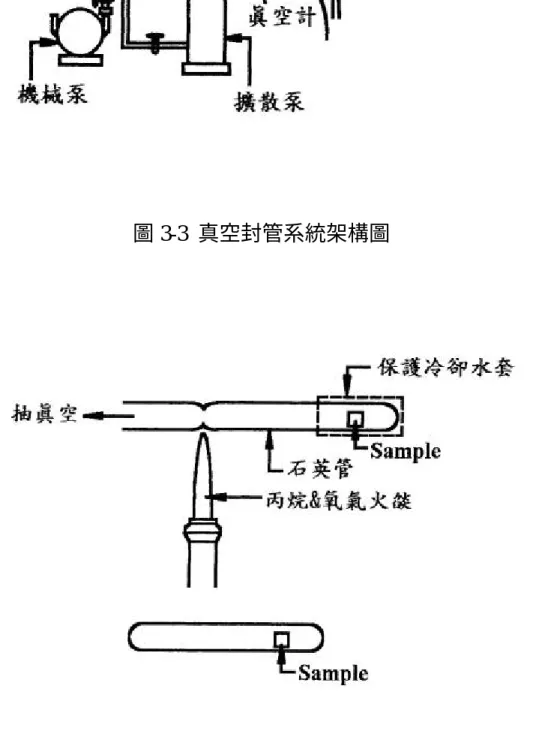
(27) 要的氮氣、氬氣流量,使進入腔體中,點燃電漿氣體至適當的射 頻功率,並調整腔室壓力至欲濺鍍之操作壓力,而後進行預濺鍍 約 10 到 20 分鐘。此預濺鍍目的是為除去靶材表面污染,並使腔 室內的氮氣與氬氣分佈均勻。 3. 濺鍍靶材:預濺鍍靶材後,將擋板(shutter)打開並開始計時, 依據不同的靶材濺鍍速率和薄膜沈積速率,至所需的薄膜厚度。 4. 破真空換銅靶:因本實驗採用單靶磁控濺鍍儀,所以在成長完鉭 基氮化物後,需要破真空取出原鉭靶並換上銅靶,為避免薄膜長 時間暴露在空氣中氧化或污染,此步驟動作需要快速進行。 5. 重複步驟 1 的粗抽與細抽。 6. 重複步驟 2 之預濺鍍 Cu 靶:此步驟因成長銅膜,故僅通入所需 的氬氣,而不需通氮氣,同樣預濺鍍 10 到 20 分鐘,以除去 Cu 靶表面的氧化物。 7. 重複步驟 3 之濺鍍 Cu 靶。 8. 破真空取出試片:薄膜成長完畢需靜置一小時以上,才能通入氮 氣破壞真空度和取出試片,以避免因殘留應力或過劇的溫差變化 造成薄膜品質的劣化剝落。. 3.2.3 封管 將濺鍍完成試片切成適當大小,使恰可放入內徑約 7mm 的石英 管,然後連接上附有機械幫浦和擴散幫浦的封管機(參見圖 3-3), 進行封管,其步驟如下: 1.用丙酮溶液、異丙醇溶液和去離子水清洗石英管。. 27.
(28) 2.將石英管插上封管機抽真空,並用火焰來回預烤石英管,以驅趕水 氣,並在適當處燒結凹槽,以置放雜物源。 3.關閉封管機閥門,取出石英管,將切割及清洗過後之試片放入石英 管內,同時在石英管內置入 Ga(20 mg)、As(20 mg)或不置放 任何雜物。 4.將內含試片的石英管插回封管機上,打開閥門續抽真空至壓力 5× 10-5 torr 後,再以瓦斯和氧氣混合產生的高溫火焰將石英管燒斷, 完成封管。 封管步驟隨不同氣氛環境退火而略有不同,如為真空氣氛封管, 則可省去第二步驟中之燒結凹槽置放雜質源步驟,如圖 3-4 所示,置 放雜質源封管如圖 3-5 所示。. 3.2.4 退火(annealing) 封管完成後,將石英管送入高溫爐內進行熱處理;為使熱處理過 程能在一恆定溫度下進行,故先將高溫爐溫度升到預定退火處理溫度 後,再放入石英管,而後分別進行不同溫度之熱處理,經一小時退火 後,將石英管從高溫爐取出。熱處理進行時,由於雜質源的蒸氣壓關 係,使得石英管內會充滿砷蒸氣或鎵蒸氣,故取出時需用濕毛巾包覆 遠離試片端,以淬冷(quenching)方式拿出高溫爐,避免砷蒸氣或 鎵蒸氣凝結在石英管壁或試片表面上,如圖 3-6 所示,完成退火步驟。. 3.3 薄膜的分析 3.3.1 X 光繞射儀分析. 28.
(29) 實驗以 Siemens D5000 型 X 光繞射分析儀進行鑑定,該儀器使用 銅靶和 Ni 質濾光片,銅靶之特性波長(CuKα射線)λ=1.5418 Å, 實驗之操作條件為電壓 40kV、電流 30mA。掃描角度 2θ由20°~ 65°, 每 0.05°掃描 3 秒。根據 JCPDS 卡之資料,鉭基氮化物繞射角度約 36.1°,鉭基氮化物和其他可能出現化合物之繞射峰皆可在 20°~65° 的範圍內觀測到。上層銅的繞射晶面主要為(111)和(200)繞射角 分別為 38.5°和 44.8°。本實驗 XRD 分析結果有關銅和鉭基氮化物 及其化合物的繞射資料詳見於表 3-1、表 3-2。. 3.3.2 X 光光電子電子能譜分析(XPS). 定量實驗以成功大學貴儀中心 Fison (VG) ESCA 210 型的 X 光電 子能譜儀分析,其工作原理與特性詳見 2.5 節。. 3.3.3 掃描式電子顯微鏡分析. 採用 JEOL—6400 掃描式電子顯微鏡來觀察薄膜之表面結構。操 作時之加速電壓為 20kV,放大倍率 10,000 倍以上,視每次薄膜成長 條件不同而變動,試片分別做表面( top-view)和截面(cross-section) 的觀察,主要乃為了觀察薄膜表面平整度和膜厚。此外另可利用能量 分散光譜儀(EDS)來做表面組成元素之定性分析。. 3.3.4 電子微探針分析儀(electron probe microanalysis, EPMA). 29.
(30) 本實驗所使用的電子微探分析儀是 JXA-8900R 型,搭配有能量 分散式電子微探儀( energy dispersive spectroscope, EDS )與波長分散 式電子微探儀( wavelength dispersive spectroscope, WDS )兩種 X 光偵 測器。在實驗中我們選擇解析度與精準度較高的 WDS,而電子束的 加速電壓與入射電流分別為 l0 kV 與 1.8×10-8A。 我們使用電子微探針分析法進行薄膜組成的量測,此法是利用電 子束激發出薄膜中的特性 X-ray,然後再經由能量分散式電子微探儀 (WDS)X 光偵測器,測得 X-ray 光子之數量將待測試片所測得的訊 號與純元素標準試片(standard)所測得的訊號相互比對,則可得知 薄膜之化學組成。而WDS 之原理乃是根據布拉格定律 (Bragg’s law) : nλ= 2dsinθ,d 為晶格的平面間距,θ為入射光及繞射 X 光與晶體 表面的夾角,n 為自然數。當各種元素受電子束轟擊之後激發出 X 光 皆有其特定之波長,如果將 X 光導入一已知平面間距的晶體中調整 入射角,不同元素會在不同入射角度出現繞射現象,繞射出來的 X 光可藉 Bragg’s law 計算出波長,即可得知何種元素。本法與 EDS 相 較有較高的解析度與低雜訊等優點,但一次只能偵測一個元素,既費 時又有對 X 光收集效率差的缺點。. 3.3.5 鉭基氮化物電性分析 30.
(31) 由於鉭基氮化物電性較接近於金屬,所以可直接採用四點探針法 (four point probe technique)量測薄膜的電阻值,再根據公式修正項 等換算成電阻率。 四點探針法可量測半導體或晶圓之電阻值。它是利用等距、共線 的四根探針,靠近兩邊外面的兩根探針通適當大小的電流(I) ,中間 內側兩根探針則可測得電壓( V) ,帶入下列公式,則可得到電阻率。 雖然此法會造成表面輕微的破壞,但仍比直接測試法易於量測。. ρ =2π ×(V/ I) ×Γ. Γ為修正因子項. 3.3.6 表面測量儀(Surface profiler). 本實驗以 Sloan DEKTAK3 0305ST 型α-step 量測薄膜的厚度。 實驗時,在基板薄膜上黏貼銅膠帶,因此無黏貼處和有黏貼處,有一 膜厚的高度落差,因此可藉由此測量儀得知其成長膜厚。α-step 是 利用探針以機械傳動的方法掃描薄膜表面,因此屬於一種破壞性量 測;探針在經過陡升區因震動產生的機械訊號經轉換成電子訊號,放 大後經過微處理器計算而得到膜厚。本機器的量測範圍為 5-10000 nm,精準度在 5 埃範園內。. 31.
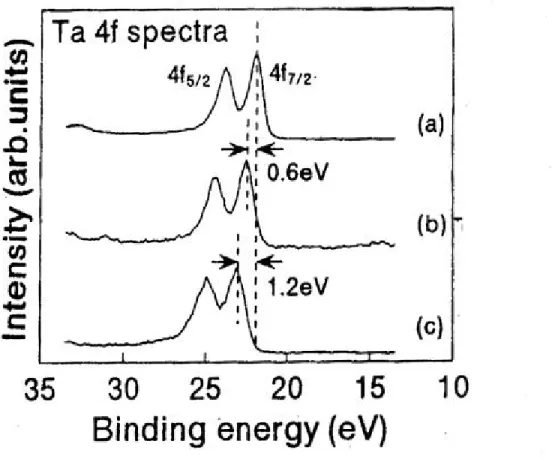
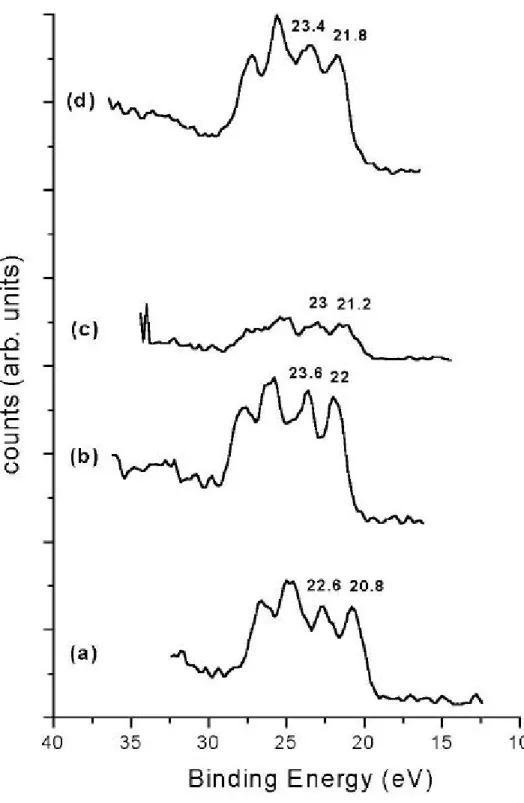
(32) 第四章 實驗結果與討論 4.1 X 光光電子能譜儀定量分析結果 4.1.1 鉭基氮化物定量分析結果 在缺乏成長鉭基氮化物薄膜之實驗參數情況下,我們藉助 X 光光 電子能譜儀來作定量分析,藉由分析定量組成來調整我們實驗的射頻 功率、氬氣和氮氣流量比等參數。表 4-1 為 X 光光電子能譜儀定量分 析的數據結果整理,A 到 H 為第一次送測試片,實驗結果顯示氮原 子的含量遠大於鉭原子,因此我們藉調高射頻功率和氬氣流量,並降 低氮氣流量,期使鉭靶被撞擊出更多的鉭原子,並和腔體氣氛中的少 量氮氣結合成鉭基氮化物。表 4-1 之 I 到 L 四組試片(成長條件參照 表 4-1)為第二次 XPS 的實驗結果,結果顯示氮原子和鉭原子的含量 比值接近於 3.2 到 4.8。 綜觀表 4-1 氮原子對鉭原子的定量分析比值,發現即使在相同的 成長條件下,不管是相同基板(E-F)或不同基板(A-B、G-H、K-L) 上,分析結果會有不同,推估其原因為薄膜表面組成成分不均勻所 致:因濺鍍時沈積基板上的成長速率不夠慢,使鉭原子和氮原子的結 合機率隨時間而不同,造成沈積在基板上的鉭原子和氮原子的組成成 分不均勻;當然也有可能是因儀器本身對濃度靈敏度、空間解析度、 能量解析度等問題所造成的影響,使數據有些許的誤差差異。. 4.1.2 鉭基氮化物束縛能偏移的探討 根據參考資料指出〔20〕,鉭元素外層電子 4f7/2 軌域和 4f5/2 軌 域的束縛能分別為 21.7eV 和 23.5eV,相差 1.8eV;鉭基氮化物的外 32.
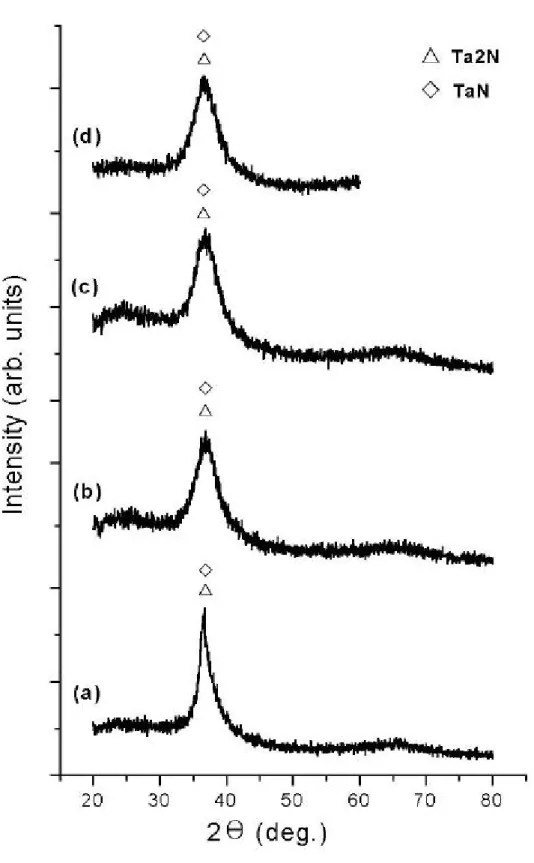
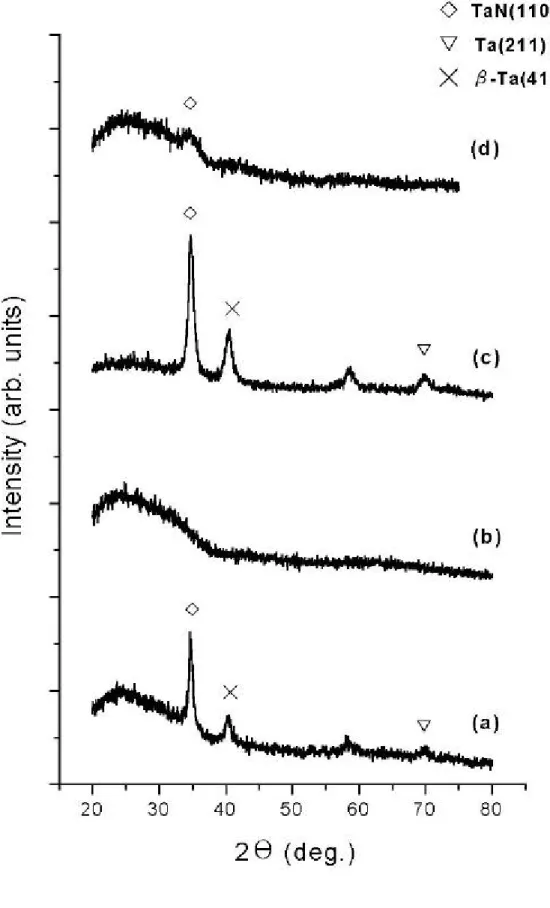
(33) 層電子 4f7/2 軌域束縛能如下:Ta2N 為 22.3eV,TaN 為 22.9eV,分 別較鉭元素外層電子 4f7/2 軌域偏移 0.6eV 和 1.2eV,如圖 4-1 所示 〔20〕。 圖 4-2 為我們實驗的 XPS 束縛能圖譜,(a)到(d)曲線分別對 應表 4-1 之 I 到 L組的結果,圖中標示數值為鉭基氮化物外層電子 4f7/2 軌域和 4f5/2 軌域的束縛能;四組外層軌域的束縛能,明顯不同於參 考文獻中鉭基氮化物(Ta2N 、TaN)的數值,而 L 組 4f7/2 軌域和 4f5/2 軌域的束縛能分別為 21.8eV 和 23.4eV,較接近鉭元素外層軌域 的束縛能,事實上在後節 X 光繞射儀分析可證實,L 組試片的成長條 件為非晶質的鉭基氮化物(a-TaNx)。另外四組圖譜的形貌也不同於 文獻,推測可能是因試片表層產生氧化情形所致〔20、44〕。. 4.2 鉭基氮化物的 X 光繞射儀分析 根據表 4-2 之 M 到 P 組的條件,在玻璃基板上成長鉭基氮化物薄 膜,其 X 光繞射圖譜如圖 4-3 所示,圖中顯示上述各組,在 33°到 40° 間皆有一寬廣波峰,此寬廣波峰的最大值約出現在 36.6° ~37°間,經 JCPDS 圖卡比對為 Ta2N (002)、TaN (101)的結構面。 根據表 4-2 之Ⅰ到Ⅳ組的條件,換成以氮化鉭靶為濺鍍靶材,同 樣在玻璃基板上成長鉭基氮化物薄膜,其 X 光繞射圖譜如圖 4-4 所 示,發現在Ⅰ、Ⅲ組實驗條件下,在 34.5° 附近出現 TaN(110)結構、 40.2°附近出現β-Ta(411)結構、69.6° 附近出現 Ta(211)結構。圖 4-5 為表 4-2 之Ⅴ到Ⅷ組條件的 X 光繞射圖譜,圖中Ⅵ、Ⅶ、Ⅷ組的 繞射圖譜,在 34.5° 附近出現 TaN(110)的結構面。. 33.
(34) 4.3 鉭基氮化物擴散阻礙層在真空環境下熱處理後之 分析 4.3.1 有無鉭基氮化物擴散阻礙層的 XRD 分析比較 為瞭解鉭基氮化物擴散阻礙層對阻礙銅金屬原子擴散的影響,我 們在砷化鎵基板上成長鉭基氮化物後(厚約 141 nm) ,再成長一層銅 膜(厚約 324 nm),另一組試片則直接濺鍍一層相同厚度的銅膜,詳 細成長參數參見表 4-3 中之 Q、R 組;試片成長完畢後,依 3.2.3 節 所述封管步驟完成封管後,再放入高溫爐進行不同溫度退火一小時, 另一組試片則不退火,以作為退火前後的比較。 在真空不退火的情況下,砷化鎵基板上直接鍍銅和多加一層阻礙 層的 X 光繞射圖譜比較,如圖 4-6 所示, (a)圖為沒有鉭基氮化物阻 礙層,除在 43.3°附近的 Cu(111)結構外,另在 45.3°附近亦有一根 尖銳波峰出現,根據 JCPDS 圖卡比對結果,推估可能結構為 CuGa2 (110)或是 Cu3As(212)/(113) ,此說明銅膜和砷化鎵基板介面接觸, 在不退火情況下就會產生介面結合反應,生成銅鎵或銅砷化合物。 (b) 圖為砷化鎵基板和銅膜間多一層阻礙層的 X 光繞射圖譜,在 33°到 40° 間有一寬廣繞射波峰,此結果顯示成長的鉭基氮化物是呈非晶質結 構,另外銅鎵化合物在 45.3°附近的繞射波峰也沒有出現,因此鉭基 氮化物阻礙層明顯可以阻礙銅金屬和砷化鎵基板的結合擴散反應。. 4.3.2 鉭基氮化物擴散阻礙層在砷化鎵基板上耐火程度之探 討分析 根據表 4-3 之 R 組的成長條件,我們希望知道鉭基氮化物熱穩定 34.
(35) 性的極限,還能穩定存在銅金屬層和砷化鎵基板間,同時且能阻礙銅 原子或砷化鎵彼此間的擴散行為。圖 4-7 為鉭基氮化物在砷化鎵基板 和銅膜間,不退火、500℃和 600℃各退火一小時後的 X 光繞射圖譜。 在(a)不退火的情況下,31.6°附近是 GaAs(200)的波峰,33°到 40°間的寬廣波峰是鉭基氮化物,43.3°附近是 Cu(111)的波峰;在 (b)500℃退火一小時後的圖譜發現 50.4°附近出現 Cu(200)的波 峰,其餘波峰則和不退火情況下相同;而在(c)600°C 退火一小時 後的繞射圖譜則出現明顯的不同,鉭基氮化物薄膜在此條件退火後, 明顯地對阻礙銅金屬和砷化鎵基板間的擴散行為,完全失去效力,產 生諸如 Cu3As(24.5°) 、CuGa2(44.6°)和 TaAs(30.7°、60.8°)等化 合物。由上述 XRD 分析結果得知,鉭基氮化物可以在 500℃溫度下 穩定存在一小時,而在 600℃時則失去阻礙層能力。 為了更進一步瞭解鉭基氮化物在幾度時,發生阻礙失效情形,於 是以同樣實驗參數再次成長一批試片(表 4-3 之 S 組),同樣在真空 下,進行 575℃及 625℃退火各一小時,圖 4-8 為其 XRD 繞射圖;發 現鉭基氮化物薄膜在 575℃退火一小時的繞射圖形,和之前 500℃退 火一小時相比較,幾乎是相同的,而 625℃退火一小時的繞射圖形則 顯示鉭基氮化物薄膜失去阻礙效能;綜合以上實驗結果,證實鉭基氮 化物擴散阻礙層可以阻礙砷化鎵和銅金屬間的擴散行為,耐熱溫度在 575℃以下。. 4.3.3 鉭基氮化物擴散阻礙層在玻璃基板上耐火程度之探討 分析. 為了對我們成長出的鉭基氮化物的特性作瞭解,我們在玻璃基板 35.
(36) 上成長一層厚約 575nm 的鉭基氮化物薄膜,然後真空封管,分別進 行 400℃、500℃、600℃的退火各一小時(表 4-3 之 T 組) ,圖 4-9 為 其 X 光繞射圖譜。同樣發現在繞射角 33°至 40°間有一寬廣波峰,顯 示成長的鉭基氮化物薄膜應是非晶質的結構,且晶粒大小不規則;不 退火情形下的 XRD 繞射結果和 400℃、500℃各退火一小時的 XRD 繞射結果是一樣的,但在退火 600℃一小時後,發現在 33.9°、36.6° 和 38.7°附近,分別出現 Ta2N (100)、Ta2N (002)和 Ta2N (101)的尖銳 波峰,另外在 50.7°附近,出現 Ta2N (102)的波峰,推估是非晶質的 鉭基氮化物薄膜在經過 600℃的高溫退火後,這些結晶面再成長的傾 向較強,使薄膜緣這些結構面快速成長的結果。 經由上述實驗結果,再參照 4.3.2 節結果,得知在真空環境下退 火,鉭基氮化物可以在 575℃熱穩定一小時,而在 600℃退火一小時 後,會出現銅砷和鉭砷等化合物,推估鉭基氮化物阻礙層行為的失 效,是源自於本身晶粒再成長的結果,使得銅原子循此晶界途徑,擴 散進入砷化鎵基板,也可能是砷化鎵本身無法承受此高溫,致使砷或 鎵原子循上述晶界往上擴散進入銅金屬層,產生銅砷或銅鎵化合物。. 4.4 不同氣氛下退火之 XRD 分析 4.4.1 鎵氣氛環境退火之 XRD 分析 我們期望在退火時加入一些氣氛,壓抑住砷化鎵基板中,砷或鎵 原子在高溫時逸出的問題。因此以 3.2.3 節所述方法進行封管,並在 石英管內加入雜質源鎵元素。 圖 4-10(b) 、 (c)為鉭基氮化物(212 nm)在砷化鎵基板和銅金 屬(284 nm)間(表 4-3 之 U 組試片),在鎵氣氛環境的石英管內, 36.
(37) 分別進行 600℃及 650℃退火一小時後的 X 光繞射圖譜。 比較 X 光繞射圖譜發現在 650℃熱處理情況後,在 40.4°出現 Cu3As(211)的尖銳波峰,另外在 30.7°與 60.8°出現 TaAs(004)與 (211)波峰,而 600℃熱處理後則無 Cu3As、TaAs 等化合物產生, 推估是砷化鎵基板的砷元素循 Ta2N(100) 、TaN(101)和 Ta2N(101) (分別在 33.9°、36.8°和 38.7°)等多晶質晶界擴散上來並與銅元素和 鉭基氮化物的鉭元素結合結果。在 35.2°、44.6°和 49.4°出現 CuGa2 (101)、CuGa2(102)和 Ga3Ta5(310)波峰,並使 Cu(111)(出 現在 43.3°)的結構面消失,而這些化合物則不出現於 600℃熱處理情 況下,推估是砷化鎵基板的鎵元素循上述多晶質晶界擴散上來,並與 銅元素和鉭基氮化物的鉭元素結合結果。 上述鉭基氮化物薄膜在鎵氣氛下退火,出現 TaAs、Cu3As、 CuG a2、Ga3Ta5 等化合物,雖然我們推估原因,但並不能確認這些化 合物是基板高溫破壞後,砷或鎵元素逸出並和銅膜結合的結果,抑或 是鎵元素在淬冷後沈積在銅膜上的結果。 為進一步瞭解這些化合物的來源,實驗進行時,我們以玻璃基板 作為實驗對照組,以完全相同的條件步驟成長薄膜、封管,並於 600 ℃熱處理一小時(表 4-3 之 W 組) 。比較玻璃基板和砷化鎵基板的繞 射圖譜,如圖 4-10(a) 、 (b)所示,結果顯示在玻璃基板上,除了鉭 基氮化物和銅膜的波峰外,並沒有銅鎵等的化合物,由此證實銅鎵化 合物的形成,不是石英管氣氛中的鎵原子在淬冷後沈積銅膜上的,而 是銅原子循高溫再結晶的鉭基氮化物晶界往下擴散到砷化鎵基板,並 結合成為銅鎵化合物。. 4.4.2 砷氣氛環境退火之 XRD 分析 37.
(38) 我們以和上節同樣的成長條件,在砷化鎵基板(表 4-3 之 V 組) 和玻璃基板上(表 4-3 之 X 組)成長鉭基氮化物和銅膜,並在砷氣氛 環境下進行封管退火,希望測試在砷氣氛環境下,能否壓抑砷原子在 高溫熱處理下,從砷化鎵基板逸出的問題。封管過程如圖 3-5 所示之 熱蝕刻(thermal etching)方式,首先對石英管預烤以趕走水氣,然 後在石英管內放入砷化物(As4 或 As2O3) ,以較低溫的火焰加熱砷 化物,慢慢驅趕不純的氧化物,使砷化物還原成銀亮顆粒的純砷元 素,並在石英管中央部位燒出一凹槽,以區隔砷元素和試片,最後再 放入試片,其後步驟同前述方式。我們分別在 575℃、600℃、650℃ 溫度下退火一小時,其 X 光繞射圖譜如圖 4-11 所示。從圖中明顯看 出砷氣氛下的退火,出現一些諸如 Cu5As2、CuAs 2、β-Cu3As 的銅 砷化合物,這些化合物在前述幾節的分析中是不曾出現的,加上在玻 璃基板上同樣條件的分析結果,我們可以證實這些銅砷化合物是石英 管中的砷元素在淬冷後沈積在銅膜表面上的結果,因此在砷氣氛環境 下,並不能改善鉭基氮化物擴散阻礙層壓制砷化鎵中砷原子先行逸出 的問題。. 4.5 掃瞄式電子顯微鏡下退火後晶粒大小的探討 晶粒大小在退火前後的變化是我們有興趣的,因此我們希望藉由 電子顯微鏡觀察銅膜和鉭基氮化物薄膜晶粒在退火前後的改變,在進 行試片觀察前,適當的試片處理是必須的。相關文獻裡曾用 Secco etching 的方法做酸蝕刻〔17〕 ,然後在光學顯微鏡下觀察蝕刻後的薄 膜表面;因為高溫退火使得一些新的介面相化合物生成,而這些新的 介面化合物一般認為是薄膜完全劣化前的先行產物,同樣擴散阻礙層 38.
(39) 在失效前也會發生如上情形,且此介面化合物對相同濃度溶液的被蝕 刻速率和原薄膜不同,因此藉觀察酸蝕刻後薄膜表面的新介面化合 物,就可推論擴散阻礙層破壞失效的溫度,大約是在幾度時發生。 銅的酸蝕刻溶液一般是用硝酸(HNO3) ,對鉭基氮化物則是採用 硫酸(H2SO4)加氫氟酸(HF);由於我們有興趣的是對退火後的晶 粒大小進行電子顯微鏡的觀測,所以並不需完全照 Secco etching 的方 法,吃掉全部薄膜進行觀測,而是希望吃出晶粒成長後的晶界即可, 因此採低濃度配置進行酸蝕刻。我們以表 4-3 之 Y 組的成長條件在矽 基板上濺鍍一層膜厚約 387 nm 的鉭基氮化物,另一組試片(表 4-3 之 Z 組)則在鉭基氮化物上繼續濺鍍一層厚約 227 nm 銅膜,而後兩 組試片分別進行真空封管並做 575℃、650℃、700℃的退火各一小時; 對於鉭基氮化物,我們以 HF:H2SO4:H2O=1:5:7 的比例蝕刻 5 分鐘;銅膜則採用 HNO3:H2O = 1:50 的比例蝕刻 3 分鐘,結果如 圖 4-12 和圖 4-13 所示。在圖 4-12 中,我們觀察蝕刻後的表面,發現 類似晶粒的塊狀物,推估 700℃退火的塊狀物大小約為 500 nm,650 ℃退火的塊狀物大小約為 350 nm,這塊狀物的大小尺寸,明顯隨著 退火溫度升高而增加,但與我們所知非晶質或多晶質的晶粒大小,有 明顯過大的傾向,我們認為這是在高溫退火下,提供鉭基氮化物分子 一個能量,讓其循著最佳排列和最低能量的狀況下,所誘發晶粒與晶 粒間逐漸結合成塊的現象。在圖 4-13 中,我們觀察銅膜被蝕刻後的 表面,並沒有吃出晶界現象,推認原因可能是蝕刻溶液比例、蝕刻時 間等配合不佳緣故。. 4.6 電子微探針分析儀(EPMA)定量分析結果. 39.
(40) EPMA 之電子束所涵蓋的試片面積,約為電子束直徑 1µm,貫穿 試片深度亦約為 1µm,因此我們用表 4-2 之 M 組和 P 組成長條件, 在矽基板上成長一層約 1μm 厚的鉭基氮化物,希望能藉由 EPMA 的 定量分析和 XPS 定量分析結果作一比較。定量分析在試片表面上每 間格 1mm 取一個分析值,共取三個實驗值。 實驗結果見表 4-4,顯示鉭原子個數和氮原子個數的比值分別為 6.87 和 4.96,明顯鉭原子個數多於氮原子個數,和先前 XPS 分析的 氮原子個數多於鉭原子個數的結果不太一致;我們認為是 EPMA 在 做定量分析時,對像鉭元素等重元素內含輕元素的分析時,解析度變 差的緣故。EPMA 對固態元素的定性和定量分析,適用於原子序大於 11(Na)的元素,分析能力約為 100 ppm,解析範圍約 1µm;單獨作定 性分析則可測至原子序 5(B)以上的元素。若分析薄膜試片有破裂、斷 面,或不規則顆粒突起,都會影響分析精度,因此試片表面越平整, 則分析精度越準確〔42〕;基於上述各原因,我們認為鉭基氮化物做 EPMA 的定量分析時,是會遭遇到瓶頸的。. 4.7 電性量測 當成長鉭基氮化物時,腔體內的氮氣流量越高,鉭基氮化物內的 氮成分則相對越高,電阻率也會隨氮含量增加而增加,但相對地,氮 含量越高,則阻礙效果越好,能承受更高溫度的熱處理〔17、30〕。 我們對表 4-2 之 M 組的鉭基氮化物進行電阻率量測,得到電阻率約 為 290~330μΩ-cm,和表 2-1 相關文獻測出的電阻率結果差不多 〔17〕。. 40.
(41) 第五章 結論 本實驗中,我們可在 2×10-3 torr 的濺鍍壓力、射頻功率 200W 和 氬氣流量 9.5 sccm、氮氣流量 0.5 sccm 下,在砷化鎵基板上成長出非 晶質的鉭基氮化物,並可歸納以下一些結論:. 1. 鉭基氮化物擴散阻礙層存在銅金屬和砷化鎵基板間,的確可以避免 銅鎵或銅砷化合物的介面相產生。 2. 非晶質的鉭基氮化物可經由 600℃左右的熱處理退火後,產生如 (100)、(002)、(101)、(102)等結構面的多晶 Ta2N。 3. 鉭基氮化物在銅膜和砷化鎵基板間,進行真空氣氛、溫度 575℃下 之退火一小時,可以壓抑銅金屬和砷化鎵間之擴散行為,而在 600 ℃以上的溫度,則發現鉭砷、銅鎵等化合物。 4. 在雜質源砷或鎵氣氛下進行熱處理,並不能有效提升耐熱溫度和壓 抑銅金屬和砷化鎵間之擴散問題。 5. 鉭基氮化物的電阻率約為 280~330μΩ-cm。. 41.
(42) 參考文獻 1. R. E. Williams, Gallium Arsenide Processing Techniques, 學風科學 圖書出版社, p.17-51, 1984. 2. M. Passlack, M. Hong, J. P. Kwo, L.W. Tu, “Recombination velocity at oxide-GaAs interfaces fabricated by in situ molecular beam epitaxy,” Applied Physics Letters, Vol. 68, p.3605-3607, 1996. 3. 莊達人, VLSI 製造技術, 高立圖書出版社, p.146-181, 1998. 4. Alexander E. Braun, “Aluminum Persists as Copper Age Dawns,” Semiconductor International, p.58-66, 1999. 5. 顧子琨, “極大型積體電路之銅連結線技術,” 電子月刊第五卷第六 期, p.117-133, 1999. 6. Y. Igarashi, T. Yamanobe, T. Ito, “Thermal stability of copper interconnects fabricated by dry-etching process,” Thin Solid films, 262, p.124-128, 1995. 7. S. Wolf, Silicon processing for the VLSI Era– Volume Ⅱ, Lattice Press. , p.84-175, 1990. 8. H. Ono, T. Nakano, T. Ohta, “Diffusion barrier effects of transition metals for Cu/M/Si multilayers (M = Cr, Ti, Nb, Mo, Ta, W),” Applied Physics Letters, 64, p.1511-1513, 1994. 9. J. Chuang, S. Tu, M. Chen, “Sputter-deposited Mo and reactively sputter-deposited Mo-N films as barrier layers against Cu diffusion,” Thin Solid Films, 346, p.299-306, 1999. 10. J. O. Olowolage, J. Li, J. W. Mayer, “Effects of oxygen in TiN x on the diffusion of Cu in Cu/TiN/Al and Cu/TiNx/Si structures,” Applied Physics Letters, 58, p.469-471, 1991. 11. S. Q. Wang, S. Suthar, C. Hoeflich, B. J. Burrow, “Reactively sputtered TiN as a diffusion barrier between Cu and Si,” J. Appl. 42.
(43) Phys., 68, p.5176-5183, 1990. 12. S. Q. Wang, S. Suthar, C. Hoeflich, B. J. Burrow, “Diffusion barrier properties of TiW between Si and Cu,” J. Appl. Phys., 73, p.2301-2306, 1993. 13. E. Kolawa, J. S. Chen, J. S. Reid, P, J. Pokelan, M.A. Nicolet, “Tantalum-based diffusion barriers in Si/Cu VLSI metallizations,” J. Appl. Phys., 70, p.1369, 1991. 14. R. Hoogeveen, M. Moske, H. Geisler, K. Samwer, “Texture and phase transformation of sputter-deposited metastable Ta films and Ta/Cu multilayers,” Thin Solid Films 275, p.203-206, 1996. 15. M. Stavrev, D. Fischer, C. Wenzel, K. Drescher, N. Mattern, “Crystallographic and morphological characterization of reactively sputtered Ta, Ta-N and Ta-N-O thin films,” Thin Solid Films, 307, p.79-88, 1997. 16. T. Oku, E. Kawakami, M. Uekubo, K. Takahiro, S. Yamaguchi, M. Murakami, “Diffusion barrier property of TaN between Si and Cu,” Appl. Surf. Sci., 99, p.265-272, 1996. 17. K. Min, K. Chun, K. Kim, “Comparative study of tantalum and tantalum nitrides (Ta2N and TaN) as a diffusion barrier for Cu metallization,” J. Vac. Sci. Technol. B, Vol. 14, No. 5, p.3263-3269, 1996. 18. M. Stavrev, C. Wenzel, A. Moller, K. Drescher, “Sputtering of tantalum-based diffusion barriers in Si/Cu metallization: effects of gas pressure and composition,” Applied Surface Science, 91, p.257-262, 1995. 19. J. Chuang, M. Chen, “Properties of thin Ta-N films reactively sputtered on Cu/SiO 2/Si substrates,” Thin Solid Films, 322, p.213-217, 1998.. 43.
(44) 20. M. Takeyama, A. Noya, T. Sase, A. Ohta, “Properties of TaNx films as diffusion barriers in the thermally stable Cu/Si contact systems,” J. Vac. Sci. Technol. B, 14, p.674-678, 1996. 21. K. Wakasugi, M. Tokunaga, T. Sumita, H. Kubota, M. Nagata, Y. Honda, “Superconductivity of reactively sputtered TaN film for ULSI process,” Physica B, 239, p.29-31, 1997. 22. X. Sun, E. Kolawa, J. Chen, J. S. Reid, M. A. Nicolet, “Properties of reactively sputter-deposited Ta-N thin films,” Thin Solid Films, 236, p.347-351, 1993. 23. M. Stavrev, D. Fischer, A. Preub, C. Wenzel, N. Mattern, “Study of nanocrystalline. Ta(N,O). diffusion. barriers. for. use. in. Cu. metallization,” Microelectronic Engineering, 33, p.269-275, 1997. 24. M. H. Tsai, S. C. Sun, C. P. Lee, H. T. Chiu, C. E. Tsai, S. J. Chuang, S. C. Wu, “Metal-organic chemical vapor deposition of tantalum nitride barrier layers for ULSI applications,” Thin Solid Films, 270, p.531-536, 1996. 25. C. Lee, Y. Shin, “Ta-Si-N as a diffusion barrier between Cu and Si,” Materials chemistry and Physics, Vol. 57, p.17-22, 1998. 26. Y. Lee, B. Suh, M. S. Kwon, C. Park, “Barrier properties and failure mechanism of Ta-Si-N thin films for Cu interconnection,” J. Appl. Phys., Vol. 85, No. 3, p.1927-1934, 1999. 27. Y. Lee, B. Suh, S. Rha, C. Park, “Structural and chemical stability of Ta-Si-N thin film between Si and Cu,” Thin Solid Films, 320, p.141-146, 1998. 28. C. Y. Chang, F. Kai, GaAs High-Speed devices, John Wiley & Sons Inc., Chap. 2, 1994. 29. N. Terao, “Structure of Tantalum Nitrides,” Japan J. Appl. Phys., 10, p.248-259, 1971. 30. 許振聲、陳錦山, “ 鉭基氮化物擴散阻礙層在銅金屬化系統之失效 44.
(45) 機制,” 真空科技卷12, p.5-17, 1999. 31. C.Y. Chang, Francis Kai, GaAs High-Speed Devices, John Wiley & Sons Inc., p.288-294, 1994. 32. K. Kim, M. Kniffin, R. Sinclair, C. R. Helms, “Interfacial reactions in the Ti/GaAs system,” J. Vac. Sci. Technol. A, Vol. 6, No. 3, p.1473-1477, 1988. 33. M. Cuziewicz, A. Piotrowska, E. Kaminska, K. Golaszewska, A. Turos, E. Mizera, A. Winiarski, J. Szade, “Characteristics of sputter-deposited TiN, ZrB2 and W2B diffusion barriers for advanced metallizations to GaAs,” Solid-State Electronics, 43, p.1055-1061, 1999. 34. T. Feng, A. Dimoulas, A. Christou, G. Constantinidis, Z. Hatzopoulos, “Failure mechanisms of GaAs MESFETs with Cu/Refractory Metallized Gates,” Microelectron. Reliab., Vol. 37, p.1699-1702, 1997. 35. G. Scherb, D. M. Kolb, “Cu deposition onto n-GaAs (100): optical and current transient studies,” Journal of Electroanalytical Chemistry, 396, p.151-159, 1995. 36. R. Beyers, K. B. Kim, R. Sinclair, “Phase equilibria in metal-gallium-arsenic systems: Thermodynamic considerations for metallization materials,” J. Appl. Phys., 61, p.2195-2202, 1987. 37. V. S. C. Len, R. E. Hurley, N. McCusker, D. W. McNeill, B. M. Armstrong, H. S. Gamble, “An investigation into the performance of diffusion. barrier. materials. against. copper. diffusion. using. metal-oxide-semiconductor (MOS) capacitor structures,” Solid-State Electronics, 43, p.1045-1049, 1999. 38. 李世鴻, 半導體工程原理, 全威圖書, p.401-404, 1998. 39. D. L. Smith, “Thin-Film Deposition Principles and Practice,” The 45.
(46) McGraw-Hill Companies Inc., p.483-499, 1999. 40. M. Ohring “The Materials Science of Thin Film,” Academic Press, p.121-123, 1992. 41. 黃振昌, “表面分析儀器-X光光電子能譜儀,” 精密儀器發展中心, p.5-6, 1998. 42. 許樹恩, 吳泰伯, 材料科學叢書1-X光繞射原理與材料結構分析, Chap. 20, p.532-533, 1996. 43. C. D. Wagner, W. M. Riggs, L. E. Davis, J. F. Moulder, G. E. muilenberg, “Handbook of X-ray Photoelectron Spectroscopy,” Perkin-elmer Corp., 1984. 44. A. O. Ibidumni, R. L. MaSaitis, R. L. Opila, A. J. Davenport, H. S. Isaacs, J. A. Taylor, “Characterization of the oxidation of tantalum nitride,” Surface and Interface analysis, Vol. 20, p.559-564, 1993.. 46.
(47) 圖 2-1 f.c.c 晶體結構. 圖 2-2 砷化鎵晶體結構(閃鋅礦結構,zincblende). 47.
(48) 圖 2-3 砷化鎵之電子漂移速度圖. 圖 2-4 砷化鎵之能隙圖 48.
(49) substrat shutte ion. valv targe. N2 Ar. MFC turbo pum. cold water power. heat 圖 3-1 濺鍍實驗系統示意圖 49. mechanica l.
(50) TaNX 膜濺鍍. XRD 分析. XPS 分析. Cu 膜濺鍍. 鎵氣氛環境封管. 真空環境封管. 砷氣氛環境封管. 不同溫度退火. XRD 分析. SEM 分析 圖 3-2 實驗流程架構示意圖. 50.
(51) 圖 3-3 真空封管系統架構圖. 圖 3-4 真空封管過程示意圖 51.
(52) 圖 3-5 置放雜質源的封管示意圖. 圖 3-6 出爐時冷卻方法. 52.
(53) 圖 4-1 鉭基氮化物外層電子 4f7/2 和 4f5/2 軌域的束縛能(a)Ta(b) Ta2N(c)TaN. 53.
(54) 圖 4-2 鉭基氮化物外層電子 4f7/2 和 4f5/2 軌域的束縛能(a)150W Si (b)150W Glass(c)200W Si(d)200W Glass 54.
(55) 圖 4-3 表 4-2 中 M、N、O、P 組的 X 光繞射圖譜(a) M (200W, 9.5/0.5) (b) N (150W, 9.5/0.5) (c) O (150W, 13/0.5) (d) P (150W, 18/0.5). 55.
(56) 圖 4-4 表 4-2 中Ⅰ、Ⅱ、Ⅲ、Ⅳ組的 X 光繞射圖譜(a)Ⅰ(50W, 5/0) (b) Ⅱ(50W, 5/0.5) (c) Ⅲ(100W, 7.5/0) (d) Ⅳ(100W, 7.5/1) 56.
(57) 圖 4-5 表 4-2 中Ⅴ、Ⅵ、Ⅶ、Ⅷ組的 X 光繞射圖譜(a)Ⅴ(150W, 7.5/1) (b)Ⅵ(175W, 6.5/1) (c)Ⅶ(175W, 8.5/1) (d)Ⅷ(200W, 9.5/0.5). 57.
(58) 圖 4-6 真空氣氛、不退火情況下的 X 光繞射圖譜(a)無鉭基氮化物 阻礙層(Cu/GaAs) (b)有鉭基氮化物阻礙層(Cu/TaNx/GaAs) 58.
(59) 圖 4-7. 真空氣氛、不同熱處理一小時下,鉭基氮化物在砷化鎵基板 和銅膜間〔Cu(324nm) / TaNx (141nm) / GaAs〕的 X 光繞射 圖譜(a)未退火(b)500℃(c)600℃ 59.
(60) 圖 4-8 鉭 基 氮 化 物 在 砷 化 鎵 基 板 和 銅 膜 間 〔 Cu(284nm)/TaNx (212nm)/GaAs〕退火一小時後的 X 光繞射圖譜(a)575℃ (b) 625℃ 60.
(61) 圖 4-9 鉭基氮化物在玻璃基板上退火一小時後的 X 光繞射圖譜(a) 未退火(b)400℃(c)500℃(d)600℃ 61.
(62) 圖 4-10 鎵氣氛環境、不同基板〔Cu (284nm)/ TaNx (212nm)/ Sub.〕、 不同溫度下退火一小時後的 X 光繞射圖譜(a)600℃玻璃基 板(b)600℃砷化鎵基板(c)650℃砷化鎵基板 62.
(63) 圖 4-11 砷氣氛環境、不同基板〔Cu (284nm)/ TaNx (212nm)/ Sub.〕、 不同溫度下退火一小時後的 X 光繞射圖譜(a)575℃砷化鎵 基板(a1)575℃玻璃基板(b)600℃砷化鎵基板(b1)600 ℃玻璃基板(c)650℃砷化鎵基板 63.
(64) (a). (b). 圖 4-12 真空氣氛下(a)不退火(b)575℃退火一小時後,鉭基氮化 物在矽基板上(TaNx/Si)經酸蝕刻後表面之 SEM 圖. 64.
(65) (c). (d). 圖 4-12(續上)真空氣氛下(c)650℃(d)700℃退火一小時後,鉭 基氮化物在矽基板上(TaNx/Si)經酸蝕刻後表面之 SEM 圖. 65.
(66) (a). (b). 圖 4-13 真空氣氛下(a)575℃(b)700℃退火一小時後,銅膜在鉭基氮化物上 (Cu/TaNx/Si)經酸蝕刻後表面之 SEM 圖. 66.
(67) 晶格常數( Lattice constant ). 5.65 Å. 密度( Density ). 5.317 g/cm3. 原子密度( Atomic density ). 4.4279×1022 atoms/cm3. 原子量( Molecular weight ). 144.642. 線性膨脹係數( Linear expansion coefficient ). 5.73×10-6 K-1. 比熱( Specific heat ). 0.327 J/g-K. 晶格熱導率( Lattice thermal conductivity). 0.55 W/cm-K. 相對介電常數( Relative dielectric constant ). 12.85. 能隙( Bandgap ). 1.423 eV. 臨限電場( Threshold field ). 3.3 KV/cm. 最大漂移速度( Peak drift velocity ). 2.1×107 cm/s. 電子遷移率( Electron mobility ). 8500 cm2/V-s. 電洞遷移率( Hole mobility ). 400 cm2/V-s. 熔點( Melting point ). 1238℃. 表 2-1 砷化鎵在室溫時(300 K)之特性. 56.
(68) 鉭金屬和鉭基氮化物 的種類. 結 構. 晶格常數 (Lattice parameter). 電阻率 (μm-cm). α- Ta. Body-centered cubic system. a = 3.305 Å. 40. β- Ta. Tetragonal system. a = 5.34 Å c = 9.94 Å. 150. TaN~0.05 (β-phase ) Ta2N ( ã-phase ). Body-centered cubic system. a = 3.370 Å. 80. Hexagonal close-packed system. ä – TaN. Hexagonal system. å - TaN. Hexagonal system. Ta5N6. Hexagonal system. Ta4N5. Tetragonal system. Ta3N5. Tetragonal system. as = 5.283 Å cs = 4.928 Å a = 2.938 Å c = 2.869 Å a = 5.188 Å c = 2.903 Å a = 5.175 Å c = 10.307 Å a = 6.835 Å c = 4.272 Å a = 10.229 Å c = 3.875 Å. 表 2-2 鉭金屬和鉭基氮化物之結構特性 57. 240 250~330 250~330 -.
(69) 金屬/砷化鎵. 穩定溫度. 不穩定 發生溫度. Au/GaAs. -. 250℃. Au causes dissociation of GaAs Ga migrates into Au. Pt/n-GaAs. -. 350℃. PtAs2、PtGa. 不穩定時產生現象. Ti/GaAs. 300℃. 400-500℃. W/GaAs. 500-550℃. 600℃. Au/TiW/GaAs. 600℃-15hr. -. WSi0.64/GaAs. 850℃. -. MoSi2/GaAs. 800℃. -. Si/Ta/Si/GaAs. 800℃. Cu/Ti/GaAs. -. 產生 Ti/TixGa1-x/TiAs/GaAs 的結構 低溫退火產生 Ti2Ga3、TiAs 高溫退火產生 Ti5Ga4、Ti5As3 介面附著力變差且存在高應力,使得鎢膜剝 落 (The W film peels off due to poor adhesion and high stress in the interface). 920℃-20min TaSi2 350-450℃. CuTi、Cu3Ti. 表 2-3 砷化鎵基板上擴散阻礙層之相關文獻整理 [ref. 31-34] 58.
(70) Cu d (A). Intensity(I/I0). (h k l). Cubic 2Θ. 2.088 1.808 1.278 1.090 1.0436 0.9038. 100 46 20 17 5 3. 111 200 220 311 222 400. 43.3 50.4 74.1 89.9 95.1 116.9. CuGa2 d (A). Intensity(I/I0). (h k l). Tetragonal 2Θ. 2.920 2.829 2.545 2.031 2.000 1.946 1.893 1.650 1.603 1.460 1.415 1.395 1.375 1.297 1.273 1.265 1.237 1.179 1.168 1.161 1.144. 7 35 80 100 80 16 3 6 1 6 30 30 <1 <1 2 5 14 10 <1 35 18. 002 100 101 102 110 003 111 112 103 004 200 113 201 104 202 210 211 114 005 212 203. 30.6 31.6 35.2 44.6 45.3 46.6 48.0 55.7 57.4 63.7 65.9 67.0 68.1 72.9 74.5 75.0 77.0 81.6 82.5 83.1 84.7. Cu3As d (A). Intensity(I/I0). (h k l). Hexagonal 2Θ. 3.63 3.21 2.56 2.37 2.23 2.08 2.02. 40 20 10 30 30 100 100. 002/110 111 112 202 211 300 113. 24.5 27.8 35.1 38.0 40.4 43.5 44.9. 表 3-1 銅、銅鎵、銅砷化合物之 X 光繞射資料 59.
數據

+7
相關文件
volume suppressed mass: (TeV) 2 /M P ∼ 10 −4 eV → mm range can be experimentally tested for any number of extra dimensions - Light U(1) gauge bosons: no derivative couplings. =>
For pedagogical purposes, let us start consideration from a simple one-dimensional (1D) system, where electrons are confined to a chain parallel to the x axis. As it is well known
The observed small neutrino masses strongly suggest the presence of super heavy Majorana neutrinos N. Out-of-thermal equilibrium processes may be easily realized around the
Define instead the imaginary.. potential, magnetic field, lattice…) Dirac-BdG Hamiltonian:. with small, and matrix
incapable to extract any quantities from QCD, nor to tackle the most interesting physics, namely, the spontaneously chiral symmetry breaking and the color confinement..
(1) Determine a hypersurface on which matching condition is given.. (2) Determine a
• Formation of massive primordial stars as origin of objects in the early universe. • Supernova explosions might be visible to the most
The difference resulted from the co- existence of two kinds of words in Buddhist scriptures a foreign words in which di- syllabic words are dominant, and most of them are the
