Dual Excited-State Intramolecular Proton Transfer Reaction in
3-Hydroxy-2-(pyridin-2-yl)-4H-chromen-4-one
Chyi-Lin Chen,† Chun-Wei Lin,†Cheng-Chih Hsieh,†Chin-Hung Lai,† Gene-Hsiang Lee,‡ Chih-Chieh Wang,§ and Pi-Tai Chou*,†
Department of Chemistry, National Taiwan UniVersity, Taipei, 106, Taiwan, R.O.C., Instrumentation Center, National Taiwan UniVersity, Taipei, Taiwan, R.O.C., and Department of Chemistry, Soochow UniVersity, Taipei, Taiwan, R.O.C.
ReceiVed: October 14, 2008; ReVised Manuscript ReceiVed: NoVember 19, 2008
The synthesis, characterization and fundamental of the dual excited-state proton-transfer properties of 3-hydroxy-2-(pyridin-2-yl)-4H-chromen-4-one (1a) are reported. In the electronic ground state, there exist two competitive hydrogen bonding (HB) isomers for 1a. Conformer 1a(O) reveals a five-membered ring HB structure between O-H and carbonyl oxygen, while conformer 1a(N) possesses a six-membered ring HB formation between O-H and pyridyl nitrogen. In a single crystal, the X-ray crystallography unveils an exclusive formation of conformer 1a(N). In solution such as CH2Cl2, 1a(O) and 1a(N) are in equilibrium, and their respective absorption chromophores are significantly different due to different degrees of hydrogen-bond induced π electron delocalization. Upon excitation, both conformers 1a(O) and 1a(N) undergo excited-state intramolecular proton transfer (ESIPT) reaction. Following ESIPT, 1a(O) gives rise to a tautomer emission maximized at 534 nm in CH2Cl2. Conversely, due to dominant radiationless quenching processes the tautomer emission for 1a(N) cannot be obtained with a steady-state manner but can be resolved from time-resolved fluorescence. Time resolved fluorescence estimates an equilibrium constant of 27 ( 5 in favor of 1a(N) in CH2Cl2. Ultrafast ESIPT also takes place for the unique 1a(N) form in the crystal. Due to the prohibition of quenching processes in the solid state, bright tautomer emission maximized at 540 nm is resolved for 1a(N) (Φf ∼ 0.3). The interplay between two HB conformers with on(1a(O))/off(1a(N)) character in tautomer emission may find future applications such as the recognition of organic Lewis acid/base in organic solvents.
1. Introduction
Proton transfer has been ubiquitously found in chemical and
biological reactions.1Among various types of proton transfer
patterns, the category relevant to excited-state intramolecular proton transfer (ESIPT) has received much attention owing to the simplicity of its reaction pattern. Numerous ESIPT molecules have been strategically designed and synthesized in an aim to
shed light on the fundamental in proton-transfer mechanism2
and/or to explore their potential applications (we cite only a
small selection of representative reports).3
In terms of hydrogen bonding (HB) structure, most ESIPT molecules possess either five- or six- membered ring hydrogen bonds between O-H (or N-H) and CdO (or pyridinic nitrogen). Recently, we have explored a new class of ESIPT molecules bearing a seven-membered ring HB structure akin
to the core chromophore of the green fluorescence protein.4
Among those ESIPT molecules possessing five-membered-ring hydrogen bond, 3-hydroxyflavone (3HF, see Scheme 1) has been considered a paradigm ever since its ESIPT property was
discovered by Kasha and co-workers.5In nonpolar solvent such
as cyclohexane, 3HF exhibits the S0 f S1 (ππ*) transition
maximized at 340 and 354 nm, while the fluorescence of 3HF shows an anomalously large Stokes shifted band maximized at
∼526 nm (Φ ) 0.36, τf) 3 ns).6Upon methylating 3HF to
form 3-methoxyflavone, 3-methoxyflavone shows normal Stokes
shifted fluorescence maximized at∼360 nm in cyclohexane.6d
It is thus concluded that ESIPT takes place from the hydroxyl proton to the carbonyl oxygen, giving rise to the proton-transfer tautomer emission. Due to its relatively weak, five-membered ring hydrogen bond (c.f. six-membered ring hydrogen bond), 3HF was once adopted as a model compound to demonstrate appreciable barrier during ESIPT. Subsequently, it was found that the retardation of ESIPT at low temperature is mainly due to the external HB interaction caused by traces of protic solvent
impurity (e.g., water, alcohols) present in the nonpolar solvents.6a
In the dry, extensively purified nonpolar solvent such as cyclohexane, the time-resolved measurement rendered an
ul-trafast time of ESIPT for 3HF (τpt< 240 fs),6f-iand ESIPT is
essentially barrierless, which is perhaps induced by the low
frequency, skeletal motions associated with the hydrogen bond.7
In yet another approach, study of ESIPT coupled excited-state intramolecular charge transfer (ESICT) by strategically functionalizing 3HF with electron donating substituents has been
an intriguing fundamental topic. Using 4′
-N,N-diethylamino-3-hydroxyflavone (see Scheme 2) as an example,8upon excitation,
adiabatic, ultrafast charge transfer takes place from the diethyl-amino moiety to the carbonyl oxygen, resulting in a charge transfer state (CT*, * denotes the electronically excited state, see Scheme 2). After solvent relaxation, the equilibrated CT*,
CTeq*, then undergoes CTeq* f PTeq* (PT denotes the proton
transfer tautomer) proton transfer. Due to the great difference
in equilibrium polarization between CTeq* and PTeq* (see
Scheme 2), a solvent polarity induced barrier is introduced,
* Corresponding author. Telephone: 33663894. Fax: +886-2-23695208. E-mail: [email protected].
†Department of Chemistry, National Taiwan University. ‡Instrumentation Center, National Taiwan University. §Department of Chemistry, Soochow University.
10.1021/jp809072a CCC: $40.75 2009 American Chemical Society
Published on Web 12/15/2008
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
which then channels into the proton transfer coordinate. Such a nuclear (hydrogen atom) motion coupled solvent polarization effect has been rationalized by a mechanism similar to the
weak-coupling electron transfer described by the Marcus theory.8c,9
Due to the relatively slow, solvent induced barrier coupled ESIPT rate, the associated dual emission and its ratiometric changes as a function of solvent polarity and/or electron donating/accepting strength have been exploited in several potential applications such as solvent polarity and metal ion
sensors, etc.10
Herein, we report the synthesis, characterization and photo-physical properties of a new class of functionalized 3HF derivatives. In this work, 3-hydroxy-2-(pyridin-2-yl)-4H-chromen-4-one (1a, see Scheme 1) was strategically designed such that the hydrogen bond could potentially be located at either OH---OdC or OH---N (pyridine) site, forming five- (1a(O)) and six-(1a(N)) membered ring HB configurations, respectively. For a comparative study, 3-hydroxy-2-(pyridin-4-yl)-4H-chromen-4-one (1b, Scheme 1) was also synthesized. Due to the lack of HB formation between the hydroxyl group and the pyridinic nitrogen, 1b is expected to possess a unique five membered ring HB configuration between the hydroxyl group and carbonyl oxygen similar to that of 3HF. In this study, our aim mainly focuses on the spectroscopy and dynamics of the competitive intramolecular HB equilibrium between 1a(O) and 1a(N) and their associated ESIPT properties. The resulting different ESIPT
pathways and hence the possible ratiometric emission may provide latent applications in e.g. molecular recognition. Details of the results and discussion are elaborated in the following sections.
2. Experimental Section
Scheme 3 depicts synthetic routes of 1a and 1b. All reactions were performed under nitrogen. Solvents were distilled from appropriate drying agents prior to use. Commercially available reagents were used without further purification unless otherwise stated. All reactions were monitored by TLC with Merck precoated glass plates (0.20 mm with fluorescent indicator UV254) and were visualized with UV light irradiation at 254/ 366 nm. Flash column chromatography was carried out with the use of silica gel from Merck (230-400 mesh). Mass spectra were obtained on a JEOL SX-102A instrument operating in electron impact (EI) or fast atom bombardment (FAB) mode.
The 1H and 13C NMR spectra were obtained on Bruker
spectrometers operating at frequencies as indicated for each
compound. Chemical shifts were reported relative to CDCl3(1H
7.24) and at 77.0 ppm in CDCl3[13C (central line of t)]. FT-IR
spectra were recorded on a Nicolet magna-IR 550 series II. Melting points were uncorrected.
General Procedure for the Synthesis of Chalcones. KOH (2.9 equiv) was added to a suspension of relevant aldehyde (1.0 equiv, see Scheme 3) and the appropriate acetophenone (1.05 equiv) in EtOH (6 mL/mmol acetophenone). The mixture was
stirred at 50°C for 3 h, then cooled down to room temperature
and left overnight. The reaction mixture was poured into water and acidified with aqueous HCl (1 M) to obtain the correspond-ing chalcones (a or b) that could be further crystallized in EtOH. Spectroscopic and analytical data for a and b are shown below. (E)-1-(2-Hydroxyphenyl)-3-(pyridin-2-yl)prop-2-en-1-one (a). Using picolinaldehyde (1.5 mL, 15 mmol) gave 2.6 g
(76%) of a, which was further recrystallized from EtOH.1H
NMR:δ 12.7 (s, 1H), 8.6 (d, J ) 4.0 Hz, 1H), 8.2 (d, J ) 15.2 Hz, 1H), 8.0-7.8 (m, 1H), 7.7 (d, J ) 1.6 Hz, 1H), 7.84-7.48 (m, 1H), 7.47-7.44 (m, 1H), 7.30-7.28 (m, 1H), 7.27-6.98 (m, 1H), 6.94-6.90 (m, 1H).13C NMR:δ 193.8, 163.3, 152.5, 150.0, 143.0, 136.8, 136.5, 130.1, 125.7, 124.5, 123.9, 119.9, 118.8, 118.3. (E)-1-(2-Hydroxyphenyl)-3-(pyridin-4-yl)prop-2-en-1-one (b). Using isonicotinaldehyde (1.5 mL, 15 mmol) gave 2.2 g
(65%) of a, which was further recrystallized from EtOH.1H
NMR: δ 7.92-7.88 (m, 2H), 7.56-7.52 (m, 1H), 7.48-7.43 (m, 2H), 7.30-7.26 (m, 1H), 7.23-7.20 (m, 1H), 6.97(d, J )
19, 1H), 6.91-6.87 (m, 1H).13C NMR:δ 161.8, 147.0, 135.8,
133.1, 130.6, 125.7, 123.3, 123.0, 122.9, 119.0, 117.4, 112.3. General Procedure for the Synthesis of 3-Hydroxy-2-(pyridinyl)-4H-chromen-4-one. Aqueous 30% H2O2 (8-11
equiv) was added to a solution of the appropriate chalcone (a or b, see Scheme 3) (1.0 equiv) and aqueous 4 M NaOH (5.0 equiv) in a 1:1 mixture of MeOH and THF (20 mL/mmol
chalcone) at 0°C. The reaction was stirred at room temperature
overnight. The mixture was then acidified with aqueous HCl (1 M) and the product filtered off. Spectroscopic and analytical data for 1a and 1b are shown below.
3-Hydroxy-2-(pyridin-2-yl)-4H-chromen-4-one (1a). The crude product (440 mg, 1.3 mmol) was recrystallized from EtOH
to yield 380 mg (82%) of 1a.1H NMR:δ 8.5 (m, 1H), 8.28
(m, 1H), 8.11 (d, J ) 8 Hz, 1H), 7.96 (m, 1H), 7.64 (d, J ) 1 Hz, 1H), 7.50 (d, J ) 0.5 Hz, 1H), 7.48-7.41 (m, 1H),
7.39-7.33 (m, 1H).13C NMR:δ 173.6, 154.3, 152.5, 146.6,
SCHEME 1: Molecular Structures of 3HF, 1a (Isomer Included), 1b, and Their Respective ESIPT Processes
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
143.3, 139.5, 138.0, 133.3, 125.9, 124.0, 123.7, 122.9, 119.8,
117.6 FAB-MS: m/z 240.1 (M++ 1).
3-Hydroxy-2-(pyridin-4-yl)-4H-chromen-4-one (1b). The crude product (500 mg, 1.3 mmol) was recrystallized from EtOH
to yield 400 mg (84%) of 1a.1H NMR:δ 7.91-7.88 (m, 1H),
7.51-7.46 (m, 1H), 7.00-6.98 (m,1H), 6.93-6.91 (m, 1H),
6.89-6.83 (m, 2H), 6.79-6.75 (m, 2H). 13C NMR: δ 173.7,
161.5, 143.3, 136.6, 130.7, 121.1, 119.5, 117.5, 115.5, 111.3,
107.94. FAB-MS: m/z 239.0 (M++ 1).
Crystallographic Data Collection and Refinement. Data collection of compound 1a(N) was carried out on a NONIUS KappaCCD diffractometer with Mo radiation (λ ) 0.71073 Å) at 150(1) K. A preliminary orientation matrix and unit cell parameters were determined from 15 frames, each frame
corresponds to the 1degreeω scan in 20 s, followed by spot
integration and least-squares refinement. Data were measured
usingω scans, 0.50 degree per frame, 100 s. per degree. Cell
parameters were retrieved and refined using DENZO-SMN11
software on all observed reflections. Data reduction was
performed with the DENZO-SMN11 software. An empirical
absorption was based on the symmetry-equivalent reflections
and applied the data using the SORTAV.12program. The structure
analysis was made by using SHELXTL program on PC
computer. The structure was solved using the SHELXS-9713
program and refined using SHELXL-9714program by full-matrix
least-squares on F2values. All hydrogen atoms were generated
geometrically (C-H ) 0.95) and refined using a riding mode with the exception of the hydrogen atom (H(2)) of hydroxyl group, which is located in the difference Fourier map with the corresponding positions. The final full-matrix, least-squares
refinement on F2was applied for all observed reflections [I >
2σ(I)]. All calculations were performed using the SHELXTL
software package.15 Crystallographic data and details of data
collections and structure refinements of 1a(N) are listed in the Supporting Information.
Measurements. Steady-state absorption and emission spectra were recorded with a Hitachi (U-3310) spectrophotometer and an Edinburgh (FS920) fluorimeter, respectively. The various solvents were of spectragrade quality (Merck Inc.) and were used upon receipt. Benzene and acetonitrile showed traces of fluorescence impurities and were fractionally distilled prior to use. Nanosecond lifetime studies were performed with an Edinburgh FL 900 photon-counting system with a hydrogen-filled or a nitrogen lamp as the excitation source. The emission decays were analyzed by the sum of exponential functions, which allows partial removal of the instrument time broadening
and thus renders a temporal resolution of∼200 ps. The setup
for picosecond dynamical measurements consisted of a femto-second Ti-Sapphire oscillator (82 MHz, Spectra Physics). The fundamental train of pulses was pulse-selected (Neos, model N17389) to reduce the repetition rate to typically 0.8-8 MHz, and then used to produce second harmonics (375-425 nm) as SCHEME 2: Relaxation Processes for Excited-State Charge Transfer Coupled Proton Transfer Demonstrated by N,N-Diethylamino-3-hydroxyflavones8
SCHEME 3: Synthetic Routes of 1a and 1b
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
an excitation light source. A polarizer was placed in the emission path to ensure that the polarization of the fluorescence was set
at the magic angle (54.7°) with respect to that of the pump laser
to eliminate fluorescence anisotropy. An Edinburgh OB 900-L time-correlated single photon counting system was used as a
detecting system, rendering a temporal resolution of∼15 ps.
Fluorescence upconversion measurements were performed with a femtosecond optically gated system (FOG-100, CDP). The fundamental of a Ti:sapphire laser (Spectra Physics) at 750-850 nm with an average power of 0.5 W and a repetition rate of 82 MHz was used to produce second harmonics (SH) at 375-425 nm by focusing onto a 0.5 mm thick BBO type-I crystal. The SH were then separated from the fundamental pulses with a dichroic mirror and used as the pump pulses. The pump
pulses were then focused onto a rotating cell, and the optical path length was 1.0 mm. The resulting fluorescence was collected by an achromatic lens and then focused on another BBO type-I crystal (0.5 mm). The optically delayed remaining fundamental pulses were also focused on the BBO crystal and used as gate pulses for the sum-frequency generation. A Berek’s variable waveplate was placed in the pump beam path to ensure that the polarization of the pump laser was set at the magic
angle (54.7°) with respect to that of the probe laser to eliminate
Figure 1. Normalized absorption and emission spectra of 1a (red -b-)
and 1b (black -f-) in CH2Cl2(∼2 × 10-5M,λex∼ 350 nm) and the
single crystal emission of 1a (blue -b-) (λex) 360 nm).
Figure 2. Absorption spectrum (black -b-) and the excitation spectrum
(red -f-, monitored at 534 nm) of 1a in CH2Cl2(∼2 × 10-5M).
Figure 3. Absorption and emission spectra of 1a prior to the addition
of HCl(g)(black -b-). The absorption and emission of 1a (∼2 × 10-5
M) in CH2Cl2purged with HCl(g)to reach the saturation (red -f-, see
text for detail). The excitation spectra of 1a in CH2Cl2purged with
HCl(g) to reach the saturation by monitoring at 430 nm (blue -O-) and
560 nm (green -4-), respectively.
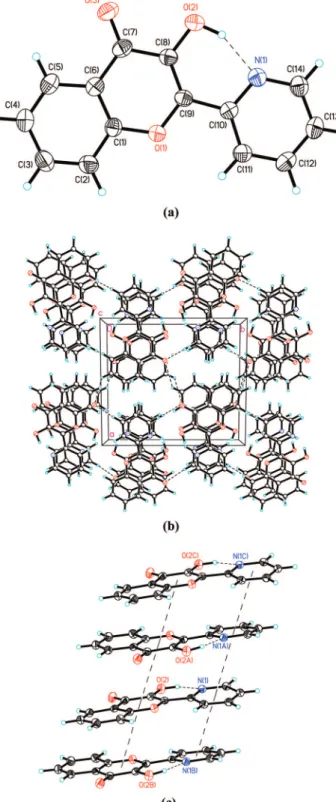
Figure 4. (a) The molecular structure of 1a with thermal ellipsoids
set at 50% probability level. (b) The molecular packing of 1a(N) viewed along the c-axis. (c) The 1D chain-like architecture of 1a(N) in an ABAB pattern (dashed line) through theπ-π intermolecular interaction
among 1a(N) molecules.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
fluorescence anisotropy. The upconverted signal was then separated by an F/4.9 (f ) 380 mm) single monochromator (CDP2022) and detected via a photon counting PMT (R1527P, Hamamatsu). The cross correlation between SH and the
fundamental had a full width at half-maximum (fwhm) of ∼
150 fs, which was chosen as a response function of the system. An integrating sphere (Laboratory sphere) was applied to measure the quantum yield in the solid state, for which the solid thin film was prepared via direct vacuum deposition and excited by an argon ion laser at 363 nm. The resulting luminescence was acquired with an intensified charge-coupled detector for subsequent quantum yield analyses according to a reported
method.16
Theoretical Approaches. The Gaussian 03 program was used
to perform the ab initio calculation on the molecular structure.17
Geometry optimizations for all structures were carried out with
the 6-31G(d′) basis set to the B3LYP functional.18,19The hybrid
DFT functional B3LYP has proven to be a suitable DFT
functional to describe hydrogen bond.20 Hessians and hence
vibrational frequencies were also performed to check whether the optimized geometrical structures for those molecules were at energy minima, transition states, or higher order saddle points. After obtaining the converged geometries, the
TD-B3LYP/6-31++G(d′,p′) was used to calculate the vertical excitation
energies.20,21
3. Results and Discussion
3.1. Steady-State Approach. As depicted in Figure 1, 1a
(2× 10-5M) in CH2Cl2exhibits an UV absorption spectrum
with the lowest lying transition maximized at 351 nm. The
absorption extinction coefficient of 1.7× 104M-1cm-1at 350
nm indicates that the S0fS1is aππ* transition. Upon 350
nm excitation, as shown in Figure 1, an anomalously long wavelength emission band was resolved, with a peak wavelength
at 534 nm. The large Stokes shift (9763 cm-1) between
absorption and emission peak frequencies is reminiscent of the
occurrence of ESIPT in 3HF and its derivatives.5,8However, in
sharp contrast to the identity indicated by absorption and fluorescence excitation spectra in 3HF, a significant difference between these two spectra was observed for 1a, in which the lowest lying peak wavelength (343 nm) of the excitation spectrum is blue-shifted around 8 nm in comparison to that of the absorption (see Figure 2). Since the intensity of the excitation
wavelength had been calibrated, such a difference cannot be attributed to the difference in the wavelength-dependent response of the instrument between absorption and emission spectra. Furthermore, as ensured by various characterizations of its purity (see experimental section), the possibility of a spectral difference originating from traces of impurity is also discarded. Realizing that 1a might exist in two HB isomers, i.e., 1a(O) and 1a(N) (see Scheme 1), in which 1a(N) is subject to protonation due to the greater basicity in pyridinic nitrogen, we thus performed
a simple test by purging HCl gas to 1a in CH2Cl2. Such a
titration procedure is supposed to protonate the pyridinic nitrogen and hence to block the formation of O-H---N HB.
Figure 3 reveals the emission spectrum of 1a in CH2Cl2purged
with HCl(g) to reach full protonation (until changes in the
emission spectra cease). In comparison to neutral 1a, the
tautomer emission (560 nm) was increased by ∼25 folds,
accompanied by a rather small portion of the normal (430 nm) emission. It is worthy to point out here that the excitation spectrum monitored at both normal (430 nm) and tautomer emission (560 nm) of the protonated 1a is exactly the same, which is also identical with respect to the absorption spectrum (see Figure 3), indicating that both 430 and 560 nm bands originate from the same ground-state species. Details of the associated relaxation dynamics for neutral versus protonated 1a will be further discussed.
In light of the above observations, we thus conclude the existence of equilibrium between 1a(N) and 1a(O) HB
con-formers in CH2Cl2, in which the protonation blocks the 1a(N)
HB conformer, leading to the formation of 1a(O)H+ HB
conformer. Note that such 1a(O) h 1a(N) equilibrium in
ground-state must be fast in terms of kinetics because1H NMR
could not resolve any O-H peak. This viewpoint of equilibrium is also supported by the absorption and emission spectra of 1b, in which only 1b(O) HB conformer exists due to the lack of an O-H---N(pyridine) hydrogen bond. As a result, 1b exhibits a unique tautomer emission maximized at 547 nm (see Figure 1), and the excitation spectrum (not shown here), which is independent of the monitored emission wavelength, is identical to the absorption profile. In brief, the results of steady-state UV absorption and fluorescence measurements imply an equilibrium between 1a(O) and 1a(N) in solution. The absorption profile for 1a(N) is red-shifted with respect to that of 1a(O), plausibly
due to different degrees of hydrogen bond inducedπ electron
delocalization (vide infra). Upon excitation, 1a(O) undergoes fast ESIPT, giving rise to a unique 534 nm tautomer emission. In sharp contrast, no steady-state emission could be resolved
for 1a(N) in CH2Cl2as well as in other polar, aprotic solvents
such as toluene and acetonitrile. Further insight into the relaxation dynamics of 1a(N) will be elaborated in the section of fluorescence up-conversion measurement.
Single crystals of 1a were also successfully grown. As shown in Figure 4a, the X-ray structural determination reveals a dominant 1a(N) population in a single crystal form, which exhibits a intramolecular O(2)---N(1) hydrogen bond with N---O
distance of 2.589(2) Å. The small dihedral angle (4.36°) of
N(1)-C(10)-C(9)-C(8) reveals that the 2-pyridyl moiety is nearly coplanar with respect to the parent chromone moiety. This result can be rational by the O(2)---N(1) hydrogen bond
formation together with partialπ bond character of C(9)-C(10)
with bond distance of 1.462(2) Å. It is important to note that adjacent 1a(N) molecules are parallel and superimposed in an ABAB pattern (Figure 4b) to form a 1D chain-like architecture
(Figure 4c) through theπ-π intermolecular interactions between
the pyridyl-pyridyl moieties and between chromone-chromone Figure 5. Time-resolved sum frequency signal of fluorescence at
different wavelength and gate pulse (760 nm) for 1a in CH2Cl2. The
solid lines express the corresponding best-fitting curve.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
moieties, respectively, with the distances of 3.713 and 3.709 Å between the ring centroids. These results are essentially in agreement with the previous report about the crystal molecular structure of 2-(1H-benzimidazol-2-yl)-3-hydroxy-4H-
chromen-4-one.22
3.2. ESIPT Dynamics. More insight into the mechanism of ESIPT for 1a may be resolved by the associated relaxation dynamics. Figures 5a-e show the time-resolved up-converted
fluorescence signal of 1a (1× 10-3M in CH2Cl2) at various
emission wavelengths (λex∼ 380 nm). Pertinent spectroscopic
and dynamic data are listed in Table 1. Upon monitoring at 450 nm, which is supposed to be in the normal emission region but could not be resolved from a steady state manner due to fast ESIPT (vide supra), the time-resolved 450 nm emission is composed of response limited rise and decay (<150 fs) components (see Figure 5a). On the other hand, when monitoring exclusively at the tautomer emission region of e.g. 660 nm, as shown in Figure 5e, multiexponential decay curves were observed. The best deconvoluted fitting of the relaxation dynamics renders a response limit risetime (<150 fs), a fast but resolvable decay, and a nearly constant value (within the acquisition time of 100 ps) with a small pre-exponential factor
contribution (<4%) compared with the fast decay (∼96%)
components. The system response limited decay for the normal emission as well as rise for the tautomer emission unambigu-ously indicates an ultrafast rate of ESIPT. The fast and resolvable decay time constant was measured to be 22.6 ( 0.3 ps. The very long decay constant indeed is attributed to a long population decay, which was further resolved to be 1.4 ns by the time-correlated single photon counting measurement.
We then took the fitted pre-exponential value of the 22.6 ps decay component at various monitored wavelengths and plotted the emission intensity versus wavelength, shown in Figure 6. Obviously, the up-converted emission of the fast decay (22.6
ps) component was maximized at∼577 nm, which is
signifi-cantly red-shifted with respect to the 534 nm peak obtained with the steady state measurement. On the other hand, upon integrat-ing the long-decay (1.4 ns) component, the plot for the emission intensity versus emission wavelength reveals a maximum at ∼530 nm (see Figure 6), which is consistent with that obtained from the steady state measurement. Thus, the relaxation dynamics, in combination with the steady state results, show that both conformers 1a(O) and 1a(N) undergo ESIPT reaction
in solution (e.g., CH2Cl2). Following ESIPT, 1a(O) gives rise
to a tautomer emission maximized at 534 nm with an observed
lifetime of 1.4 ns in CH2Cl2, while the tautomer emission for
1a(N) undergoes a fast decay constant of 22.6 ps and its intensity is too weak to be resolved in a steady state manner. Note that the normal emission of 1a(N), although it could not be resolved with a steady state means, is expected to be red-shifted compared to that of 1a(O). This viewpoint is supported by the time-resolved fluorescence monitored at shorter wavelengths such as 540 nm (see Figure 5b), in which in addition to the 22.6 ps decay component, the time-resolved profile also exhibits an ultrafast, unresolvable decay component (<150 fs). Finally, ultrafast ESIPT also takes place for 1a(N) in the crystal, as
TABLE 1: Photophysical Properties of 1a, 1a(O)H+in CH2Cl2, and 1a in Single Crystala
λabs/nm λem/nm relaxation dynamics/psb
1a CH2Cl2 351 N: - 450 nm [τ1) 0.13 (0.98), τ2) 28.9 (0.02) ] T: 534 540 nm [τ1) 0.35 (0.37), τ2) 22.8 (0.63) ] 580 nm [τ1) 0.24 (0.18), τ2) 22.6 (0.82) ] 620 nm [τ1) 0.42 (0.03), τ2) 22.6 (0.97) ] 660 nm [τ) 22.4 ] 660 nm [τ1) 24(0.96), τ2) 1400 (0.04) ]c single crystal N: -T: 540 540 nm [τ) 3500 ]c 77 K CH2Cl2 T: 528 530 nm [τ) 4900 ]c 1a(O)H+ CH2Cl2 361 N: 430 430 nm [τ) 15.7 ] T: 560 580 nm [τ1) 14.8 (-0.48), τ2) 647 (0.52) ] a
The experimental error for the fitted time constant is less than ∼10%.b
Data in parentheses are the fitted pre-exponential factor.c
The relaxation dynamics were measured by time-correlated single photon counting.
Figure 6. (Blue-solid line) Steady state emission spectrum. (b) Spectrum taken by plotting the pre-exponential value of the 1.4 ns decay component versus the monitored wavelength. (O) Spectrum taken by plotting the pre-exponential value of the 22.6 ps decay component versus the monitored wavelength for 1a in CH2Cl2. (Dashed line) The
best-fitted spectrum of the 22.6 ps (O) component.
Figure 7. Time-resolved sum frequency signal of fluorescence and
gate pulse (780 nm) for 1a in CH2Cl2. The solid lines express the
corresponding best-fitting curve. The black solid line denotes the decay monitored at the emission of 430 nm and the blue solid line represents the rise and decay of the 580 nm emission.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
supported by the large Stokes shifted fluorescence with a system response risetime (<150 fs), while the prohibition of the nonradiative decay process (vide infra) in the single crystal leads
to a bright 540-nm tautomer emission (Φf∼ 0.3) with a lifetime
as long as 3.5 ns.
One intriguing feature lies in the relaxation dynamics of the
protonated form, i.e., 1a(O)H+ in CH2Cl2. Dual emission
ascribed to normal (430 nm) and tautomer (560 nm) species
was resolved for 1a(O)H+in CH2Cl2(see Figure 3, vide supra).
Upon monitoring at the normal emission of 430 nm, the time-resolved signal is composed of a response-limited rise (<150 fs) and a single exponential decay of 15.7 ps (see Figure 7). On the other hand, the relaxation dynamics monitored at the tautomer emission of 560 nm are established by a resolv-able rise (14.8 ps) and a rather long decay component (647 ps). Note that the latter data are acquired by the time-correlated single photon counting technique. Within experimental and fitting uncertainty, the decay of the normal emission is matched with respect to the rise of tautomer emission, consistent with a precursor-successor type of ESIPT relationship. Although the
dynamics are clearly resolved, in comparison to the > (150 fs)-1
rate of ESIPT in 1a(O) (or 1a(N)) in neutral form, the much
slower ESIPT time scale (∼15 ps) in 1a(O)H+is intriguing.
On one hand, it may imply the rise of an intrinsic, appreciable
barrier during ESIPT for 1a(O)H+. However, since the driving
force of the forming oxy-anion dual hydrogen bonds (see Figure 8) is greater than that of the single hydrogen bond in 1a(O),
this hypothesis, in our view, may be groundless. Alternatively,
it is more plausible that the cationic form 1a(O)H+ and its
proton transfer tautomer possess drastically different dipole moments in the excited state, such that the solvent polarity induced barrier is appreciable, which then channels into the
ESIPT reaction.8 Nevertheless, this viewpoint requires more
rigorous examination, including solvent polarity dependent studies with both steady-state and dynamics approaches, which is not the main focus of this study. Details will be addressed in future work.
We have also made attempts to extract the associated thermodynamic parameters of the HB equilibrium via a tem-perature-dependent absorption study. Unfortunately, owing to
the relatively high freezing point (223 K) of CH2Cl2and great
similarities in spectral profiles between 1a(O) and 1a(N), we failed to obtain the thermodynamics via the steady state approach. Alternatively, we realize that the ratios of the pre-exponential factors for 22.6 ps versus 1.4 ns component are within 22 to 32 in the monitored emission range of 540-580 nm. Assuming that 1a(O) and 1a(N) exhibit very similar radiative lifetime, i.e., the same transition dipole due to the similarities in spectral features (in terms of spectral profile and absorptivity), we qualitatively estimate the ground-state
equi-librium constant for 1a(O)h1a (N) in 298 K CH2Cl2to be
within 27 ( 5 M-1. The thermally favorable 1a(N) population
can be rationalized by the greater basicity of pyridinic nitrogen Figure 8. Proposed ESIPT mechanism of 1a(O)H+. Note that a large change of the dipole moment is expected during the ESIPT process (see text for detailed discussion).
SCHEME 4: Calculated Energy Potential Diagram of 1a with Dual ESIPT Pathways after Applying Solvation in CH2Cl2a
a
The absorption and emission gaps were taken from the experimental data. See detailed discussion in the text.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
(c.f. carbonyl oxygen), together with the associated six mem-bered ring HB geometry (c.f. five memmem-bered-ring HB structure in 1a(O)).
3.3. Theoretical Approach. Supplementary support of the ground-state thermodynamics is provided by the computational approach (see Experimental Section). The full geometry
opti-mization based on the B3LYP/6-31G(d′) theoretical level reveals
that 1a(N) is more stable than 1a(O) by 3.79 kcal/mol in gas phase. It is worthy to note that if considering the polarization
function on hydrogen such as 6-31G(d′,p′), the optimized
geometry of the tautomeric form of 1a(O) is converged to 1a(O). The result implies that the tautomer form of 1a(O) in the ground-state is very unstable with respect to the normal forms, such that the 1a(O) proton transfer tautomer f 1a(O) reverse proton transfer may be rather small or even barrierless. After applying
solvation in CH2Cl2based on Onsager theory, the difference in
energy is reduced to 3.59 kcal/mol, still in favor of 1a(N), qualitatively in agreement with that estimated experimentally. Further calculations show that the corresponding proton-transfer tautomer is higher in energy than the respective 1a(O) and 1a(N) by 15.10 and 7.47 kcal/mol in the ground state. To each
ground-state we then simply add absorption and emission peak frequency (see Figures 1) to mark the energy level of the
corresponding S1state. As a result, the overall energetics is
depicted in Scheme 4. The results clearly indicate that ESIPT for both 1a(O) and 1a(N) is thermodynamically favorable, consistent with the experimental results.
Another interesting feature lies in the frontier orbital analysis.
On the basis of time dependent DFT (TD-DFT), the S0f S1
ππ* transition was calculated to be 3.49 eV (355 nm) and 3.43 eV (361 nm) for 1a(O) and 1a(N), respectively, the results of which are well-correlated with the peak (343 nm) of excitation spectrum for 1a(O) and an expected red-shifted absorption of 1a(N). The frontier orbital analyses reveal that theπ-electron density of the proton-transfer tautomer form of 1a(O) in the excited-state is mainly delocalized at the chromone moiety, while that of the tautomer form of 1a(N), in sharp contrast, is largely spread around the pyridine moiety and extended to the carbonyl oxygen. As expected, the former is similar to the electronic configuration of 1b tautomer (see Table 2). The results manifest two different electronic configurations of tautomers resulting from ESIPT of two respective isomers. Due to the
TABLE 2: Theoretical Results on Energy and Frontier Orbitals of Various Electronic States of 1a(O), 1a(N), and 1b
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
drastically different lifetimes measured in this study (vide supra), the interconversion between these two proton-transfer tautomers in the excited-state is not likely to occur. This can be rationalized by the necessity of breaking the strong hydrogen bond for interconversion, the time scale of which seems to be incompa-rable with the respective relaxation dynamics. Likewise, the conversion between excited 1a(O) and 1a(N) species is also prohibited due to the ultrafast rate of ESIPT for each isomer in the excited state (see Scheme 4).
3.4. Relaxation after ESIPT. Last but not least, after ESIPT, the dominant radiationless deactivation process for 1a(N) tautomer in solution is also of fundamental interest. As supported by the above X-ray analysis, the associated photophysics of 1a in a single crystal clearly originate from the 1a(N) conformer. As depicted in Figure 1, independent of the excitation wave-length, the 1a(N) conformer in solid exhibit a unique, bright emission maximized at 540 nm. In comparison to the onset of
the absorption spectrum of∼400 nm, the large Stokes shifted
emission warrants the occurrence of ESIPT in 1a(N) solid crystal, resulting in the 540 nm tautomer emission. This is in sharp contrast to the lack of steady-state emission for 1a(N) in
CH2Cl2and other solvents. The results are reminiscent of many
ESIPT molecules exhibiting extremely weak tautomer emission in solution due to cis-trans isomerization, which induces a dominant deactivation channel for the emission quenching in
the excited state.4 For example, in the case of 2-(2′
-hydrox-yphenyl)benzothiazole, it has been well-known that following
ESIPT, cis-tran isomerization takes place in the excited state.23
In the case of 1a(N) in solution, once forming the keto-tautomer via ESIPT (see Scheme 1), it seems that the cis-trans isomerization along C(9)-C(10) rotation may play a key role to account for the dominant radiationless deactivation process. As for an extreme case, crossover of the ground and excited-state potential energy surface alone rotational coordinate may
lead to the formation of conical intersections.24 It is thus
plausible that radiationless decay of 1a(N) proton-transfer tautomer would be associated with the seam of conical intersec-tion with a sloped topology lying parallel to the C(9)-C(10) rotational pathway.
Alternatively, it is also possible that the tautomer is subject to a dynamical quenching effect, induced via an excited-state charge transfer complex between solute and solvent, as has been demonstrated that was acting for a dual intramolecular hydrogen bonded 3-hydroxyflavone derivative, 2,8-diphenyl-3,7,-dihy-droxy-4H,6H-pyrane[3,2-g]chromene-4,6dione, a diflavonol mol-ecule which has the possibility of single-proton transfer of
double-proton transfer.25To examine this, luminescence of 1a
in 77 K CH2Cl2solid matrix was performed and the
photo-physical data are listed in Table 1. Due to the thermally favorable 1a(N) (vide infra), one expected 1a(N) to be the
dominant species at 77 K. Evidently, in 77 K CH2Cl2, solid
matrix, a bright 1a(N) tautomer emission maximized at 528 nm
was resolved (Φf∼0.41, τf) 4.9 ns, see Table 1). Since both
solvent diffusion and cis-trans rotation are prohibited in 77 K
CH2Cl2solid matrix, the results cannot single out the associated
quenching mechanism. At current stage, however, the observa-tion of highly emissive 1a(N) tautomer emission in a single crystal leads us to be favorable on the deactivation pathway incorporating cis-trans isomerism.
4. Conclusion
In conclusion 1a has been synthesized and characterized and its dual ESIPT channels explored. In the solid state, 1a exists exclusively in 1a(N) HB conformer, which, upon excitation,
undergoes ESIPT from the hydroxyl proton to the pyridinic nitrogen, forming a keto-tautomer emitting at 540 nm. In
solution such as CH2Cl2, 1a exists in two HB isomers 1a(O)
and 1a(N) with an equilibrium constant of∼22-32 in favor of
1a(N). Both isomers undergo ultrafast (<150 fs) ESIPT, forming a zwitterion and keto-tautomer, respectively, in the excited state. A fast radiationless pathway, possibly induced by the C(9)-C(10) rotational deactivation, then takes place in the keto-tautomer, resulting in the lack of steady-state emission. To our best knowledge, this is the first time that such a dual channel ESIPT pathway with comprehensive reaction dynamics is investigated in the case of 3-HF relevant ESIPT molecules. As shown in
the proton titration, the resulting on (with H+)/off (without H+)
emission property for the 1a(O) versus 1a(N) tautomer fluo-rescence may lead to future studies in e.g. reversible Lewis acid/ basis detection in organic solvents.
Acknowledgment. We thank the National Science Council (grant numbers 99-1989-2004).
Supporting Information Available: A cif file containg the crystallographic data for 1a(N). This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) For instance: Studies in physical and theoretical Chemistry; Mu¨ller, A., Ratajack, H., Junge, W., Diemann, E., Eds.; Electron and Proton Transfer in Chemistry and Biology 78; Elsevier: Amsterdam, 1992.
(2) (a) Scheiner, S. J. Phys. Chem. A 2000, 104, 5898. (b) Waluk, J. Conformational Aspects of Intra- and Intermolecular Excited-State Proton Transfer In Conformational Analysis of Molecules in Excited States; Waluk, J., Ed.; Wiley-VCH: New York, 2000. (c) Chou, P. T. J. Chin. Chem. Soc.
2001, 48, 651. (d) Wu, K. C.; Cheng, Y. M.; Lin, Y. S.; Yeh, Y. S.; Pu,
S. C.; Hu, Y. H.; Yu, J. K.; Chou, P. T. Chem. Phys. Lett. 2004, 382, 203. (e) Paterson, M. J.; Robb, M. A.; Blancafort, L.; DeBellis, A. D. J. Am.
Chem. Soc. 2004, 126, 2912. (f) Lochbrunner, S.; Wurzer, A. J.; Riedle, E. J. Phys. Chem. A 2003, 107, 10580. (g) de Vivie-Riedle, R.; De Waele,
V.; Kurtz, L.; Riedle, E. J. Phys. Chem. A 2003, 107, 10591. (h) Cheng, C. C.; Chang, C. P.; Yu, W. S.; Hung, F. T.; Liu, Y. I.; Wu, G. R.; Chou, P. T. J. Phys. Chem. A 2003, 107, 1459. (i) Lukeman, M.; Wan, P. J. Am.
Chem. Soc. 2003, 125, 1164.
(3) For example, see: (a) Chou, P. T.; McMorrow, D.; Aartsma, T. J.; Kasha, M. J. Phys. Chem. 1984, 88, 4596. (b) Ernsting, N. P.; Nikolaus, B.
Appl. Phys. B: Laser Opt. 1986, 39, 155. (c) Acun˜a, A. V.; Amat-Guerri,
F.; Cataln˜, J.; Costella, A.; Figuera, J.; Munoz, J. Chem. Phys. Lett. 1986,
132, 576. (d) Kasha, M. In Molecular Electronic DeVices; Carter, F. L.,
Siatkowski, R. E., Wohltjen, H., Eds.; Elsevier Science Publishers: New York, 1988; pp 107-121. (e) Cataln˜, J.; del Valle, J. C. J. Am. Chem. Soc.
1993, 115, 4321. (f) Ferrer, M. L.; Acun˜a, A. U.; Amat-Guerri, F.; Costela,
A.; Figuera, J. M.; Florido, F.; Sastre, R. Appl. Opt. 1994, 33, 2266. (g) Jones, G.; Rahman, M. A. J. Phys. Chem. 1994, 98, 13028. (h) Liphardt, M.; Gooneskera, A.; Jones, B. E.; Ducharme, S.; Takacs, J. M.; Zhang, L.
Science (Washington, D.C.) 1994, 263, 367. (i) Kuldova´, K.; Corval, A.;
Trommsdorff, H. P.; Lehn, J. M. J. Phys. Chem. A 1997, 101, 6850. (j) Sytnik, A.; Gormin, D.; Kasha, M. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 8267. 11968. (k) Sytnik, A.; del Valle, J. C. J. Phys. Chem. 1995, 99, 13028. (l) Roshal, A. D.; Grigorovich, A. V.; Doroshenko, A. O.; Pivovarenko, V. G.; Demchenko, A. P. J. Phys. Chem. A 1998, 102, 5907. (m) Tarkka, R. M.; Zhang, X.; Jenekhe, S. A. J. Am. Chem. Soc. 1996, 118, 9438.
(4) Chen, K. Y.; Cheng, Y. M.; Lai, C.-H.; Hsu, C. C.; Ho, M. L.; Lee, G. H.; Chou, P. T. J. Am. Chem. Soc. 2007, 129, 4534.
(5) (a) Sengupta, P. K.; Kasha, M. Chem. Phys. Lett. 1979, 68, 382. (6) (a) McMorrow, D.; Kasha, M. J. Phys. Chem. 1984, 88, 2235. (b) Chou, P.; McMorrow, D.; Aartsma, T. J.; Kasha, M. J. Phys. Chem. 1984,
88, 4596. (c) Kasha, M. J. Chem. Soc., Faraday Trans. 2 1986, 82, 2379.
(d) Etter, M. C.; Urban´czyk-Lipkowska, Z.; Baer, S.; Barbara, P. F. J. Mol.
Struct. 1986, 144, 155. (e) Brucker, G. A.; Kelley, D. F. J. Phys. Chem. 1988, 92, 3805. (f) Schwartz, B. J.; Peteanu, L. A.; Harris, C. B. J. Phys. Chem. 1992, 96, 3591. (g) Ameer-Beg, S.; Ormson, S. M.; Brown, R. G;
Matousek, P.; Towrie, M.; Nibbering, E. T. J.; Foggi, P.; Neuwahl, F. V. R.
J. Phys. Chem. A 2001, 105, 3709. (h) Bader, A. N.; Ariese, F.; Gooijer,
C. J. Phys. Chem. A 2002, 106, 2844. (i) Bader, A. N.; Picocarenko, V. G.; Demchenko, A. P.; Ariese, F.; Gooijer, C. J. Phys. Chem. B 2004, 108, 10589.
(7) Chudoba, C.; Riedle, E.; Pfeiffer, M.; Elsaesser, T. Chem. Phys.
Lett. 1996, 263, 622.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
(8) (a) Chou, P. T.; Martinez, M. L.; Clements, J. H. J. Phys. Chem.
1993, 97, 2618. (b) Chou, P. T.; Martinez, M. L.; Clements, J. H. Chem. Phys. Lett. 1993, 204, 395. (c) Swinney, T. C.; Kelley, D. F. J. Chem. Phys. 1993, 99, 211. (d) Parsapour, F.; Kelley, D. F. J. Phys. Chem. 1996, 100, 2791. (e) Ormson, S. M.; Brown, R. G.; Vollmer, F.; Rettig, W. J. Photochem. Photobiol. A 1994, 81, 65. (f) Chou, P. T.; Huang, C. H.; Pu,
S. C.; Cheng, Y. M.; Liu, Y. H.; Wang, Y.; Chen, C. T. J. Phys. Chem. A.
2004, 108, 6452. (g) Cheng, Y. M.; Pu, S. C.; Yu, Y. C.; Chou, P. T.;
Huang, C. H.; Chen, C. T.; Li, T. H.; Hu, W. P. J. Phys. Chem. A. 2005,
109, 3777.
(9) (a) Marcus, R. A. J. Chem. Phys. 1965, 43, 379. (b) Marcus, R. A.
Annu. ReV. Phys. Chem. 1964, 15, 155. (c) Marcus, R. A. J. Chem. Phys. 1965, 43, 679. (d) Marcus, R. A. ReV. Mod. Phys. 1993, 65, 599.
(10) (a) Bublitz, G. U.; Boxer, S. G. Annu. ReV. Phys. Chem. 1997, 48, 213. (b) Klymchenko, A. S.; Demchenko, A. P. J. Am. Chem. Soc. 2002,
124, 12372. (c) Klymchenko, A. S.; Demchenko, A. P. Phys. Chem. Chem. Phys. 2003, 5, 461.
(11) DENZO-SMN, Otwinowski & Minor, 1997.
(12) SORTAV Blessing, R. H. Acta Crystallogr., Sect. A 1995, A51, 33–38.
(13) SHELXS-97 Sheldrick, G. M. Acta Crystallogr., Sect. A 1990, A46, 467–473.
(14) Sheldrick, G. M. SHELXL-97; University of Go¨ttingen: Go¨ttingen, Germany, 1997.
(15) SHELXTL: Structure analysis program, Version 6.10; Bruker-axs: Madison, WI, 2000.
(16) de Mello, J. C.; Wittmann, H. F.; Friend, R. H. AdV. Mater. 1997,
9, 230.
(17) Gaussian 03, ReVision C.02 Frisch, M. J. ; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.;
Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian, Inc.: Wallingford, CT, 2004.
(18) Petersson, G. A.; Al-Laham, M. A. J. Chem. Phys. 1991, 94, 6081. (19) (a) Becke, A. D. Phys. ReV. A 1988, 38, 3098. (b) Lee, C.; Yang, W.; Parr, R. G. Phys. ReV. B 1988, 37, 785.
(20) Kone´, M.; Illien, B.; Graton, J.; Laurence, C. J. Phys. Chem. A
2005, 109, 11907.
(21) Stratmann, R. E.; Scuseria, G. E.; Frisch, M. J. J. Chem. Phys.
1998, 109, 8218.
(22) Svechkarev, D. A; Baumer, V. N.; Syzova, Z. A.; Doroshenko, A. O. J. Mol. Struct. 2008, 882, 63.
(23) (a) Brewer, W. E.; Martinez, M. L.; Chou, P. T. J. Phys. Chem.
1990, 94, 1915. (b) Ikegami, M.; Arai, T. J. Chem. Soc., Perkin Trans. 2002, 2, 1296.
(24) Weber, W.; Helms, V.; McCammon, J. A.; Langhoff, P. W. Pro.
Natl. Acad. Sci. U.S.A. 1999, 96, 6177.
(25) (a) Falkovskaia, E.; Pivovarenko, V. G.; del Valle, J. C. Chem.
Phys. Lett. 2002, 352, 415. (b) Falkovskaia, E.; Pivovarenko, V. G.; del
Valle, J. C. J. Phys. Chem. A 2003, 107, 3316. JP809072A
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009