學]
On: 27 April 2014, At: 22:19 Publisher: Taylor & Francis
Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK
Journal of Biomaterials
Science, Polymer Edition
Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/tbsp20
Synthesis and biological
properties of
antitumor-active conjugates of ADR with
dextran
Jan-An Guu , Ging-Ho Hsiue & Tzuoh-Miin Juang Published online: 02 Apr 2012.
To cite this article: Jan-An Guu , Ging-Ho Hsiue & Tzuoh-Miin Juang (2002) Synthesis and biological properties of antitumor-active conjugates of ADR with dextran , Journal of Biomaterials Science, Polymer Edition, 13:10, 1135-1151, DOI: 10.1163/156856202320813846
To link to this article: http://dx.doi.org/10.1163/156856202320813846
PLEASE SCROLL DOWN FOR ARTICLE
Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) contained in the publications on our platform. However, Taylor & Francis, our agents, and our licensors make no representations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of the Content. Any opinions and views expressed in this publication are the opinions and views of the authors, and are not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon and should be independently verified with primary sources of information. Taylor and Francis shall not be liable for any losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoever
relation to or arising out of the use of the Content.
This article may be used for research, teaching, and private study purposes. Any substantial or systematic reproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in any form to anyone is expressly forbidden. Terms & Conditions of access and use can be found at http://www.tandfonline.com/page/terms-and-conditions
Also available online -www.vsppub.com
Synthesis and biological properties of antitumor-active
conjugates of ADR with dextran
JAN-AN GUU1, GING-HO HSIUE2;¤and TZUOH-MIIN JUANG1
1Department of Applied Chemistry, National Chiao Tung University, Hsinchu, 300, Taiwan 2Department of Chemical Engineering, National Tsing Hua University, Hsinchu, 300, Taiwan
Received 10 February 2002; revised 10 May 2002; accepted 5 July 2002
Abstract—Three kinds of polymeric adriamycin (ADR) conjugates of dextran were synthesized, namely a dextran– Gly-Leu-Gly-ADR (DGLGA) conjugate with a lysosomally degradable tripeptide spacer group, a dextran– Gly-Leu-Gly-ADR-galactosamine (DGLGA-Ga) conjugate with a targeting moiety of galactosamine on DGLGA, and a dextran– C6H10-ADR (DC6A) conjugate with a
hexam-ethylen spacer group. The content of the ADR moiety in the polymeric-drug conjugate was about 3 mol%. Enzyme hydrolysis of DGLGA and DC6A was carried out by incubation with papain. The
total amount of ADR released after 48 h was 43 mol% for DGLGA and less than 1 mol% for DC6A.
In an in vitro cytotoxicity experiment, the DGLGA-Ga conjugate has higher cytotoxic ef cacy than the other conjugates for incubation with Hep-3B cells and consequently, the capability of targeting he-patoma cells of the galactosamine residue was determined. In contrast, for the incubation with SiHa cells of these conjugates, there was no signi cant cytotoxicity effect. The in vivo cytotoxic ef cacy of each conjugate (20 mg ADR equiv./ kg) against CT-26 mice colon cells implanted subcutaneously in Balb-C mice was studied. The DGLGA conjugate generated the best therapeutic effect with the presence of long-term survival (LTS) at day 50 (2/6).
Key words: Dextran; polymeric pro-drug; antitumor activity; adriamycin; galactosamine.
INTRODUCTION
The anthracycline antibiotic adriamycin (ADR) shows a valuable broad spectrum of antitumour activity in humans [1]. However, its clinical use is often limited by peripheral toxicity [2]. Just like the problems caused by the use of ADR in cancer chemotherapy, low-molecular-weight antitumor drugs generally have serious side-effects, due to a high concentration of toxicity in normal tissue and the short duration of activity. Hence, the development of a potential drug delivery system capable of
¤To whom correspondence should be addressed.
selectively concentrating anticancer agents at the targeting tissue and prolonging the treatment duration of anticancer agents is much needed.
One strategy to improve chemotherapy is to prepare new drug forms by using water-soluble synthetic or natural polymers as carriers of biologically active com-pounds. Biocompatible water-soluble synthetic and/ or natural polymers have been extensively utilized as drug carriers to improve the therapeutic ef cacy of anticancer drugs by prolonging the length of their residence in human bodies and increasing the selectivity to the desired sites. Several macromolecular compounds have been suggested as drug carriers, including serum albumin, brinogen, immunoglobulin, lectins, polysaccharides, DNA, liposomes, and synthetic polymers [3– 5]. In 1974, De Duve et al. [6] developed the ‘lysosomotropic agent’ concept based on drug-carrier conjugates that are selectively taken up into lysosomes. The drugs then demonstrated their pharmacological activity by releasing bioactive agents from the polymeric carriers and penetrating through lysosomal membranes into cytoplasm. Accordingly, Ringsdorf presented a model of the use of a polymer as a targetable drug carrier [7]. The model combines the concept of site-speci c drug release with site-speci c recognition and predicts some important bene cial properties of polymer– drug conjugates, compared with their low-molecular-weight analogues. Based on this model, it is reasonable to argue that dextran conjugates containing ADR are a promising approach. The drug is covalently bound to the polymer via oligopeptide spacers that are designed speci cally for stability in the circulation [8] and terminal cleavage by lysosomal enzymes following pinocytic internaliza-tion of the conjugate [9]. Dextran funcinternaliza-tions as a polymer carrier because it is a water-soluble, biodegradable, and non-antigenetic natural polysaccaride [10]. The hydroxy groups of dextran can be replaced by pendant modi able carboxylic deriv-atives for the grafting of drug derivderiv-atives. It can also be covalently attached to both the parent drug and the targeting unit. Furthermore, between the polymer carrier and the drug, a well-designed oligopeptide spacer could be degraded by enzymes, which could prompt the release of the drug. Kopecek and co-workers synthesized a macromolecular pro-drug, i.e. HPMA copolymers xing p-nitroaniline (NAp) residues as model drugs through various peptidyl spacer groups, and investigated the enzymatic release behavior of NAp from HPMA copolymer by lysosomal pro-teases in vitro. They found that the release rates were signi cantly accelerated by increasing the length of the oligopeptidyl spacer and also depended on the speci c sequence of the oligopeptide. Because the tripeptide Gly-Leu-Gly possesses the es-sential requisite that it can be degraded by lysosome enzymes, it was used in this study as the spacer far drug binding [11].
In addition, targeted drug deliver may improve the chemotherapy of tumors. Liver-speci c targeting can be achieved intravenously by the use of vehicles de-signed to interact with liver-associated receptors, such as the hepatocyte galactose receptor. Galactosamine, a well-known compound that can be bound with a hepato-cyte receptor [12] to achieve active targeting, is introduced into the dextran– ADR conjugate for liver targeting.
The main aim of the present study was the synthesis of dextran– ADR conju-gates and to investigate their in vitro and in vivo properties. The preparation of a dextran– ADR conjugate with a tripeptide spacer (Gly-Leu-Gly) and a hexam-ethylene spacer is described. Furthermore, galactosamine, a terminal moiety that can be used to target polymer conjugates to liver hepatocytes, was incorporated into one dextran conjugate. The hydrolysis of these conjugates to release ADR in the presence of papain and pH 7.4 buffer solutions was carried out. The cytotoxicity effect of the conjugates against Hep-313 and SiHa human tumor cells and the ther-apeutic effect on CT-26 tumor cells implanted in mice were also investigated.
MATERIALS AND METHODS
Materials
Dextran was purchased from Pharmacia Fine Chemicals Co., Uppsala, Sweden, and had an average molecular weight of 40 000 (T-40). Adriamycin was from Pharmacia & Upjohn S.p.A. N-t-Boc-glycine-N-hydroxysuccinimide ester (Boc-Gly-Osu), pa-pain, glutathione (GSH), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from the Sigma Chemical Co., USA. Tri uo-roacetic anhydride (TFAA) was provided by Merck, Darmstadt, Germany, while all the other chemicals were of commercial grade and were used without further puri cation.
Synthesis of Boc-glycyl-L-leucine (Boc-Gly-Leu) (1)
Boc-Gly-OSu (5 g, 18.4 mmol) and leucine (2.74 g, 20.8 mmol) were added to a dimethyl sulfoxide (DMSO; 10 ml) solution and the reaction was conducted by stirring for 3 days at room temperature. The DMSO was then removed and ethyl acetate was added to dissolve the dry residue. The undissolved residue was ltered. The reaction mixture was extracted with 10% citric acid solution (3 £ 20 ml). The organic layer was isolated and the water layer extracted with ethyl acetate (2 £ 20 ml). The ethyl acetate extracts were combined and washed with water and then dried with MgSO4. Ethyl acetate was removed using a rotary evaporator under
reduced pressure. The crude product was puri ed further by recrystallization from ethyl acetate and n-hexane to produce a white powder product. The preparation procedure of this reaction is presented as equation (1) in Scheme 1. Yield 80.6%; m.p. 136– 137±C.1H-NMR [(CD
3/2SO]: ± 0.84 (dd, 6H, CH3), ± 1.36 (s, 9H, CH3),
± 1.48 (m, 2H, CH2), ± 1.59 (m, 1H, CH), ± 3.53 (d, 2H, CH2), ± 4.21 (m, 1H, CH),
± 6.89 (t, 1H, NH), ± 7.92 (d, 1H, NH).
Synthesis of Boc-glycyl-L-leucyl-4-nitrophenol ester (Boc-Gly-Leu-ONp) (2) Boc-Gly-Leu (3 g, 10.4 mmol) and p-nitrophenol (1.74 g, 12.5 mmol) were dissolved in dimethyl formamide (DMF; 10 ml). The mixture was cooled in an ice
Scheme 1. Synthesis of the tripeptide spacer Gly-Leu-Gly.
bath, dicyclohexylcarbodiimide (DCC) (2.79 g, 13.5 mmol) was added with stirring, and the mixture was then stirred at room temperature for 24 h. DMF was removed using a rotary evaporator and ethyl acetate was added to dissolve the dry residue; the precipitate of the mixture was removed by ltration. The residue was extracted with a 10% citric acid solution (3 £ 20 ml) and then with saturated sodium bicarbonate (3 £ 20 ml). The organic layer was isolated and the water layer extracted with ethyl acetate (2 £ 20 ml). The ethyl acetate extracts were combined and washed with water, dried with MgSO4, and evaporated to dryness in vacuo. The crude product
was puri ed further by recrystallization from ethyl acetate and n-hexane to produce a white powder product [equation (2) in Scheme 1]. Yield 76%; m.p. 123– 125±C. 1H-NMR [(CD
3/2SO]: ± 0.96 (t, 6H, CH3), ± 1.40 (s, 9H, CH3), ± 1.67 (m, 2H,
CH2), ± 1.75 (m, 1H, CH), ± 3.82 (d, 2H, CH2), ± 4.74 (d, 1H, NH), ± 5.31 (t, 1H,
NH), ± 6.88 (m, 1H, NH), ± 7.25 (d, 2H, CH), ± 8.22 (d, 2H, CH).
Synthesis of Boc-glycyl-L-leucyl-glycine (Boc-Gly-Leu-Gly) (3)
Boc-Gly-Leu-ONp (2 g, 4.9 mmol) was dissolved in dry DMSO (5 ml) and then glycine (0.4 g, 5.4 mmol) was added. The mixture was stirred for 3 days at room temperature. DMSO was removed and ethyl acetate was added to dissolve the dry residue; the precipitate was removed by ltration. The reaction mixture was extracted with a 10% citric acid solution (3£20 ml). The organic layer was isolated and the water layer extracted with ethyl acetate (2 £ 20 ml). The ethyl acetate extracts were combined and washed with water, dried with MgSO4, and evaporated
to dryness in vacuo. The crude product was puri ed by recrystallization from ethyl acetate and n-hexane to produce a white solid product [equation (3) in Scheme 1]. Yield 36%; m.p. 146– 147±C.1H-NMR [(CD
3/2SO]: ± 0.83 (t, 6H, CH3), ± 1.36
(s, 9H, CH3/, ± 1.43 (m, 2H, CH2), ± 1.60 (m, 1H, CH), ± 3.52 (d, 2H, CH2), ± 3.71
(d, 2H, CH2), ± 4.32 (m, 1H, CH), ± 6.95 (t, 1H, NH), ± 7.84 (d, 1H, NH), ± 8.25 (t, 1H, NH).
Synthesis of dextran 4-nitrophenyl carbonate[dextran– COO(C6H4)NO2]
The activation of dextran was carried out according to a similar method de-scribed by Vandoorne et al. [13]. Dextran (5 g) was dissolved in 20 ml of DMSO/pyridine 1/ 1 to (v/ v) mixture. The solution was cooled to ¡10±C and
then 4-dimethylaminopyridine (DMAP) (0.2 g, 1.6 mmol) and 4-nitrophenyl chlo-roformate (2.3 g, 11.4 mmol) were added. The reaction mixture was stirred at a low temperature (¡10±C) for 30 min and then slowly dropped into ethanol (300 ml)
with stirring to obtain a white precipitate. The precipitate was washed with a large excess of ethanol (3 £ 100 ml) and evaporated to dryness in vacuo to produce a white dextran– COO(C6H4)NO2 powder. The degree of actvation of dextran was
determined by the hydrolysis of activated dextran in NaOH solution. Activated dex-tran (100 mg) was dissolved into 10 ml of 0.1Nsodium hydroxide solution and the absorption of the carbonate residue was monitored by UV–visible spectroscopy at 402 nm for the p-nitroaniline group. The content of carbonate was determined using Beer’s law with " D 18 400 [13].
Synthesis of the dextran– Gly-L-Leu-Gly conjugate (6)
Boc-Gly-Leu-Gly (1 g, 2.9 mmol) (3), prepared previously as shown in Scheme 1, was placed into a 25 ml round-bottomed ask. Tri uoroacetic acid (2 ml) was added and the reaction was conducted under stirring at room temperature for 45 min. The mixture was evaporated in vacuo to produce the oily glycyl-L-leucyl-glycine (Gly-Leu-Gly). The oily residue of Gly-Leu-Gly (0.2 g, 0.8 mmol) was dissolved in a DMSO solution in which dextran– COO(C6H4/NO2(4.3 g) was dissolved and then
triethylamine (0.2 ml) was added. The mixture was stirred at room temperature for 3 days and then slowly dropped into ethanol (300 ml) with stirring to obtain a white precipitate. The precipitate was washed with a large excess of ethanol (3 £ 100 ml) and evaporated to dryness in vacuo to produce a white dextran–Gly-Leu-Gly conjugate powder (see Scheme 2).
Synthesis of the dextran– C5H10COOH conjugate
N-t-Boc-6-aminohexanoic acid (Boc-C5H10COOH) (1 g, 4.3 mmol) was placed
into a 25 ml round-bottomed ask. Tri uoroacetic acid (2 ml) was added and the reaction was conducted by stirring at room temperature for 45 min. After removal of excess tri uoroacetic acid and by-products by evaporation, the oily
Scheme 2. Synthesis of the dextran– Gly-Leu-Gly-galactosaminederivative.
NH2C5H10COOH residue (0.2 g, 1.52 mmol) was dissolved in a DMSO solution
in which dextran– COO(C6H4/NO2 (4.3 g) was dissolved and then triethylamine
(0.2 ml) was added. The mixture was stirred at room temperature for 3 days, then slowly dropped into ethanol (300 ml) and stirred to achieve a white precipitate. The precipitate was washed with a large excess of ethanol (3 £ 100 ml) and evaporated to dryness in vacuo to produce a white dextran– C5H10COOH conjugate.
Synthesis of the dextran– Gly-Leu-Gly-ADR (DGLGA) conjugate (7)
For the synthesis of the dextran– Gly-ADR conjugate, dextran– Gly-Leu-Gly (88 mg, 0.54 mmol), adriamycin (10 mg, 0.016 mmol), and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (363 mg, 1.62 mmol) were mixed and stirred in 15 ml of puri ed water, and the pH of tie mixture was adjusted to 8 using HCl (1% w /w). The solution was under reaction by stirring for 48 h at room temperature. Puri cation of the mixture was carried out by dialysis with an 8000– 10 000 molecular weight dialysis membrane. The puri ed solution was freeze-dried to obtain the red-colored DGLGA conjugate. The content of ADR in DGLGA was determined by UV–visible spectroscopy. The maximum absorption wavelength of ADR and DGLGA has been detected to be about 254 nm. A calibration curve was made by detecting different concentration of ADR solution at 254 nm. The absorption of DGLGA was measured at 254 nm and then the ADR content of DGLGA was found by comparison with the calibration curve of ADR. In this study, the ADR content of DGLGA was about 3 mol%. Moreover, the synthesis dextran– C6H10-ADR (DC6A)(7) followed the same procedure of the synthesis of
DGLGA.
Release of ADR from the dextran– ADR conjugate
The enzymatic hydrolysis of DGLGA was carried out by incubation with papain. DGLGA was dissolved in a buffer solution containing 0.10 Mcitric acid, 0.10 M disodium hydrogen phosphate, and 1 mM EDTA and the mixture was adjusted to pH 5.50 with NaOH. After DGLGA was dissolved, glutathione (23 mg) and papain (12 mg) were added. The mixture was incubated in a shaking bath at 37±C. A 1 ml
sample of the mixture was removed every hour in the rst 24 h and every 12 h thereafter. The samples were analyzed by HPLC with a Phenomenex LUNA 5u C18 (2) column. The eluant was prepared by mixing PBS (pH 7.4) and acetonitrile (80/ 20, v/ v) and the analysis was run with a ow rate of 1 ml/ min. The eluate was monitored at 254 nm with a TSP SPECTRA 100 VARIABLE UV/visible CE detector. The enzymatic hydrolysis experiment of DC6A was conducted using the
same method.
Cytotoxicity of DGLGA against Hep-3B cells
A semi-automated tetrazolium-based colorimetric (NTT) assay [14] was employed to examine the cytotoxicity effect of DGLGA and DC6A. Exponentially growing
cells were cultured in 96-well plates and treated with various concentrations (5, 2.5, 0.5, 0.05, 0.005 ¹M) of DGLGA, DC6A, and ADR for 3 days at 37±C. After
exposure of the cells to the dextran– ADR conjugates, the cells were washed twice with a serum-free medium and once with water. 0.5 ml of a medium solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was added to each well and the MTT assay was performed 4 h later. The entire assay was repeated twice and growth inhibition was calculated using the following equation:
growth inhibition (%) D [T ] ¡ [B]
[C] ¡ [B] £ 100%:
In this equation, [T ] is the amount of cells in the treatment group after incubation for 72 h; [C] is the number of cells in the control group after incubation for 72 h; and [B] is the initial number of cells.
In vivo cytotoxicity of DGLGA against CT-26 cells in mice
The experiments of conjugates against Balb-C mice (males, 7– 8 weeks old, 20– 30 g) were inoculated subcutaneously at day 0 with 106 viable CT-26 cells.
ADR or polymeric ADR conjugates were administered intraperitoneally as a single dose of 20 mg ADR-equiv./ kg at day 4. Five groups of mice were tested, with six mice in each group. The weight and tumor size of the animals were measured daily. The survival time and number of long-term survivors (LTS) at day 50 were monitored.
RESULTS AND DISCUSSION
Preparation of the dextran– ADR conjugate
Dextran is a water-soluble, biodegradable, and non-antigenetic natural polysac-caride; thus, it has many essential properties to be a polymer carrier in a conjuga-tion pro-drug system. However, the hydroxy groups of dextran cannot connect with spacer and drug directly. Hence, pre-activation of dextran is required, to replace the hydroxy group with pendant modi able carboxylic derivatives before being cou-pled with the spacer and drug. A number of methods can be used to transform the polysaccaride hydroxyl group into suitable derivatives that can react with amines or thiols [15]. In this study, the activation of dextran was conducted by mixing dextran with 4-nitrophenyl chloroformate to produce the corresponding carbonate moiety [13] (Scheme 2). The carbonate content in the activated dextran was determined by hydrolyzing the activated dextran that dissolves in NaOH aqueous solution. When the activated dextran was dissolved into 0.1 N NaOH.aq/, a yellow color solution was obtained. Then the absorption of the solution was monitored by UV–visible spectroscopy and the amount of carbonate in dextran was calculated using Beer’s law. For the purpose of realizing the reaction conditions, the time dependence of the degree of activation of dextran at 0±C was determined. As the time proceeded,
the proportion of p-nitrophenyl carbonates increased accordingly. After reaching the maximal value at about 24 h, it slowly decreased. Moreover, dextran activation was carried out at 0±C for 30 min at the beginning and then the dextran was warmed
back to room temperature. As a result, the content of p-nitrophenyl carbonate in-creased with time until 30 min and then started to decrease; afterwards, the amount remained at less than half of the maximal yalue at 12 h. These results reveal that the reaction time and temperature affects the activation degree of dextran. They also possibly suggest the presence of side-reactions, i.e. slow hydrolysis of the active species, or reaction of the active carbonate with polymeric hydroxyls with the for-mation of inter- or intra-molecular carbonate structures [13]. In order to avoid the excessive formation of those side-products, the activation reaction of dextran was carried out at ¡10±C for 30 min and the amount of carbonate in dextran, calculated
with Beer’s law, was about 11 mol%. We were also able to produce dextran of a higher activation degree by controlling the reaction time and temperature. However, the highly activated product was almost insoluble in water, so it could not be used for the subsequent reaction.
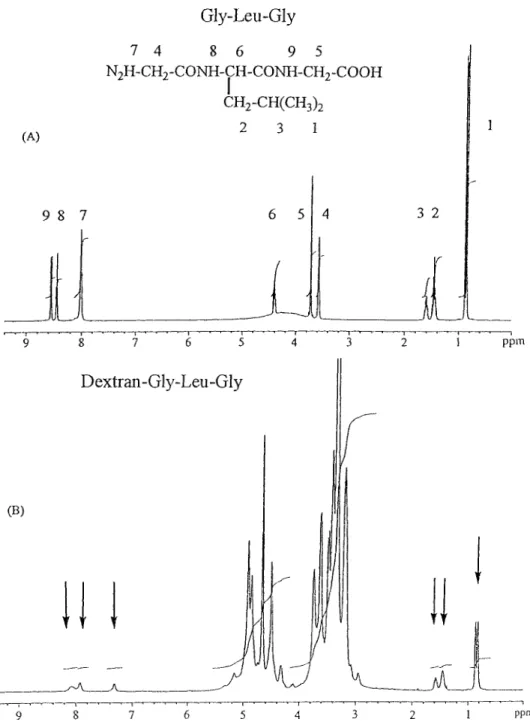
The tripeptide spacer Gly-Leu-Gly was synthesized based on solution-phase peptide synthesis methods. The NH group protected glycine (Boc-Gly) was used as the starting material, to which leucine and the other glycine were connected to it one-by-one via amide bonding. The protection group Boc was then disposed of by tri uoroacetic acid. After Gly-Leu-Gly was produced, it was coupled with the activated dextran though amide bonding. The NMRl spectra of the spacer component Gly-Leu-Gly and dextran– Gly-Leu-Gly are shown in Fig. 1. In Fig. 1B, besides the strong absorptions of the hydroxyl group between 4.2 and 5.2 ppm,
Figure 1. 1H-NMR spectra [(CD
3/2SO] of (A) Gly-Leu-Gly and (B) dextran– Gly-Leu-Gly.
some smaller absorption peaks approximately match the absorption of Gly-Leu-Gly, as shown in Fig. 1A. These peaks are thought to be due to the tripeptide spacer residue. The absorption of amino groups of the tripeptide spacer between 7 and 9 ppm had shifted slightly from low eld (Fig. 1A) to high eld (Fig. 1B); this
Scheme 3. Synthesis of the dextran– Gly-Leu-Gly-ADR-galactosamine conjugate.
should be in uenced by the hydrogen bonding interaction between the amide group of Gly-Leu-Gly and the hydroxy groups of dextran. The content of Gly-Leu-Gly in the dextran derivative was approximately 6 mol%, which was determined by measuring the ratio of the peak area of Gly-Leu-Gly versus dextran in the NMR spectrum.
The attachment of ADR to the dextran derivative was accomplished using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) as the cou-pling reagent under aqueous conditions [16] (Scheme 3). The amount of ADR, determined by detection and calculation via UV spectra, was about 3 mol%. In a previous report, this reaction was allowed to proceed for 48 h at room temperature and the pH value was adjusted to between 5 and 6 [16]. However, in the present study we found that the bonding ratio at pH 8 (3.1 mol%) was higher than that at pH 5.5 (1.2 mol%). Such a result was probably due to the process of reduction of ADR from the reagent that had to be performed under basic conditions. Thus, the reaction of attaching ADR to dextran was carried out at pH 8 in this study.
Release of ADR from the dextran– ADR conjugate
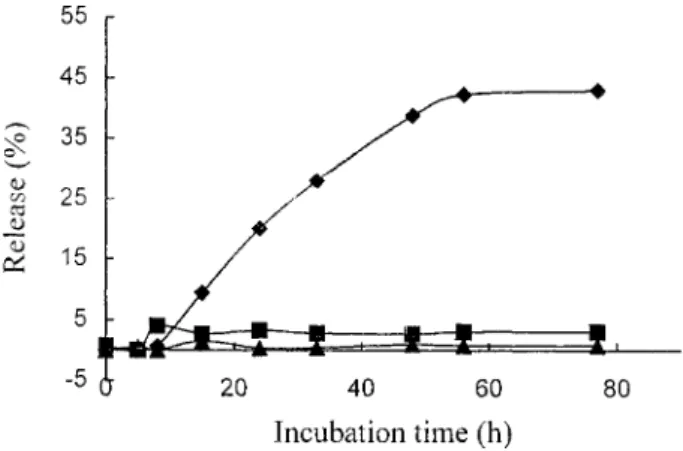
The hydrolysis of the dextran– ADR conjugate was carried out by incubating the conjugate with papain (or PBS) at 37±C. ADR residues were analyzed once an hour
by HPLC using UV detection at 204 nm and the time dependende of the release of ADR was plotted. The results of the hydrolysis of DGLGA by papain and PBS is shown in Fig. 2. As time proceeded, ADR was released under treatment with papain; the total release was approximately 43 mol% at 48 h and then the release of ADR slowed down. When treated with PBS, the release of ADR was almost negligible. DC6A was also incubated with papain and the results are also shown in
Fig. 2, in comparison with DGLGA. DC6A presented a rather low releasing effect
in the degradation of papain.
In the development of targetable lysosomotropic drug delivery systems, the drug carrier linkage is of utmost importance. All macromolecular carriers ultimately
Figure 2. Time dependence of the release of ADR after incubation of the DGLGA conjugate with papain (u ), in pH 7.4 buffer solution (f), and the incubation of DC6A with papain (s).
arrive in the lysosomal compartment of the cell following their pinocytic capture. Drug– polymer linkages are designed to be hydrolyzed by lysosomal enzymes and to be resistant to attack in other body compartments. Kopecek et al. reported in a detailed study that lysosomal enzyme degraded the oligopeptide side-chain in HPMA copoymer. They concluded that in the lysosome of liver cells, cathepsin B is the most important thiol protease that plays a major role in degrading natural and synthetic polypeptides [17]. Papain is a cysteine protease and has enzymatic speci city similar to cathepsin B [18]. Therefore, papain could be used as a model enzyme to carry out the enzymatic hydrolysis of dextran– ADR conjugates, as was used in this study.
An extensive study on the release of a model compound from poly[N-(2-hydroxy-propyl) methacrylamide] (HPMA) by changing the peptide spacers has also been reported [19]. The release rate of p-nitrophenol (a model compound) from the polymer was strongly dependent on the length and sequence of the peptide spacers between the main chain and p-nitrophenol. Compared with the case of a dipeptide spacer, conjugates with tripeptide sequences led to much higher release rates (approximately 3–8 times) and their rates were dependent on the speci c sequence. For tetrapeptides, the release rates were much accelerated and also dependent on the sequence. In the present study, we introduced a tripeptide chain into the dextran– ADR conjugate as the lysosomal enzyme degradable spacer. The tripeptide spacer Gly-Leu-Gly has suf cient length and the sequence followed the rules of oligopeptide design [20]. Therefore, DGLGA released ADR by the speci c hydrolysis of papain but did not release ADR in pH 7.4 phosphate buffer solution, which is analogous situation in the blood stream. DC6A, with a hexamethylene
spacer that could not be digested by enzyme, shows an insigni cant release of ADR under incubation with papain.
Cytotoxicity of the dextran– ADR conjugate against tumor cellsin vitro
The lysosomotropic agent concept [6] explains how the polymeric-drug conjugate enters cells through the endocytosis route and is digested by the enzymes in lysosome to release the drug. Therefore, dextran– drug conjugates, after pinocytosis into the cells, will eventually end up within the lysosomal compartment. The cytotoxicity effect of these conjugates was determined by MTT assay against Hep-3B and SiHa cells. Hep-Hep-3B (or SiHa) cells were cultured for 72 h under exposure to the polymeric conjugates. The results for Hep-3B are shown in Fig. 3. ADR shows the highest growth-inhibitory effect against Hep-3B cells among the ve conjugates, for it enters cells by diffusion, or facilitated the transport pathway without driven energy. The cytotoxicity effect of the other conjugates follows the order DGLGA-Ga > DGLGA > DC6>dextran. The intensity of the cytotoxicity
of these conjugates could also be separated into two groups; the higher cytotoxicity group conjains DGLGA-Ga and DGLGA, and the lower cytotoxicity group has DC6A and dextran. DGLGA-Ga and DGLGA have a tripeptide spacer between the
polymer carrier and ADR. Such a tripeptide spacer with the sequence Gly-Leu-Gly was designed to match the active subsite of the lysosomal enzyme. Chiu et al. [11] investigated this tripeptide as the spacer in a poly(amino acid)– drug conjugate, in which the model drug was released at moderate rate. Thus, the enzyme-degradable spacer promoted the drug release effect of DGLGA-Ga and DGLGA. However, the spacer that connects dextran and ADR of DC6A is a hexamethylene group,
which could not be digested by the lysosomal enzyme at all. Therefore, it shows no signi cant growth inhibition effect towards tumor cells and the result is similar to that of dextran (Fig. 3). In addition, the macromolecular carrier itself may in uence the activity of the conjugate. Even when it is not connected with any drugs, dextran shows a slight effect on the growth inhibition of tumor cells. Forster and Lloyd [21] also point out that the permeability limit of lysosomal membranes lies approximately in the size of dipeptides. The accumulation of dextran molecules in lysosomes gives rise to problems of osmotic balance of lysosomal membranes and, consequently, malfunction of the cells. As a result, the accumulation of dextran has little effect on the growth inhibition of tumor cells.
Hepatocytes have a receptor for galactoseterminated glycoproteins, which ef -ciently takes them up from the circulation [22, 23]. Thus, the galactosamine residues in an anticancer pro-drug are usually utilized as the targeting moiety, based on the ability of acutely targeting at the surface of the hepatoma cell [24]. In the present study, the higher cytotoxicity effect of DGLGA-Ga than DGLGA might be due to its faster rate of cellular uptake (Fig. 3).
SiHa cells are a squamous carcinoma cell line of the cervix [25], in which there is no lysosomal enzyme, as in hepatocytes. In the cytotoxicity experiments against SiHa cells, free ADR drug displayed the highest growth-inhibitory effect; DGLGA-Ga, DGLGA, DC6A, and dextran have similar cytotoxicity effects without a
signi cant effect of cell killing (Fig. 4). This shows that regardless of whether there is an oligopeptidyl spacer in the drug– polymer conjugate or not, the cytotoxicity
Figure 3. Cytotoxicity effect of DGLGA-Ga (f), DGLGA (s), DC6A ({), free ADR drug (E), and
the control group (F) against Hep-3B hepatoma cells in vitro. The cells were exposed for 72 h to the conjugate. Untreated cells were used as the control group.
Figure 4. Cytotoxicity effect of DGLGA-Ga (e), DGLGA (n ), DC6A ({), free ADR drug (E), and
the control group (F) against SiHa cells in vitro. The cells were exposed for 72 h to the conjugate. Untreated cells were used as the control group.
effects are all the same. This result re ects the requirement of a suitable enzyme for the digestion of oligopeptide, which helps to release ADR from dextran– drug conjugates.
Cytotoxicity of the dextran– ADR conjugatein vivo
The in vivo cytotoxicity effect of DGLGA, DGLGA-Ga, DC6A, and free ADR drug
was examined in order to compare their ability to suppress the growth of tumor cells in Balb-C mice and to prolong the survival time of mice. The murine colon carcinoma cell line CT-26 was implanted in mice. This cell line contains many
enzymes with similar capability to the cells existing in liver and lung [26] and its distinct shape is easy to detect. A single dose (5 mg ADR-equiv./ kg) of each polymeric ADR conjugate, as well as free ADR, was administered intraperitoneally on day 4 after tumor cells were implanted in the mice. The survival time of the mice was monitored and is shown in Fig. 5. Tumor growth was not suppressed by any of the conjugates and no conjugate was able to extend the survival time of the mice. This is possibly due to an insuf cient drug dosage.
Figure 5. Effect of the treatment of Balb-C mice carrying i.p. CT-26 with a single dose (5 mg ADR eqiv./kg) of DGLGA-Ga ({), DGLGA (n ), DC6A (E), free ADR drug (e), and the control group
(E) on the survival time. The mice carrying CT-26 in the control group were not treated with any conjugate.
Figure 6. Effect of the treatment of Balb-C mice carrying i.p. CT-26 with a single dose (20 mg ADR eqiv./kg) of DGLGA-Ga ({), DGLGA (n ), DC6A (E), free ADR drug (f), and the control group
(F) on the survival time. The mice carrying CT-26 in the control group were not treated with any conjugate.
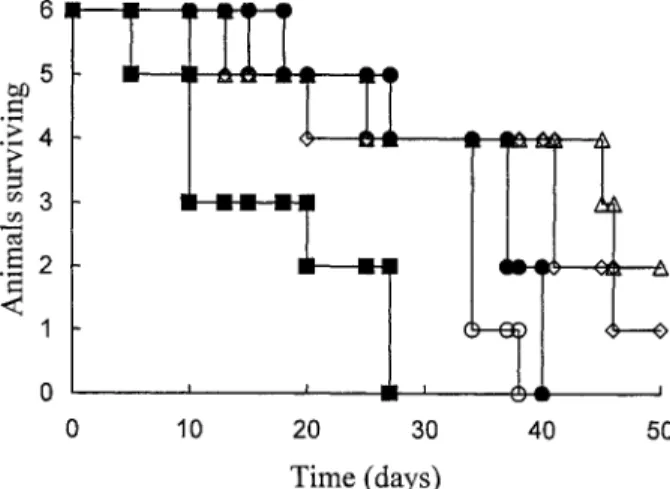
Therefore, the dosage was increased from 5 mg to 20 mg in each polymeric ADR conjugate, and a single dose was administered intraperitoneally to mice after the tumor cells were implanted. The survival time and the tumor size were monitored as shown in Figs 6 and 7, respectively. The mean survival time (MST) and long-term survivors (LTS) are given in Table 1. The survival time of the mice treated by free ADR is shorter than that of the untreated control group. This indicates that ADR exhibits high toxicity to mice in such a dosage. Animals treated with DC6A
had increased in tumor size and had a similar survival time to the untreated control group. This is because DC6A does not possess high toxicity. The poor therapeutic
effect of DC6A revealed that ADR is not released from the polymer– drug conjugate.
Moreover, the toxicity of the ADR component is reduced by the shielding effect of the polymer chain attached to dextran. The tumor growth of animals treated with DGLGA was suppressed to only a small degree (Fig. 7), yet their survival time was much longer than that of the control group and the free ADR drug-treated group. In fact, two mice reached a LTS longer than 50 days. This implies that DGLGA
Figure 7. Effect of the treatment of Balb-C mice carrying i.p. JZT-26 with a single dose (20 mg ADR eqiv./kg) DGLGA-Ga ({), DGLG (n ), DC6A (E), free ADR drug (f), and the control group (F)
on the tumor size. The mice carrying CT-26 in the control group were not treated with any conjugate. Table 1.
Effect of i.p. administration of PGA– ADR conjugates on Balb-C mice (nD 6) bearing 106CT-26
cells inoculated i.p. on day 0 (dose 20 mg ADR /kg on day 4)
Compound Survival time (days) MST (SE) T /C (%) LTS (day 50) (days) Control 18, 27, 37, 37, 40, 40 33.2 (3.6) — 0/ 50 ADR 5, 10, 10, 20, 27, 27 16.5 (3.9) 49.7 0/ 50 DGLGA 13, 25, 45, 46 32.3 (8.0) 97.3 2/ 50 DGLGA-Ga 10, 20, 41, 41, 46 31.6 (7.0) 95.2 1/ 50 DC6A 15, 25, 34, 34, 34, 38 30 (3.5) 90.4 0/ 50
exhibits a certain degree of activity on therapeutics. This is probably due to the fact that ADR released from the Gly-Leu-Gly spacer of the polymer– drug conjugate is able to eliminate some of the tumor cells. The survival result of mice treated with DGLGA-Ga is similar to that of the DGLGA-treated group. The targeting function of the galactosyl group did not have an effect in the DGLGA-Ga therapeutic case. This is because the galactosyl– dextran conjugates have high af nity only to liver cells, without any af nity to other tissues [30].
CONCLUSION
A dextran– ADR conjugate, DGLGA, having a Gly-Leu-Gly tripeptide spacer was synthesized. In addition, DGLGA-Ga, which is a conjugate that contains a tripeptide spacer and a liver-targeting galactosamine moiety, was prepared. Gly-Leu-Gly was synthesized using the solution-phase peptide synthesis method. The biologically active agent ADR was xed onto dextran under aqueous conditions in which the pH was adjusted to 8. The hydrolysis results show that DGLGA releases iree ADR speci cally in response to papain; in other words, the dextran– ADR conjugate containing the Gly-Leu-Gly spacer is not expected to release ADR in the blood stream (pH D 7.4), but to release effectively after uptake into tumor cells through hydrolysis of the oligopeptide spacer by lysosomal enzyme. Since DGLGA-Ga has the highest cytotoxicity effect compared with the other conjugates in vitro, it is responsible for the active targeting of cell-speci c af nity of the conjugate for hepatoma cells via galactose receptor-mediated endocytosis. Although the targeting effect of the polymer– drug conjugate was not observed using CT-26 as the cell line in the in vivo experiment, the polymer– drug conjugates DGLGA and DGLGA-Ga do exhibit a therapeutic effect on mice. In summary, it is reasonable to propose that dextran– ADR conjugates containing oligopeptide and a targeting moiety can be used for cancer chemotherapy.
REFERENCES
1. R. H. Blum, Cancer Chemother Rep. 6, 247 (1975).
2. B. Neri, G. Cini-Neri, M. Bandinelli, S. Bartalucci and A. Ciapio, Int. J. Clin. Pharmacol. Ther. Toxicol21, 217 (1989).
3. Y. Tsukada, W. K. D. Bischof, N. Hibi, H. Hirai, E. Hurwitz and M. Sela,Proc. Natl Acad. Sci. USA79, 621 (1982).
4. T. Kojima, M. Hashida, S. Muranishi and H. Sezaki,J. Pharm. Pharmacol.32, 30 (1980). 5. W. C. Shen and G. J. P. Ryser,Proc. Natl Acad. Sci. USA75, 1872 (1978).
6. C. De Duve, T. de Barsy, B. Poole, A. Trouet, P. Tulkens and F. van Hoof,Biochem. Pharmacol. 23, 2495 (1974).
7. H. Ringsdorf, J. Polym. Sci. 51, 135 (1975).
8. P. Rejmanova, J. Kopecek, R. Duncan and J. B. Lloyd,Biomaterials6, 45 (1895).
9. R. Duncan, H. C. Cable, J. B. Lloyd, P. Rejmanova and J. Kopecek,Makromol. Chem.184, 1997 (1984).
10. L. Molteni, in: Drug Carrier in Biology and Medicine, G. Gregoriadis (Ed.), pp. 107– 128. Academic Press, London (1979).
11. H. C. Chiu, C. Konak, P. Kopeckova and J. Kopecek, J. Bioact. Compat. Polym. 9, 389 (1994). 12. J. Lunney and G. Ashwell,Proc. Natl Acad. Sci. USA73, 341 (1976).
13. F. Vandoorne, R. Vercauteren, D. Permentier and E. Schacht,Makromol. Chem. 186, 2455 (1985).
14. J. G. Park, B. S. Kramer, S. M. Steinberg and J. Carmichael, Cancer Res. 47, 4875 (1987). 15. S. Vansteenkiste, A. D. Marre and E. Schacht,J. Bioact. Chmpat. Polym.7, 4 (1992). 16. Y. Song, H. Onishi and T. Nagai,Chem. Pharm. Bull.40, 2822 (1992).
17. J. Kopecek,Biomaterials5, 19 (1984).
18. L. A. McCormick-Thomson and R. Duncan,J. Bioact. Compat. Polym.4, 242 (1989). 19. J. Kopecek, P. Rejmanova and V. Chytry,Makromol. Chem.182, 799 (1981). 20. P. Rejmanova and J. Kopecek,Makromol Chem.184, 2009 (1983).
21. S. Forster and J. B. Lloyd,Biochim. Biophys. Acta947, 465 (1988). 22. G. Ashwell and J. Harford,Annu. Rev. Biochem. 51, 531 (1982). 23. G. Ashwell and A. G. Morell,Adv. Enzymol.41, 99 (1974).
24. Y. Ohya, H. Kobayashi and T. Ouchi,Reactive Polym.15, 153 (1991).
25. F. Friedl, I. Kimura, T. Osato and Y. Ito,Proc. Soc. Exp. Biol. Med.135, 543 (1970).
26. H. K. Schackert, T. Itaya, G. Schackert, E. Fearon, B. Vogelstein and P. Frost,Int. J. Cancer43, 823 (1989).
27. M. Nishikawa, A. Kamijo, T. Fujita, Y. Takakura, H. Sezaki and M. Hashida,Pharm. Research 10, 1253 (1993).