Reactions of Ruthenium Acetylide Complexes with
Isothiocyanate
Chao-Wan Chang, Ying-Chih Lin,* Gene-Hsiang Lee, Shou-Ling Huang, and
Yu Wang
Department of Chemistry, National Taiwan University, Taipei, Taiwan 106, Republic of China
Received December 2, 1997
Treatment of Cp(PPh
3)[P(OMe)
3]RuCtCPh (2; Cp )
η
5-C
5H
5) with PhNdCdS at room
temperature affords the [2 + 2] cycloaddition product Cp(PPh
3)[P(OMe)
3]RuCdC(Ph)C-(dNPh)S (3a), containing a four-membered ring, and the neutral vinylidene phosphonate
complex Cp(PPh
3)[P(dO)(OMe)
2]RudCdC(Ph)C(SH)dNPh (4a) in a 9:1 ratio. Formation
of 4a results from an Arbuzov-like dealkylation reaction possibly after addition of PhNdCdS.
The same reaction at 40 °C affords a higher yield of 4a and Cp(PPh
3)[P(OMe)
3]RuCdC-(Ph)C(dS)N(R)C(dNR)S (5a; R ) Ph) which results from addition of a second isothiocyanate
to the four-membered ring of 3a. The reaction of 2 with PhCH
2NdCdS at room temperature
directly affords the six-membered-ring product 5b (R ) CH
2Ph). Trimerization of phenyl
isothiocyanate is catalyzed by Cp(dppe)RuCtCPh (1
′
; dppe ) Ph
2PCH
2CH
2PPh
2) in refluxing
CH
2Cl
2. This catalytic reaction proceeds through a pathway in which the first two steps
are the same as those observed in the reaction of 2a. An attempt to purify the precursor of
the trimerization product gave the cocrystallization of 1
′
and (PhNCS)
3(8). The structures
of 3a, 4a, 5b, and the cocrystallization product of 1
′
and 8 have been determined by
single-crystal X-ray diffraction analysis.
Introduction
Metal acetylide complexes have been the focus of
recent study due to their application in organometallic
1and materials
2chemistry. The acetylide ligand is quite
reactive toward electrophiles, undergoing either
alky-lation or protonation at the
β-carbon to give stable
vinylidene complexes. Another common reaction
ob-served for this ligand is the [2 + 2] cycloaddition of the
triple bond with unsaturated organic substrates.
3Cy-cloadditions of organic substrates such as CS
2,
4(NC)
2CdC(CF
3)
2, and (NC)
2CdC(CN)
25to the acetylide
ligand in various metal complexes have been reported.
Nickel(0) complexes promote the cyclocoupling of alkynes
with isocyanates.
6This reaction may proceed through
a metallacycle in which one alkyne and one isocyanate
have been coupled. Herein we report that the reaction
of isocyanate and isothiocyanate with two ruthenium
acetylide complexes results in sequential additions of
the organic substrate to the acetylide ligand to produce
novel heterocyclic ligands. With Cp(dppe)RuCtCPh, [2
+ 2] cycloaddition is the first step and is followed by
further addition of two isothiocyanate molecules to give
a trimerization product. When a trimethyl phosphite
ligand is present, the cycloaddition is accompanied by
an Arbuzov-like dealkylation reaction to give a useful
side product from which the mechanism of the
trimer-ization reaction could be delineated. Structural
char-acterization of several relevant complexes is reported
herein.
Results and Discussion
Synthesis of Acetylide Complexes. Treatment of
Cp(PPh
3)
2RuCtCPh (1)
7with P(OMe)
3in n-decane at
reflux temperature affords a racemic mixture of
Cp-(PPh
3)[P(OMe)
3]RuCtCPh (2) in high yield.
8Complex
2 is soluble in polar solvents such as CH
2Cl
2, CHCl
3,
acetone, and THF. In the
31P NMR spectrum, two
doublet resonances at
δ 158.3 and 56.6 are assigned to
the phosphite and phosphine ligands, respectively.
Complex 2 could also be prepared in lower yield from
(1) (a) Beck, W.; Niemer, B.; Wieser, M. Angew. Chem., Int. Ed. Engl.1993, 32, 923. (b) Hegedus, L. S. In Organometallics in Synthesis;
Schlosser, M., Ed.; Wiley: New York, 1994; p 383. (c) Bartik, T.; Bartik, B.; Brady, M.; Dembinski, R.; Gladysz, J. A. Angew. Chem., Int. Ed. Engl. 1996, 35, 414. (d) Ting, P. C.; Lin, Y. C.; Lee, G. H.; Cheng, M. C.; Wang, Y. J. Am. Chem. Soc. 1996, 118, 6433.
(2) (a) Myers, L. K.; Langhoff, C.; Thompson, M. E. J. Am. Chem. Soc. 1992, 114, 7560. (b) Kaharu, T.; Matsubara, H.; Takahashi, S. J. Mater. Chem. 1992, 2, 43. (c) Lavastre, O.; Even, M.; Dixneuf, P. H.; Pacreau, A.; Vairon, J. P. Organometallics 1996, 15, 1530. (d) Wu, I. Y.; Lin, J. T.; Luo, J.; Sun, S. S.; Li, C. S.; Lin, K. J.; Tsai, C.; Hsu, C. C.; Lin, J. L. Organometallics 1997, 16, 2038.
(3) Bruce, M. I.; Hambley, T. W.; Leddell, M. J.; Snow, M. R.; Swincer, A. G.; Tiekink, E. R. T. Organometallics 1990, 9, 96.
(4) (a) Selegue, J. P. J. Am. Chem. Soc. 1982, 104, 119. (b) Birdwhistell, K. R.; Templeton, J. L. Organometallics 1985, 4, 2062. (c) Selegue, J. P.; Young, B. A.; Logan, S. L. Organometallics 1991, 10, 1972.
(5) (a) Davison, A.; Solar, J. P. J. Organomet. Chem. 1979, 166, C13. (b) Bruce, M. I.; Hambley, T. W.; Snow, M. R.; Swincer, A. G. Organometallics 1985, 4, 501. (c) Barrett, A. G. M.; Carpenter, N. E.; Mortier, J.; Sabat, M. Organometallics 1990, 9, 151.
(6) Hoberg, H.; Oster, B. W. J. Organomet. Chem. 1983, 252, 359. (7) (a) Bruce, M. I.; Windsor, N. J. Aust. J. Chem. 1977, 32, 1471. (b) Bruce, M. I.; Humphrey, M. G. Aust. J. Chem. 1989, 42, 1067.
(8) Bruce, M. I. Aust. J. Chem. 1977, 30, 1602.
S0276-7333(97)01056-X CCC: $15.00 © 1998 American Chemical Society Publication on Web 05/20/1998
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
treatment of Cp(PPh
3)
2RuCl with P(OMe)
3followed by
the reaction with HCtCPh. Treatment of 1 with dppe
affords Cp(dppe)RuCtCPh (1
′
) in high yield.
[2 + 2] Cycloaddition and Arbuzov-like
Dealky-lation.
Treatment of 2 with a 10-fold excess of
PhNdCdS in CH
2Cl
2at room temperature for 3 days
affords the yellow [2 + 2] cycloaddition product
Cp-(PPh
3)[P(OMe)
3]RuCdC(Ph)C(dNPh)S (3a) and the
neutral red-orange phosphonate vinylidene complex
Cp-(PPh
3)[P(dO)(OMe)
2]RudCdC(Ph)C(SH)dNPh (4a) in
a 9:1 ratio (75% total yield). The two complexes can be
separated by column chromatography. Complex 3a is
derived from a [2 + 2] cycloaddition of the CtC triple
bond with the CdS double bond. This neutral complex
is stable in air but, in CHCl
3solution, decomposes to
give 2. The
31P NMR spectrum has two doublet
reso-nances at
δ 56.2 and 151.7 with J
P-P) 68.8 Hz, which
are assigned to the PPh
3and the P(OMe)
3ligands,
respectively. The air-stable complex 4a is formed by
an Arbuzov-like dealkylation reaction of the phosphite
ligand.
9The two OMe groups in 4a are diastereotopic
and occur in the
1H NMR spectrum at
δ 3.18 and 3.03
with J
P-H) 11.5 Hz. The
31P NMR spectrum of 4a has
resonances at
δ 48.0 and 93.4, the latter due to the
phosphonate.
Interestingly, if the reaction is carried out at 40 °C
in the presence of excess PhNdCdS, the yield of 4a
increases and the new product Cp(PPh
3)[P(OMe)
3]-RuCdC(Ph)C(dS)N(Ph)C(dNPh)S (5a) could be isolated
in moderate yield. Complex 5a was also prepared
directly from the reaction of 3a with excess PhNCS at
40 °C. In 5a two PhNCS molecules and the acetylide
ligand are incorporated to form a six-membered ring.
In analogy to this, we note that the heterocyclic
com-pound 2-thiopyridone can be prepared by the addition
of MeNCS to a cobaltacyclopentadiene complex, possibly
through CdN bond insertion into a Co-C bond followed
by reductive elimination.
10Other organic compounds
with similar heterocyclic ring structures have been
reported.
11By treatment of complex 2 with excess
PhCH
2NdCdS in CH
2Cl
2at room temperature, the
analogous red-orange complex Cp(PPh
3)[P(OMe)
3]-RuCdC(Ph)C(dS)N(CH
2Ph)C(dNCH
2Ph)S (5b) was
di-rectly obtained. Spectroscopic data for 5b are consistent
with this formulation and are comparable with those
for 5a. This reaction also yields the corresponding
phosphonate complex 4b.
For PhCH
2NdCdS, the
analogous four-membered-ring compound Cp(PPh
3)-[P(OMe)
3]RuCdC(Ph)C(dNCH
2Ph)S (3b), a precursor
of 5b, is obtained at 5 °C but transforms, in the presence
of PhCH
2NCS, to 5b over 20 min at room temperature.
Isolation of the phosphonate complex indicates that
the CR-S bond is labile. Thus, 3 may easily form the
zwitterionic vinylidene complex A (Scheme 1).
Struc-ture A has a negative charge localized on the N atom,
which may attack the carbon atom of a second
isothio-cyanate to give B. Subsequent ring closure gives the
six-membered-ring complex 5.
Structure Determination. Complex 3a was
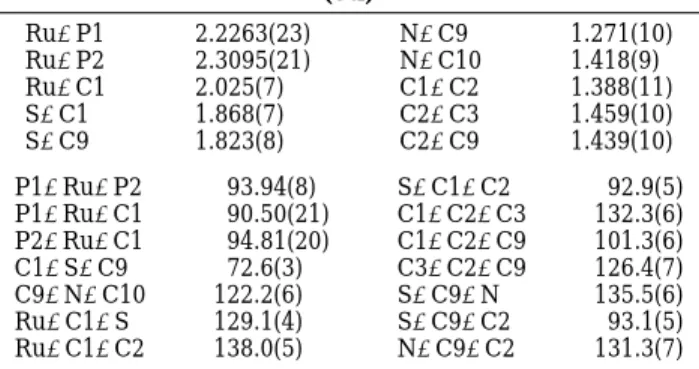
char-acterized by a single-crystal X-ray diffraction analysis;
an ORTEP drawing is shown in Figure 1. Crystal and
intensity collection data are given in Table 1, and
selected bond distances and angles are given in Table
2. The central coordination sphere of the ruthenium
atom contains an
η
5-cyclopentadienyl ring, the
phos-phorus atoms of phosphite and phosphine ligands, and
the carbon atom (C1) of the organic ligand. The four
atoms of the four-membered ring formed by the [2 + 2]
cycloaddition are essentially planar with the C1-C2
distance of 1.388(10) Å typical of a CdC double bond.
The bond distances for C1-S and C9-S (1.868(7) and
1.823(8) Å) are typical of C-S single bonds.
12The
(9) (a) Brill, T. B.; Landon, S. Chem. Rev. 1984, 84, 577. (b) Nakazawa, H.; Miyoshi, K. Trends Organomet. Chem. 1994, 1, 295.
(10) Wakatsuki, Y.; Yamazaki, H. J. Chem. Soc., Chem. Commun.
1973, 280.
(11) Wagner, G.; Richter, P. Pharmazie 1967, 22, 610.
(12) Frank, G. W.; Degen, P. J. Acta Crystallogr., Sect. B 1973, B29, 1815.
Scheme 1
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
C9dN bond distance of 1.271(10) Å confirms an imino
group. The dihedral angle between the phenyl plane
(C10-C15) on the imino group and the plane (S, C1,
C2, C9) formed by the four-membered ring is 37.8(3)°.
The two ruthenium-phosphorus bonds in 3a are
Ru-P1 ) 2.226(2) Å and Ru-P2 ) 2.310(2) Å, with the
shorter distance belonging to the phosphite ligand.
Organic compounds containing similar four-membered
rings with an imino group have been observed as stable
products from the phosphine-induced elimination of a
sulfur atom from 1,2-dithio 3-imines.
13The structural
features of the 2-imino-2H-thiete portion, including the
dihedral angle between the planes of the four-membered
ring and the phenyl substituent in the imino group, are
similar to that in 3a. The [2 + 2] cycloaddition of a CtC
bond of an iron acetylide with CS
2, yielding
2H-thiete-2-thione (
β-dithiolactone), has been reported.
4A similar
process has been proposed for the first stage of the
reaction of alkynes with CS
2.
4The reaction of
phos-phenium complexes with isocyanates leading to [2 + 2]
addition via the NdC bond to give four-membered
phosphametallacycles has been reported.
14Interest-ingly, such a reaction does not take place with the
bis-(triphenylphosphine) analogue of 2. The reaction of
vinylidene complexes with metal acetylide leading to [2
+ 2] addition via the terminal CdC bond of the
vin-ylidene to give unusual cyclic C
4bridging has also been
reported.
15The molecular structure of 4a was determined by an
X-ray diffraction study; an ORTEP drawing is shown
in Figure 2. Crystal and intensity collection data are
given in Table 1, and selected bond distances and angles
are given in Table 3.
With the formation of the
phosphonate ligand, the two ruthenium-phosphorus
bonds (Ru-P1(phosphite) ) 2.303(2) Å and Ru-P2 )
2.323(2) Å) are now comparable. The
ruthenium-carbon bond has a formal bond order of 2, consistent
with a short Ru-C1 bond (1.798(6) Å). The
carbon-carbon double bond of the vinylidene ligand is 1.337(9)
Å, typical for a C(sp
2)-C(sp) allene bond.
16The
ruthe-nium-vinylidene linkage is very nearly linear
(Ru-C1-C2 ) 175.2(5)°). The N-C9 and S-C9 bond lengths of
1.344(8) and 1.641(6) Å, respectively, both display
partial double-bond character, indicative of several
resonance contributions.
The molecular structure of 5b was determined by an
X-ray diffraction study; an ORTEP drawing which
emphasizes the heterocyclic six-membered ring is shown
in Figure 3. Crystal and intensity collection data are
given in Table 1, and selected bond distances and angles
are given in Table 4. As observed in 3a, the
Ru-P1-(phosphite) bond length of 2.239(3) Å is shorter than
(13) Goerdeler, J.; Yunis, M.; Puff, H.; Roloff, A. Chem. Ber. 1986, 119, 162.
(14) Malisch, W.; Hahner, C.; Gru¨ n, K.; Reising, J.; Goddard, R.; Kru¨ ger, C. Inorg. Chim. Acta 1996, 244, 147.
(15) Fischer, H.; Leroux, F.; Roth, G.; Stumpf, R. Organometallics
1996, 15, 3723.
(16) Maki, A. G.; Toth, R. A. J. Mol. Spectrosc. 1965, 17, 136. Table 1. Crystal and Intensity Collection Data for Cp(PPh3)[P(OMe)3]RuCdC(Ph)C(S)dNPh (3a),
Cp(PPh3)[P(dO)(OMe)2]RudCdC(Ph)C(SH)dNPh (4a), Cp(PPh3)[P(OMe)3]RuCdC(Ph)C(S)NPhC(NPh)S (5b),
and Cp(dppe)RuCtCPh‚(PhNCS)3
mol formula C41H38NO3P2SRuCl3(3a) C41H39NO3P2SRu (4a) C52H53N2O3.5P2S2Ru (5b) C60H49N3SRu
space group P1h P1h P21/c Cc a, Å 10.316(3) 10.970(3) 18.798(8) 18.904(5) b, Å 11.237(6) 12.064(5) 13.714(3) 15.951(2) c, Å 18.010(4) 14.595(3) 19.934(4) 35.173(8) R, deg 100.64(3) 90.39(4) 90.00 90.00 β, deg 94.02(2) 101.59(2) 114.76(4) 94.82(2) γ, deg 93.59(3) 100.51(4) 90.00 90.00 V, Å3 1040.6(13) 1858.5(10) 4666.5(24) 10 568(4) Z 2 2 4 8 cryst dimens, mm 0.20× 0.20 × 0.30 0.25× 0.25 × 0.30 0.50× 0.40 × 0.20 0.20× 0.20 × 0.30 radiation Mo KR,λ ) 0.7107 Å 2θ range, deg 2-45 2-45 2-45 2-60 scan type θ-2θ total no. of rflns 5303 4848 6069 8099
no. of unique reflns, I > 2σ(I) 3077 2958 3685 4983
R 0.042 0.041 0.051 0.054
Rw 0.040 0.042 0.047 0.052
Figure 1. ORTEP drawing (50% thermal ellipsoids) of 3a. Three phenyl groups on the triphenylphosphine ligand and all hydrogen atoms are eliminated for clarity.
Table 2. Selected Bond Distances (Å) and Angles (deg) of Cp(PPh3)[P(OMe)3]RuCdC(Ph)C(dNPh)S
(3a) Ru-P1 2.2263(23) N-C9 1.271(10) Ru-P2 2.3095(21) N-C10 1.418(9) Ru-C1 2.025(7) C1-C2 1.388(11) S-C1 1.868(7) C2-C3 1.459(10) S-C9 1.823(8) C2-C9 1.439(10) P1-Ru-P2 93.94(8) S-C1-C2 92.9(5) P1-Ru-C1 90.50(21) C1-C2-C3 132.3(6) P2-Ru-C1 94.81(20) C1-C2-C9 101.3(6) C1-S-C9 72.6(3) C3-C2-C9 126.4(7) C9-N-C10 122.2(6) S-C9-N 135.5(6) Ru-C1-S 129.1(4) S-C9-C2 93.1(5) Ru-C1-C2 138.0(5) N-C9-C2 131.3(7)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
that of Ru-P2(phosphine) (2.341(3) Å). The Ru-C1
bond length of 2.105(7) Å is typical of a Ru-C single
bond, and the C1-C2 bond length of 1.356(10) Å is
typical of a double bond. The carbon-sulfur double
bond in 5b is 1.635(8) Å, which is comparable to that in
ethylene trithiocarbonate (1.652(2) Å).
17The two
C(sp
2)-S single bonds (C1-S2 ) 1.749(8) and C17-S2
) 1.750(8) Å) in the six-membered ring are short enough
to reflect some double-bond character.
18The
hetero-cyclic six-membered ring is essentially planar.
Reactions of 2 with Isocyanate. Treatment of 2
with PhNCO at -20 °C for 7 days afforded the two
products Cp(PPh
3)[P(OMe)
3]RuCdC(Ph)C(dNPh)O (6)
and
Cp(PPh
3)[P(OMe)
3]RuCdC(Ph)C(dO)N(Ph)C-(dNPh)O (7) in a 1:1 ratio, as indicated by
1H and
31P
NMR spectra. These products are unstable at room
temperature and were only characterized by
spectro-scopic methods. In the
31P NMR spectrum of the crude
mixture, the two doublet resonances at
δ 155.3 and 56.9
with J
P-P) 66.9 Hz are assigned to the P(OMe)
3and
PPh
3ligands of 6 and another set of
31P resonances at
δ 151.7 and 55.0 with J
P-P) 69.1 Hz are assigned to 7.
The FAB mass spectrum of the crude mixture displayed
parent peaks at m/e 774.2 and 893.2 for 6 and 7,
respectively. By comparing the spectroscopic data for
the two products with those for 3 and 5, it is plausible
to conclude that 3 and 6 have similar structures, as do
complexes 5 and 7. The dealkylation phosphonate
product was not observed at -20 °C. However, when
the reaction was carried out at room temperature, more
than three phosphonate complexes were observed by the
31
P NMR spectra. Separation of these complexes by
chromatography caused decomposition, and no further
characterization was attempted.
(17) Klewe, F.; Seip, H. M. Acta Chem. Scand. 1972, 26, 1860. (18) Waters, J. M.; Ibers, J. A. Inorg. Chem. 1977, 12, 3273. Figure 2. ORTEP drawing (50% thermal ellipsoids) of 4a.
Three phenyl groups on the triphenylphosphine ligand and all hydrogen atoms are eliminated for clarity.
Figure 3. ORTEP drawing (50% thermal ellipsoids) of 5b. Three phenyl groups on the triphenylphosphine ligand and all hydrogen atoms are eliminated for clarity.
Table 3. Selected Bond Distances (Å) and Angles (deg) of Cp(PPh3)[P(dO)(OMe)2
]RudCdC(Ph)-C(SH)dNPh (4a) Ru-P1 2.3027(20) N-C9 1.344(8) Ru-P2 2.3234(20) N-C10 1.417(8) Ru-C1 1.799(6) C1-C2 1.336(8) P1-O1 1.582(4) C2-C3 1.503(9) P1-O2 1.617(5) C2-C9 1.478(8) P1-O3 1.491(4) C9-S 1.641(6) P1-Ru-P2 92.95(7) P1-O2-C17 120.0(4) P1-Ru-C1 91.13(19) C9-N-C10 129.2(5) P2-Ru-C1 90.57(21) Ru-C1-C2 175.2(5) Ru-P1-O1 107.81(19) C1-C2-C3 116.8(5) Ru-P1-O2 110.00(19) C1-C2-C9 124.2(6) Ru-P1-O3 119.06(19) C3-C2-C9 119.0(5) O1-P1-O2 96.8(3) N-C9-C2 114.0(5) O1-P1-O3 112.8(3) N-C9-S 125.4(5) O2-P1-O3 108.0(3) C2-C9-S 120.5(5) P1-O1-C16 122.8(5)
Table 4. Selected Bond Distances (Å) and Angles (deg) of Cp(PPh3)[P(OMe)3 ]-RuCdC(Ph)C(dS)N(CH2Ph)C(dNCH2Ph)S (5b) Ru-P1 2.2389(24) N1-C9 1.410(10) Ru-P2 2.341(3) N1-C10 1.487(9) Ru-C 2.105(7) N1-C17 1.399(9) S1-C9 1.635(8) N2-C17 1.272(10) S2-C1 1.749(8) N2-C18 1.456(10) S2-C17 1.750(8) C1-C2 1.356(10) P1-O1 1.594(6) C2-C3 1.514(10) P1-O2 1.596(6) C2-C9 1.477(10) P1-O3 1.595(6) C10-C11 1.510(12) P2-C28 1.863(8) C18-C19 1.498(11) P2-C34 1.864(8) C46-C47 1.443(12) P2-C40 1.825(8) C46-C50 1.415(12) O1-C25 1.430(11) C47-C48 1.384(12) O2-C26 1.409(11) C48-C49 1.397(11) O3-C27 1.446(11) C49-C50 1.357(12) P1-Ru-P2 94.61(9) C10-N1-C17 113.9(6) P1-Ru-C1 94.84(21) C17-N2-C18 118.8(6) P2-Ru-C1 96.78(21) Ru-C1-S2 106.2(4) C1-S2-C17 108.6(4) Ru-C1-C2 135.6(6) Ru-P1-O1 116.75(24) S2-C1-C2 117.5(6) Ru-P1-O2 121.81(24) C1-C2-C3 120.4(7) Ru-P1-O3 108.70(24) C1-C2-C9 128.0(7) O1-P1-O2 97.6(3) C3-C2-C9 111.5(6) O1-P1-O3 106.8(3) S1-C9-N1 120.0(6) O2-P1-O3 103.6(3) S1-C9-C2 121.1(6) Ru-P2-C28 122.0(3) N1-C9-C2 118.9(6) Ru-P2-C34 117.1(3) N1-C10-C11 114.2(6) Ru-P2-C40 115.7(3) S2-C17-N1 118.1(5) C28-P2-C34 100.2(4) S2-C17-N2 124.3(6) C28-P2-C40 99.7(4) N1-C17-N2 117.6(6) C34-P2-C40 98.0(4) N2-C18-C19 113.3(7) P1-O1-C25 126.0(6) C47-C46-C50 106.6(7) P1-O2-C26 121.9(6) C46-C47-C48 106.8(7) P1-O3-C27 125.3(7) C47-C48-C49 108.6(7) C9-N1-C10 118.8(6) C48-C49-C50 109.5(7) C9-N1-C17 126.9(6) C46-C50-C49 108.4(7)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
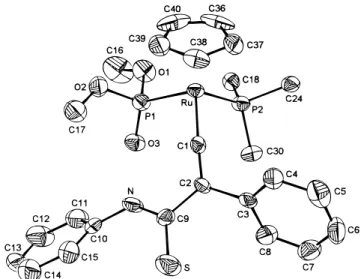
Trimerization of Isothiocyanate. Our efforts to
prepare the dppe analogues of 3 and 5 by reacting
Cp-(dppe)RuCtCPh (1
′
) with PhNCS led to isolation of a
cocrystallization product of 1
′
and
1,3,5-triphenyl-1,3,5-triazinane-2,4,6-trithione (PhNCS)
3(8),
19a
trimeriza-tion product of isothiocyanate. In the reactrimeriza-tion, the [2
+ 2] cycloaddition product
Cp(dppe)RuCdC(Ph)C(dNPh)-S (9a) and Cp(dppe)RuCdC(Ph)C(dCp(dppe)RuCdC(Ph)C(dNPh)-S)N(Ph)C(dNPh)Cp(dppe)RuCdC(Ph)C(dNPh)-S
(10a) were also isolated and identified. The reaction of
1
′
with a 10-fold excess of PhNCS at room temperature
for 3 days afforded the orange-yellow complex 9a in 72%
yield. When the reaction was carried out for 7 days at
room temperature, the brown complex 10a was isolated
in 65% yield. This transformation was monitored by
31
P NMR spectroscopy. At the beginning of the reaction,
the
31P resonance of 1
′
appeared at
δ 86.0; within 3 days
the resonance attributed to 9a appeared at
δ 94.1, and
after 4 more days the resonance of 10a appeared at
δ
99.3. With longer reaction time at room temperature,
a complex mixture containing several organometallic
compounds was obtained.
When the reaction was
carried out at 40 °C for 2 days, a mixture composed of
the major organometallic product 11, which showed a
resonance at
δ 79.4 in the
31P NMR spectrum, and 8
were obtained. Compounds 11 and 8 were separated
by column chromatography. Complex 11 is dark brown
and stable in air. Attempts to obtain single crystals of
11 by recrystallization led to the yellow cocrystallization
product of 1
′
and 8. The mass spectrum of 11 displays
only the parent peaks attributed to 1
′
. However, the
31
P NMR resonance of 11 (
δ 79.6) is different from those
of 1
′
and 10a. On the basis of these data, we believe
that 11 is a precursor of the trimerization product. The
31
P NMR chemical shift of 11 falls in the region for that
of a ruthenium vinylidene dppe complex. A possible
structure of 11 is depicted in Scheme 2. The formation
of 11 may be initiated by opening of the six-membered
ring of 10a to give the zwitterionic vinylidene complex
C, with the anionic charge localized at the N atom. The
nucleophilic attack of this N atom on a free
isothio-cyanate molecule followed by ring closure regenerates
1
′
and releases the six-membered trimerization product
8. It is less likely that the N atom will attack the CR
atom because of the lower stability of an
eight-membered ring.
The crystal structure of the cocrystallization product
of 1
′
and 8 is shown in Figure 4. Crystal and intensity
collection data are given in Table 1, and selected bond
distances and angles are given in Table 5. The two
molecules are packed together with no significant
(19) Tripolt, R.; Nachbaur, E. Phosphorus, Sulfur Silicon Relat.Elem. 1992, 65, 173.
Scheme 2
Figure 4. ORTEP drawing (50% thermal ellipsoids) of the cocrystallization product of 1′and 8. Four phenyl groups on the dppe ligand and all hydrogen atoms are eliminated for clarity.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
intermolecular contacts. The two bonds Ru-P1 and
Ru-P2 are 2.286(5) and 2.288(5) Å, respectively. The
C3 bond length of 1.89(2) Å is typical for a
Ru-C(sp) single bond, and the C3-C4 bond length of
1.186(2) Å is that of a triple bond. In the organic
portion, all three carbon-sulfur double bonds are
comparable in length: 1.64(2), 1.63(2), and 1.69(2) Å.
The heterocyclic six-membered ring is essentially planar
(the distances of all constituent atoms to the plane are
within the 0.029(10) and -0.038(22) Å range), with a
slight double-bond character for all C-N bonds.
The reaction of 1
′
with PhCH
2NCS at room
temper-ature afforded Cp(dppe)RuCdC(Ph)C(dS)N(CH
2Ph)C-(dNCH
2Ph)S (10b) in moderate yield. At 5 °C, the
product was Cp(dppe)RuCdC(Ph)C(dNCH
2Ph)S (9b),
which resulted from [2 + 2] cycloaddition of PhCH
2NCS
with the acetylide ligand. Like 2, 1
′
can differentiate
phenyl isothiocyanate and benzyl isothiocyanate.
In-terestingly, no trimerization product was observed in
this reaction. Decomposition of 10b under thermal
conditions gave a complex mixture from which no
isolable product was obtained.
When the reaction of 1
′
with PhNCO was carried out
at 5 °C, the yellow six-membered-ring complex
Cp-(dppe)RuCdC(Ph)C(dO)N(Ph)C(dNPh)O (12) was
iso-lated in moderate yield. Complex 12 is stable at 5 °C
but decomposes at room temperature. The
31P NMR
resonance of 12 appears at
δ 96.4, and a weak resonance
at
δ 98.2, assignable to the precursor of 12, appears in
the initial stage of the reaction and disappears at the
end of the reaction. We believe that this species is the
[2 + 2] cycloaddition product. In CH
2Cl
2, trimerization
of phenyl isocyanate occurs in the presence of 1
′
, giving
(PhNCO)
3.
20After 2 days, a
31P NMR resonance at
δ
76.0 indicates a major organometallic product,
presum-ably a vinylidene complex with a trimer unit bound to
the acetylide ligand. The acetylide complex is a catalyst
in this reaction. Without this catalyst, the thermolysis
of phenyl isocyanate in CH
2Cl
2yields diphenylurea,
(PhNH)
2CO.
Various metal-promoted coupling modes of
isothio-cyanates have been studied using a dirhenium
com-plex
21and a dirhodium complex.
22In our system,
transformation of isothiocyanates uses a
metal-coordi-nated acetylide ligand. However, as can been seen in
Schemes 1 and 2, the other ligands, such as dppe and a
combination of P(OMe)
3and PPh
3, play a crucial role
in differentiating the conversion of 10 to 8.
Conclusion. The reactions of ruthenium acetylide
complexes with isothiocyanate or isocyanate yielded a
series of addition products. Addition of one RNCS
molecule to the acetylide ligand via a [2 + 2]
cycload-dition gave a four-membered-ring product. Adcycload-dition of
a second RNCS molecule generated a complex with a
heterocyclic six-membered ring. For the cationic
vin-ylidene complex with a P(OMe)
3ligand, an
Arbuzov-like dealkylation of P(OMe)
3resulted in the formation
of a neutral vinylidene complex with a phosphonate
ligand. Complete characterization of this phosphonate
complex assisted in elucidating the mechanism of the
sequential addition processes. For the acetylide
com-plex with a bidentate dppe ligand, other than the two
addition processes mentioned above, a third addition led
to an organic trimerization product and regenerated the
acetylide complex.
Experimental Section
General Procedures. All manipulations were performed
under nitrogen using vacuum-line, drybox, and standard Schlenk techniques. CH3CN and CH2Cl2were distilled from
CaH2and diethyl ether and THF from Na/ketyl. All other
solvents and reagents were of reagent grade and were used without further purification. NMR spectra were recorded on Bruker AC-200 and AM-300WB FT-NMR spectrometers and are reported in units ofδ with residual protons in the solvent as an internal standard (CDCl3,δ 7.24; CD3CN,δ 1.93; C2D6
-CO,δ 2.04). FAB mass spectra were recorded on a JEOL SX-102A spectrometer and are reported in m/z units. Complexes Cp(dppe)RuCtCPh (1′) and Cp(PPh3)2RuCtCPh23were
pre-pared by following the methods reported in the literature. Elemental analyses and X-ray diffraction studies were carried out at the Regional Center of Analytical Instrument located at the National Taiwan University.
Preparation of Cp(PPh3)[P(OMe)3]RuCtCPh (2). In
a Schlenk flask charged with P(OMe)3(7.70 mL, 63.30 mmol)
and Cp(PPh3)2RuCtCPh (1; 10.00 g, 12.66 mmol), n-decane
(80 mL) was added. The resulting solution was heated to reflux for 1 h to give a yellow solution. The solvent was reduced in volume to about 5 mL under vacuum, and a yellow precipitate formed. After filtration, the precipitate was washed with 2 × 20 mL of pentane to give the product Cp(PPh3
)-[P(OMe)3]RuCtCPh (2; 7.60 g, 11.64 mmol, 92% yield).
Spectroscopic data for 2 are as follows. 1H NMR: 7.69-6.86
(m, 20 H, Ph); 4.64 (s, 5H, Cp); 3.37 (d, JP-H) 11.6 Hz, 9 H,
P(OMe)3). 31P NMR: 158.3 (d, JP-P) 64.5 Hz, P(OMe)3); 56.6
(d, JP-P) 64.5 Hz, PPh3). 13C NMR: 139.2-122.9 (Ph); 130.3
(CR); 112.6 (Cβ); 84.2 (Cp); 51.9 (P(OMe)3). Mass: 654.0 (M+),
553.1 (M+- C
2Ph); 428.9 (M+- C2Ph,P(OMe)3). Anal. Calcd
for C34H34O3P2Ru: C, 62.47; H, 5.24. Found: C, 62.68; H, 5.32.
(20) (a) Hong, P.; Sonogashira, K.; Hagihara, N. Tetrahedron Lett.
1970, 1633. (b) Schwetlick, K.; Noack, R. J. Chem. Soc., Perkin Trans.
2 1995, 395. (c) Verardo, G.; Giumanini, A. G.; Gorassini, F.; Straz-zolini, P.; Benetollo, F.; Bombieri, G. J. Heterocycl. Chem. 1995, 32, 995.
(21) Adams, R. D.; Huang, M. Organometallics 1996, 15, 3644. (22) Gibson, J. A. E.; Cowie, M. Organometallics 1984, 3, 984. (23) Bruce, M. I.; Humphrey, M. G. Aust. J. Chem. 1989, 42, 1067. Table 5. Selected Bond Distances (Å) and Angles
(deg) of Cp(dppe)RuCCPh‚(PhNCS)3 Ru-P1 2.286(5) N1-C40 1.417(21) Ru-P2 2.288(5) N1-C41 1.437(19) Ru-C3 1.893(18) N1-C43 1.491(21) Ru-C5 2.224(17) N2-C41 1.433(21) Ru-C6 2.185(16) N2-C42 1.406(21) Ru-C7 2.210(15) N2-C49 1.527(20) Ru-C8 2.247(17) N3-C40 1.415(20) Ru-C9 2.238(16) N3-C42 1.373(20) S1-C40 1.644(15) N3-C55 1.519(23) S2-C41 1.627(16) C3-C4 1.183(22) S3-C42 1.687(19) C4-C10 1.395(19) P1-Ru-P2 84.81(17) Ru-C3-C4 170.9(15) P1-Ru-C3 88.5(5) C3-C4-C10 170.5(16) P2-Ru-C3 88.6(5) S1-C40-N1 123.7(11) C40-N1-C41 123.0(13) S1-C40-N3 119.3(11) C40-N1-C43 120.3(12) N1-C40-N3 116.9(12) C41-N1-C43 116.7(13) S2-C41-N1 125.7(12) C41-N2-C42 123.8(12) S2-C41-N2 119.4(10) C41-N2-C49 118.0(12) N1-C41-N2 114.8(13) C42-N2-C49 117.9(13) S3-C42-N2 119.3(12) C40-N3-C42 123.6(13) S3-C42-N3 122.7(13) C40-N3-C55 118.3(12) N2-C42-N3 117.6(15) C42-N3-C55 117.9(13)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
Reaction of 2 with PhNCS. To a Schlenk flask charged
with 2 (0.50 g, 0.76 mmol) was added CH2Cl2(20 mL), and
PhNCS (0.92 mL, 7.66 mmol) was injected by a syringe. The resulting mixture was stirred at room temperature for 3 days while the color changed from bright yellow to brown. The solvent was removed under vacuum. The residue was redis-solved in ether and passed through a silica gel packed column. Hexane eluted the starting material, ether eluted an orange-yellow band, and methanol eluted a brown band. The solvent of the orange-yellow band was removed to give a yellow oil which was washed with hexane to give a yellow powder and further washed with 20 mL of pentane to give the product Cp-(PPh3)[P(OMe)3]RuCdC(Ph)C(dNPh)S (3a). The yield of 3a
after recrystallization from hexane/CH2Cl2(1:1) is 0.38 g (63%
yield). After a similar workup procedure, the brown band gave the orange-red phosphonate complex Cp(PPh3)[P(dO)(OMe)2
]-RudCdC(Ph)C(SH)dNPh (4a; 0.030 g, 5% yield). Spectro-scopic data for 3a are as follows. 1H NMR: 7.29-7.00 (m, 20
H, Ph); 4.75 (s, 5H, Cp); 3.37 (d, JP-H) 11.0 Hz, 9 H, P(OMe)3). 31P NMR: 151.7 (d, J P-P) 68.9 Hz, P(OMe)3); 56.2 (d, JP-P) 68.9 Hz, PPh3). 13C NMR: 134.1-122.7 (Ph, Cβ, Cγ); 84.8 (Cp); 52.3 (d, JC-P) 7.5 Hz, P(OMe)3). Mass: 790.2 (M+), 553.1 (M+- C
2Ph); 429.1 (M+- C2Ph, P(OMe)3). Anal. Calcd for
C41H39NO3P2SRu: C, 62.42; H, 4.98; N, 1.78. Found: C, 61.99;
H, 5.12; N, 1.73. Spectroscopic data for 4a are as follows.1H
NMR: 7.70-6.70 (m, 20 H, Ph); 5.26 (s, 5H, Cp); 3.18 (d, JP-H ) 11.5 Hz, 3 H, OMe); 3.03 (d, JP-H) 11.5 Hz, 3 H, OMe).13C NMR: 345.2 (t, JC-P) 17.1 Hz, CR); 137.9-123.4 (Ph, Cβ, Cγ); 93.0 (Cp); 52.2 (d, JC-P) 8.6 Hz, OMe); 51.8 (d, JC-P) 8.4 Hz, OMe). 31P NMR: 95.3 (d, J P-P) 45.9 Hz, P(OMe)2); 48.0 (d, JP-P) 45.9 Hz, PPh3). Mass: 776.0 (M+), 744.0 (M+- S);
539.0 (M+ - CdC(Ph)C(SH)CdNPh). Anal. Calcd for C40H37NO3P2SRu: C, 62.00; H, 4.81; N, 1.81. Found: C, 61.74;
H, 5.01; N, 1.70. The31P NMR spectrum of the crude product
(after 3 days of reaction time) displayed the resonances attributed to 3a and 4a in a 9:1 ratio.
The reaction of 2 with PhNCS in refluxing CH2Cl2 was
carried out under nitrogen for 4 days. The workup procedure was similar to that mentioned above. The reaction gave 4a and Cp(PPh3)[P(OMe)3]RuCdC(Ph)C(dS)N(Ph)C(dNPh)S (5a)
after purification in 40% total yield. The31P NMR spectrum
of the crude product (after 4 days of reaction time) displayed the resonances attributed to 5a and 4a in a 3:2 ratio. Spectroscopic data for 5a are as follows. 1H NMR: 7.84-6.52
(m, 30 H, Ph); 4.40 (s, 5H, Cp); 3.06 (d, JP-H) 11.0 Hz, 9 H,
P(OMe)3). 31P NMR: 148.6 (d, JP-P) 72.5 Hz, P(OMe)3); 53.9
(d, JP-P) 72.5 Hz, PPh3). Mass: 925.1 (M++ 1), 553.1 (M+
- C2Ph); 429.1 (M+ - C2Ph, P(OMe)3). Anal. Calcd for
C48H44N2O3P2S2Ru: C, 62.39; H, 4.80; N, 3.03. Found: C,
62.52; H, 4.95; N, 3.11. Complex 5a can also be prepared from the reaction of 3a with PhNCS in refluxing CH2Cl2for 3 days.
In the crude mixture, a small amount of 4a was observed.
Reaction of 2 with PhCH2NCS. In a Schlenk flask
charged with 2 (0.50 g, 0.76 mmol), PhCH2NCS (1.01 mL, 766
mmol) and CH2Cl2(20 mL) were added and the mixture was
stirred at room temperature for 3 days with the color changing from bright yellow to brown. The solvent was removed under vacuum and the residue was subjected to a silica gel packed column chromatograph. Hexane eluted the organic com-pounds, a 1:1 hexane/ether solution eluted a brown band, and methanol eluted an orange band. The brown band was dried under vacuum and the residue washed with 2× 15 mL of hexane to give the solid product Cp(PPh3)[P(OMe)3
]-RuCdC(Ph)C(S)N(CH2Ph)C(dNCH2Ph)S (5b; 0.42 g, 58%
yield). The orange band, after the same treatment, gave the orange phosphonate complex Cp(PPh3)[P(dO)(OMe)2
]-RudCdC(Ph)C(SH)dNCH2Ph (4b; 0.06 g, 10% yield).
Spec-troscopic data for 5b are as follows. 1H NMR: 7.35-6.99 (m,
30 H, Ph); 6.19 (d, JH-H) 12.7 Hz, 1H, CH2Ph); 6.02 (d, JH-H ) 12.7 Hz, 1H, CH2Ph); 4.34 (s, 5H, Cp); 3.92 (d, JH-H) 16.9 Hz, 1H, CH2Ph); 3.84 (d, JH-H) 16.9 Hz, 1H, CH2Ph); 3.35 (d, JP-H) 10.9 Hz, 9 H, P(OMe)3). 31P NMR: 148.6 (d, JP-P) 73.3 Hz, P(OMe)3); 57.9 (d, JP-P) 73.3 Hz, PPh3). 13C NMR: 134.1-122.7 (Ph, Cβ, Cγ); 84.8 (Cp); 52.3 (d, JC-P) 7.5 Hz,
P(OMe)3). Mass: 790.2 (M+), 553.1 (M+ - organic ligand);
429.1 (M+ - organic ligand, P(OMe)3). Anal. Calcd for
C50H48N2O3P2S2Ru: C, 63.08; H, 5.08; N, 2.94. Found: C,
62.95; H, 5.04; N, 3.12. Spectroscopic data for 4b are as follows. 1H NMR: 10.86 (s, 1H, SH); 7.63-6.67 (m, 20 H, Ph); 5.19 (s, 5H, Cp); 3.07 (d, JP-H) 11.2 Hz, 3 H, OMe); 2.97 (d, JP-H) 11.5 Hz, 3 H, OMe); 2.44 (d, JH-H) 7.3 Hz, 1H, -NCH2 -Ph); 2.39 (d, JH-H) 7.3 Hz, 1H, -NCH2Ph). 13C NMR: 343.5 (t, JC-P) 17.0 Hz, CR); 142.4-126.8 (Ph, Cβ, Cγ); 92.9 (Cp); 50.5 (d, JC-P) 9.4 Hz, OMe); 50.1 (d, JC-P) 8.4 Hz, OMe). 31P NMR: 95.2 (d, J P-P) 45.8 Hz, P(OMe)2); 49.3 (d, JP-P) 45.8 Hz, PPh3). Mass: 790.0 (M+), 758.0 (M+- S), 539.0 (M+
- organic ligand); 428.9 (M+- organic ligand, P(OMe)
3). Anal.
Calcd for C41H39NO3P2SRu: C, 62.42; H, 4.98; N, 1.78.
Found: C, 62.57; H, 4.64; N, 1.95.
The [2 + 2] cycloaddition product Cp(PPh3)[P(OMe)3
]-RuCdC(Ph)C(dNCH2Ph)S (3b) could be observed when the
same reaction was carried out in CDCl3at 5 °C for 5 days and
monitored by NMR spectra. Complexes 3b and 5b formed simultaneously in a 1:3 ratio at this temperature and at room temperature 3b transformed to 5b in ca. 2 h. Spectroscopic data for 3b are as follows. 1H NMR: 7.73-6.85 (m, 25 H, Ph);
4.73 (s, 5H, Cp); 4.49, 4.39 (two d, JH-H ) 13.6 Hz, 2H,
-NCH2); 3.42 (d, JP-H) 11.0 Hz, 9 H, OMe). 31P NMR: 151.8
(d, JP-P) 67.8 Hz, P(OMe)2); 56.0 (d, JP-P) 67.8 Hz, PPh3).
Mass: 804.1 (M++ 1), 553.1 (M+- organic ligand); 429.1 (M+
- organic ligand, P(OMe)3).
Reaction of 2 with PhNCO. This reaction was monitored
by NMR spectroscopy. Complex 2 (0.05 g, 0.08 mmol) and PhNCO (0.10 mL, 0.76 mmol) were dissolved in CDCl3(1 mL)
in an NMR tube under nitrogen. The resulting mixture was stored at -20 °C for 7 days. The1H and31P NMR spectra of
the mixture indicated formation of the two major products Cp-(PPh3)[P(OMe)3]RuCdC(Ph)C(dNPh)O (6) and Cp(PPh3
)-[P(OMe)3]RuCdC(Ph)C(dO)N(Ph)C(dNPh)O (7). The total
NMR yield of 6 and 7 is estimated to be about 70%, on the basis of the integration of the Cp resonances and the 31P
resonances. Since these two complexes are unstable at room temperature, no attempt was made to isolate the products. The
31P NMR spectrum of the crude product (after 7 days of
reaction time) displayed the resonances attributed to 6 and 7 in a 1:1 ratio. Spectroscopic data for 6 are as follows. 1H
NMR: 8.03-6.84 (m, 25 H, Ph); 4.63 (s, 5H, Cp); 3.62 (d, JP-H
) 11.1 Hz, 9 H, P(OMe)3). 31P NMR: 155.4 (d, JP-P ) 66.9
Hz, P(OMe)3); 56.9 (d, JP-P) 66.9 Hz, PPh3). Mass: 774.2
(M++ 1), 654.1 (M+- C(O)CdNPh); 429.1 (M+- CdC(Ph)C-(O)CdNPh, P(OMe)3). Spectroscopic data for 7 are as follows. 1H NMR: 7.85-6.83 (m, 30 H, Ph); 4.43 (s, 5H, Cp); 3.14 (d,
JP-H) 11.0 Hz, 9 H, P(OMe)3). 31P NMR: 151.7 (d, JP-P)
69.1 Hz, P(OMe)3); 54.6 (d, JP-P) 69.1 Hz, PPh3). Mass: 893.2
(M++ 1), 654.1 (M+- organic ligand + C
2Ph); 553.1 (M+
-organic ligand); 429.1 (M+- organic ligand, P(OMe) 3).
Reaction of Cp(dppe)RuCtCPh (1′) with PhNCS. In
a Schlenk flask charged with 1′(0.30 g, 0.46 mmol), PhNCS (0.55 mL, 4.59 mmol) and CH2Cl2(20 mL) were added; the
mixture was stirred at room temperature for 4 days and the color of the solution changed from bright yellow to brown. The solvent was removed under vacuum and the residue was washed with 2× 30 mL of hexane to give the product. After filtration, the solid was further washed with 20 mL of pentane, giving the product Cp(dppe)RuCdC(Ph)C(dNPh)S (9a; 0.26 g, 72% yield). Spectroscopic data for 9a are as follows. 1H
NMR: 7.80-6.30 (m, 30 H, Ph); 4.50 (s, 5H, Cp); 2.60-2.47 (m, 4 H, PCH2). 13C NMR: 133.7-122.6 (Ph, Cβ, Cγ); 84.6 (Cp);
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
29.3 (t, JC-P) 21.5 Hz, CH2). 31P NMR: 93.9 (PPh2). FAB
mass: 801.0 (M+), 565.0 (M+- organic ligand). Anal. Calcd
for C46H39NP2SRu: C, 68.98; H, 4.91; N, 1.75. Found: C,
68.99; H, 4.79; N, 1.88. If the same reaction was carried out for 7 days at room temperature, the complex Cp(dppe)RuCdC-(Ph)C(S)N(Ph)C(dNPh)S (10a; 0.28 g, 65% yield) was isolated. Spectroscopic data for 10a are as follows. 1H NMR:
7.75-6.94 (m, 35 H, Ph); 3.68 (s, 5H, Cp); 2.65-2.30 (m, 4 H, PCH2). 13C NMR: 151.0-121.5 (Ph, C
β, Cγ); 85.1 (Cp); 30.7 (t, JC-P)
22.0 Hz, CH2). 31P NMR: 99.3 (PPh2). Mass: 936.0 (M++
1), 565.0 (M+- organic ligand). Anal. Calcd for C
53H44N2P2S2
-Ru: C, 68.00; H, 4.74; N, 2.99. Found: C, 67.79; H, 5.01; N, 2.85.
Reaction of 1′(0.30 g, 0.46 mmol) with PhCH2NCS (0.61
mL, 4.58 mmol) was carried out in CH2Cl2(20 mL) at room
temperature for 3 days. The solvent was reduced under vacuum to about 1 mL, and the residue was passed through a silica gel packed column. A brown band was eluted with ether, and after removal of ether, the product was washed with 2× 20 mL of hexane to give the brown-yellow product Cp(dppe)-RuCdC(Ph)C(S)N(CH2Ph)C(dNCH2Ph)S (10b; 0.27 g, 62%
yield). Spectroscopic data for 10b are as follows. 1H NMR:
7.62-6.75 (m, 35 H, Ph); 5.96 (s, 2H, CH2Ph); 3.56 (s, 2H, CH2
-Ph); 3.84 (s, 5H, Cp); 2.90-2.58 (m, 4 H, PCH2). 13C NMR:
184.8, 151.4, 143.3-125.6 (Ph, Cβ, Cγ); 84.8 (Cp); 52.4, 51.6 (2
× CH2Ph); 30.0 (JC-P ) 22.2 Hz, CH2). 31P NMR: 88.4 (s,
PPh2). Mass: 965.1 (M++ 1), 565.0 (M+- organic ligand).
Anal. Calcd for C55H48N2P2S2Ru: C, 68.52; H, 5.01; N, 2.91.
Found: C, 68.75; H, 4.84; N, 3.18. If the same reaction is carried out at 5 °C for 5 days, the two products Cp(dppe)-RuCdC(Ph)C(dNCH2Ph)S (9b) and 10b in a 1:4 ratio are
observed in the NMR spectrum. 9b is unstable and is
converted to 10b in about 2 h at room temperature. Spectro-scopic data for 9b are as follows. 1H NMR: 7.69-6.85 (m, 30
H, Ph); 4.43 (s, 2H, CH2Ph); 3.91 (s, 5H, Cp); 2.90-2.60 (m, 4
H, PCH2). 31P NMR: 94.5 (s, PPh2). Mass: 816.1 (M++ 1),
565.0 (M+- organic ligand).
Trimerization of PhNCS on Cp(dppe)RuCtCPh. A
Schlenk flask was charged with 1′(0.07 g, 0.11 mmol), and the atmosphere was replaced with nitrogen; then PhNCS (0.55 mL, 4.59 mmol) and CH2Cl2(20 mL) were introduced and the
mixture was heated to reflux. The31P NMR spectrum revealed
a complex mixture at the initial stage of the reaction, but after 2 days, only a single product was obtained. The mixture was heated for 4 days, and the color of the solution changed from bright yellow to brown. The solvent was removed under vacuum, and the residue was washed with 2 × 30 mL of hexane to give a brown-black product. After filtration, the solid was further washed with 20 mL of pentane, giving the product (PhNCS)3(8; 0.26 g, 72% yield). Spectroscopic data
are consistent with those in the literature.14
Trimerization of PhNCO on Cp(dppe)RuCtCPh. A
Schlenk flask was charged with 1′(0.10 g, 0.15 mmol), and the atmosphere was replaced with nitrogen; then PhNCO (1.00 mL, 1.50 mmol) and CH2Cl2(20 mL) were introduced and the
mixture was stored at 5 °C for 2 days. The solvent was removed under vacuum, and the residue was subjected to silica gel packed column chromatography. The organic portion (PhNHCONHPh) was eluted with hexane, and the yellow organometallic compound was eluted with ether. Removal of ether solvent followed by addition of hexane gave a yellow precipitate. After filtration, the solid was further washed with 20 mL of pentane, giving the product Cp(dppe)RuCdC(Ph)C-(O)N(Ph)C(dNPh)O (12; 0.10 g, 73% yield). Spectroscopic data for 12 are as follows. 1H NMR: 8.10-6.90 (m, 30 H, Ph); 3.94
(s, 5H, Cp); 2.45-2.10 (m, 4 H, PCH2). 13C NMR: 153.3, 140.8,
135.0-121.6 (Ph, Cβ, Cγ); 86.2 (Cp); 29.4 (JC-P) 19.5 Hz, CH2). 31P NMR: 96.4 (PPh
2). FAB mass: 905.0 (M+), 565.0 (M+
-C2Ph). Anal. Calcd for C53H44N2O2P2Ru: C, 70.42; H, 4.91;
N, 3.10. Found: C, 70.63; H, 4.78; N, 3.34.
A Schlenk flask was charged with 1′(0.20 g, 0.31 mmol), and the atmosphere was replaced with nitrogen; then PhNCO (1.00 mL, 9.21 mmol) and CH2Cl2(20 mL) were introduced
and the mixture was heated to reflux for 2 days. The31P NMR
spectrum revealed a complex mixture at the initial stage of the reaction, but after 1 day, only a single product was obtained. The mixture was heated for 2 days and the color of the solution changed from bright yellow to brown. The solvent was removed under vacuum, and the residue was subjected to silica gel packed column chromatography. Hexane eluted the starting material, ether eluted the organometallic complex
12, and methanol eluted the trimer. Removal of methanol
under vacuum followed by addition of hexane gave a light yellow precipitate. After filtration, the solid was further washed with 20 mL of pentane, giving the product (PhNCO)3
(8a; 0.45 g, 41% yield). Spectroscopic data for 8a are consistent with those in the literature.14
X-ray Analysis of 3a. Single crystals of 3a suitable for
an X-ray diffraction study were grown as mentioned above. A single crystal of dimensions 0.20× 0.20 × 0.30 mm3was glued
to a glass fiber and mounted on an Enraf-Nonius CAD4 diffractometer. Initial lattice parameters were determined from a least-squares fit to 25 accurately centered reflections with 10.0° < 2θ < 25°. Cell constants and other pertinent data are collected in the Supporting Information. Data were collected using the θ/2θ scan method. The scan angle was determined for each reflection according to the expression 0.8 + 0.35 tan θ. The final scan speed for each reflection was determined from the net intensity gathered during an initial prescan and ranged from 2 to 7° min-1.
The raw intensity data were converted to structure factor amplitudes and their esd’s by correction for scan speed, background, and Lorentz-polarization effects. An empirical correction for absorption based on the azimuthal scan was applied to the data set. Crystallgraphic computations were carried out on a Microvax III computer using the NRCC structure determination package.24 Merging of equivalent and
duplicate reflections gave a total of 5303 unique measured data, from which 3077 were considered observed (I > 2.0σ(I)). The structure was first solved by using the heavy-atom method (Patterson synthesis), which revealed the position of the metal, and then refined via standard least-squares and difference Fourier techniques. The quantity minimized by the least-squares program was w(|Fo| - |Fc|)2. The analytical forms of
the scattering factor tables for the neutral atoms were used.25
All other non-hydrogen atoms were refined by using anisotro-pic thermal parameters. Hydrogen atoms were included in the structure factor calculations in their expected positions on the basis of idealized bonding geometry but were not refined in least squares. Final refinement using full-matrix least squares converged smoothly to values of R ) 0.042 and Rw)
0.040. The procedures for 4a, 5b, and the cocrystallization product of 1′and 8 were similar. The final residuals of the refinement were R ) 0.041 and Rw) 0.042; R ) 0.051 and Rw
) 0.047, and R ) 0.054 and Rw) 0.052 for 4b, 5b, and the
cocrystallization product of 1′and 8, respectively. Final values of all refined atomic positional parameters (with esd’s) and tables of thermal parameters are given in the Supporting Information.
Acknowledgment. We are grateful for support of
this work by the National Science Council, Taiwan,
Republic of China.
(24) Gabe, E. J.; Lee, F. L.; Lepage, Y. In Crystallographic Comput-ing 3; Sheldrick, G. M., Kruger, C., Goddard, R., Eds.; Clarendon Press: Oxford, England, 1985; p 167.
(25) International Tables for X-ray Crystallography; Reidel Publish-ing Co.: Dordrecht, Boston, 1974; Vol. IV.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
Supporting Information Available: Details of the
struc-tural determination for complexes 3a, 4a, and 5b and the cocrystallization product of 1′and 8, including tables of crystal data and structure refinement parameters, positional and anisotropic thermal parameters, and bond distances and
angles (32 pages). Ordering information is given on any current masthead page.
OM971056D
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009