Invited critical review
Human DExD/H RNA helicases: Emerging roles in stress
survival regulation
Jing-Wen Shih
a,b, Yan-Hwa Wu Lee
c,⁎
aIntegrated Laboratory, Center of Translational Medicine, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan
bGraduate Institute of Translational Medicine, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan c
Department of Biological Science and Technology, National Chiao Tung University, Hsinchu 300, Taiwan
a b s t r a c t
a r t i c l e i n f o
Article history: Received 20 March 2014
Received in revised form 5 May 2014 Accepted 6 May 2014
Available online 13 May 2014 Keywords:
Cell death DExD/H RNA helicase Nucleic acid sensing RIG-I-like receptor Stress granule Stress response
Environmental stresses threatening cell homeostasis trigger various cellular responses ranging from the activation of survival pathways to eliciting programmed cell death. Cellular stress response highly depends on the nature and level of the insult as well as the cell type. Notably, the interplay among all these responses will ultimately deter-mine the fate of the stressed cell. Human DExD/H RNA helicases are ubiquitous molecular motors rearranging RNA secondary structure in an ATP-dependent fashion. These highly conserved enzymes participate in nearly all aspects of cellular process involving RNA metabolism. Although numerous functions of DExD/H RNA helicases are well documented, their importance in stress response is only just becoming evident. This review outlines our current knowledge on major mechanistic themes of human DExD/H RNA helicases in response to stressful stimuli, especially on emerging molecular models for the functional roles of these enzymes in the stress survival regulation.
© 2014 The Authors. Published by Elsevier B.V. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Contents
1. Cellular stress response: adaption, survival or death . . . 46
2. Human DExD/H RNA helicases . . . 47
3. DExD/H RNA helicase-mediated regulation of cell survival under stress . . . 47
3.1. Defense against pathogen infections— cell death induction by DExD/H RNA helicases . . . 47
3.1.1. RIG-I-like receptors (RLRs) . . . 48
3.1.2. Non-RLR helicases . . . 50
Abbreviations: 5′ppp, 5′ triphosphate; AMD, ARE-mediated mRNA decay; ARE, AU-rich element; ARE-BP, ARE-binding protein; CARD, caspase activation and recruitment domain; CCCP, carbonyl cyanide m-chlorophenyl hydrazone; CDN, cyclic dinucleotide; CRM1, Chromosome Maintenance Region 1; CTD, C-terminal domain; ds, Double Stranded; eIF, eukaryotic initiation factor; ER, endoplasmic reticulum; FCCP, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone; G3BP1, GTPase-activating protein SH3 domain binding protein 1; HCV, Hepatitis C Virus; HIV, Human Immunodeficiency Virus; IFN, interferon; IFNB, Interferon Beta 1; IKK, IκB Kinase; IL, interleukin; IPS-1, IFNB-promoter stimulator 1; IRES, internal ribosome entry site; IRF, Interferon Regulatory Factor; KSRP, K homology Splicing Regulatory Protein; LGP2, Laboratory of Genetics and Physiology 2; MBNL1, muscleblind-like splicing regulator 1; MDA5, melanoma differentiation-associated antigen 5; mDCs, myeloid dendritic cells; mTOR, mammalian target of rapamycin; MyD88, myeloid differentiation primary response gene 88; NF-kB, Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells; NLRP3, NLR Pyrin-domain containing 3; ODN, oligodeoxynucleotide; PABP, poly(A) binding protein; PAMP, pathogen-associated molecular pattern; PB, processing body; pDC, plasmacytoid dendritic cells; Poly(I:C), polyinosinic–polycytidylic acid; PQBP, polyglutamine-binding protein 1; RACK1, The Receptor for Activated C Kinase 1; RD, repressor domain; RecA, recombinase A; RHA, RNA Helicase A; RHAU, RNA Helicase Associated with AU-rich element; RhoA, Ras homolog gene family member A; RIG, retinoic acid-inducible gene 1; RISC, RNA-induced silencing complex; RLR, RIG-I-like receptor; RNP, ribonucleoprotein; ROCK1, Rho-associated, coiled-coil contain-ing protein kinase 1; ROS, reactive oxygen species; SF, superfamily; SG, stress granule; ss, scontain-ingle stranded; STING, stimulator of IFN genes; TAP, Tip Associated Protein; TBK1, TANK bindcontain-ing kinase; TIR, Toll-interleukin receptor; TRAF2, TNF receptor-associated factor 2; TRAF3, TNF receptor-associated factor 3; TRIF, TIR-domain-containing adapter-inducing Interferon-β; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; uORF, upstream open reading frame; uPA, urokinase-type plasminogen activator; USP10, ubiquitin-specific protease 10; UTR, untranslated region; VACV, vaccinia virus; VEGF, Vascular Endothelial Growth Factor; VSV, vesicular stomatitis virus; WDR62, WD repeat domain 62.
⁎ Corresponding author at: Department of Biological Science and Technology, National Chiao Tung University, 75 Bo-Ai Street, Hsinchu 300, Taiwan. Tel.: +886 3 5718083; fax: +886 3 5721500.
E-mail address:[email protected](Y.-H.W. Lee).
http://dx.doi.org/10.1016/j.cca.2014.05.003
0009-8981/© 2014 The Authors. Published by Elsevier B.V. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Contents lists available atScienceDirect
Clinica Chimica Acta
3.2. Stress granule formation to suppress apoptosis under stress . . . 51
3.2.1. Translation silencing, stress granule assembly and cell survival upon stress . . . 51
3.2.2. Stress granule-associated RNA helicases . . . 52
3.3. Multifaceted function of DExD/H RNA helicases in stress survival regulation . . . 54
4. Conclusions and future perspectives . . . 55
Acknowledgements . . . 55
References . . . 55
1. Cellular stress response: adaption, survival or death
To maintain homeostasis, eukaryotic cells must respond continuously tofluctuations in external conditions. Beyond a certain threshold, such environmental changes can be considered‘stresses’ to the cell. Stresses can be further broadly classified into abiotic and biotic insults. Tempera-ture shift, ultraviolet light, ion concentrations, pH, oxygen tension, redox potentials and metabolite concentrations are common abiotic stress conditions. Meanwhile, biotic stresses include perturbations caused by various microbial pathogens.
Exposure to environmental stress can damage existing macromole-cules, including proteins, mRNAs, DNA and lipids. Therefore, in response to sub-lethal stress, cells mount different defense mechanisms and pro-survival strategies to adapt their metabolism, re-establish cellular homeostasis and protect themselves against potential damage. These stress responses are orchestrated through a multifaceted cellular program, such as nutrient uptake, intermediary metabolism, induction of molecular chaperones[1,2], rapid clearance of damaged macromole-cules[3], cell cycle and growth control, cell fate and lineage decisions, as well as activation of certain gene expression programs[4]. However, if the stress remains unresolved, then the eventual cell death programs are activated to eliminate these damaged cells[5]. Cell death is a neces-sary part of tissue homeostasis enabling the removal of dysfunctional cells. This phenomenon is not merely incidental during stress response but, rather, a controlled and modifiable complex process. In a multicellu-lar organism, completion of the proper pathway is critical to ensure that the appropriate outcome is ultimately achieved. Failure to die in response to stress stimuli can result in tissue pathology, organ dysfunction and ini-tiation of diseases. Nowadays, several types of programed cell death, such as apoptosis, necrosis and autophagic cell death, are defined and charac-terized by distinct morphological and molecular features.
Among all the forms of cell death, apoptosis (or type I cell death) is common to the vast majority of physiological cell death and the best char-acterized. The key apoptotic proteins, caspases, a cysteine endo-proteases family, act as death effectors in apoptosis. Caspases are synthesized as in-active zymogens that are subsequently activated by specific proteolytic cleavage. The morphologic features of apoptotic cell death include polynucleosomal DNA fragmentation, loss of overall cell shape, nuclear shrinkage, and phagocytosis of cell fragments without accompanying in-flammatory responses[6]. Various cellular stress conditions have been shown to trigger apoptosis, including irradiation, chemotherapeutic agents, viral infection, oxidative and ER stress[5,7].
Autophagy (or type II cell death) is an evolutionarily conserved mul-tistep catabolic process characterized by formation of autophagosomes, double membrane-bound vesicles surrounding long-lived cytoplasmic proteins and organelles, destined for recycling[8]. In recent years, au-tophagy has received considerable attention due to its dual roles in medi-ating decisions between life and death. The ability of autophagy to recycle old components makes it essential for normal cellular homeostasis and stress adaptation to prevent cell death. However, to cope with excessive stress, autophagic cells may commit suicide by undergoing a type of caspase-independent cell death, which differs from apoptosis and pro-grammed necrosis[9,10]. The molecular events eventually determining the protective/destructive fate of autophagy remain elusive. Interestingly, in certain conditions, apoptosis and autophagy can exert synergetic
effects, while under other circumstances inhibition of apoptosis can trigger autophagic cell death[11,12]. Recent evidence suggests cross-talk between apoptosis and autophagy at the molecular level[13]. Typically, autophagy is observed in cells that are exposed to various metabolic and therapeutic stresses, including nutrient starvation, growth factor deprivation, inhibition of the receptor tyrosine kinase/ Akt/mTOR (mammalian target of rapamycin) signaling, ischemia/ reperfusion, inhibition of proteasomal degradation, the accumulation of intracellular calcium, and ER stress.
Necrosis (or type III cell death) refers to cell deaths morphologically characterized by cytoplasmic granulation, organelle/cell swelling and cell lysis. This lysis releases intracellular constituents, leading to an in flamma-tory response provoked by immune cells. Necrotic cell death has been ob-served in response to different harsh cellular stresses, including heat shock, osmotic pressure, ischemia, glutamate excitotoxicity in neurons or cancer cells exposed to alkylating DNA damaging agents[5]. For many years, apoptosis was considered as the only form of regulated cell death, whereas necrosis was mostly considered as an accidental, random, passive and uncontrolled process. However, recent evidence has greatly changed this view and revealed the existence of‘regulated necrosis’ con-trolled by a set of signaling transduction cascades[14–17]. During the last decade, additional cell death modes (such as necroptosis, parthanatos, oxytosis, ferroptosis, ETosis, NETosis, pyronecrosis and pyroptosis) and their underlying pathways have been characterized and classified. As these cell death subroutines have been shown to share a necrotic mor-phological hallmarks and to be responsive to specific pharmacological or genetic interventions, these alternative cell death types with unique features are proposed to be considered as different forms of regulated necrosis[16,17]. For example, pyroptosis, initially introduced to describe the atypical demise of macrophages infected by Salmonella enterica serovar Typhimurium, is associated with anti-microbial responses during inflammation and a cell death subroutine mediated by the activation of caspase-1, but rather than depending on the classical apoptotic caspases [18]. During pyroptosis, caspase-1 is activated by a large supramolecular complex termed the inflammasome (also known as pyroptosome). In addition to allowing for the maturation and secretion of the inflammatory cytokines IL-1β and IL-18, the activation of caspase-1 also induces the for-mation of the plasma membrane pores rapidly leading to osmotic cell lysis, release of the cytosolic contents and cytokines that activate pro-inflammatory immune cell mediators[18]. Moreover, pyroptotic cells un-dergo DNA fragmentation with retained integrity and nuclear condensa-tion, showing positive TUNEL staining.
In addition to the modes of cell death described above, it is well ac-cepted that other pathways that have not yet been explored may exist. In general, the dominant death pathway invoked by a particular stress stimulus largely depends on the nature and level of the insult, the acti-vation state or differentiation state of individual cells and the ability of the stressed cell to handle the conditions to which it is exposed[5]. Interestingly, multiple types of death pathways could be activated in single dying cells and crosstalk between these death programs may allowfine control over the ultimate outcome. Inhibiting the dominant cell death route may not lead to cellular survival but, instead, induce the alternate cell death programs[11–13]. Therefore, understanding how these processes are interconnected could predict cellular suscepti-bility to stresses and reveal novel therapeutic targets. Additionally,
examining the molecular program occurring in dying cells could lead to the characterization of novel pathways of cell death and further our un-derstanding of the physiological and pathological processes underlying a variety of human diseases. As a detailed description of these cell death processes is beyond the scope of this article, we would like to refer the reader to a number of excellent reviews that provide more in depth in-sights into different forms of programmed cell death[6,8,10,16–20]. 2. Human DExD/H RNA helicases
RNA helicases are highly conserved molecular motors that remodel RNA or ribonucleoprotein (RNP) complexes in an ATP-dependent manner [21–23]. With only a few exceptions, RNA helicase genes are encoded in prokaryotic, archaea, eukaryotic and viral genomes[24]. These enzymes are potentially associated with the entire lifespan of RNA, from transcrip-tion and translatranscrip-tion to degradatranscrip-tion[22,24]. Indeed, a growing number of these enzymes are linked with specialized regulatory processes, and many of them are essential for viability. Although much remains elusive, fascinating models are emerging to link structural, mechanistic, and cellular function for RNA helicases in the last two decades.
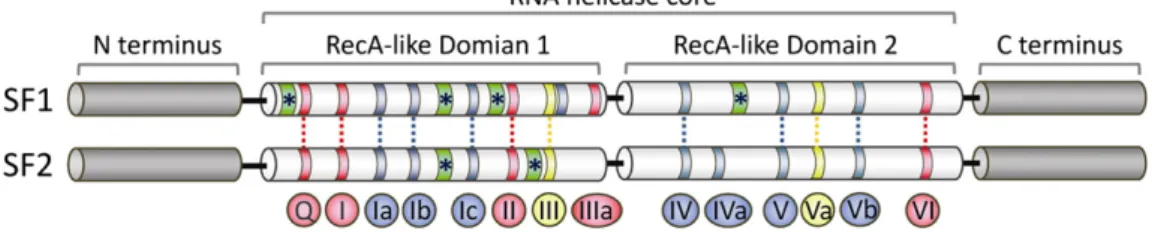
On the basis of conserved sequence motifs and structural features, nucleic acid helicases are broadly grouped into six superfamilies (SFs) [25,26]. The monomeric helicases comprise SFs 1 and 2, while the olig-omeric (mostly hexameric) ring-forming ones comprise SFs 3 to 6[26]. As the ring-shaped RNA helicases will not be discussed here, readers are referred to recent excellent reviews on these proteins[27,28]. Meanwhile, all eukaryotic RNA helicases belong to the largest groups, SFs 1 and 2. These enzymes contain a structurally conserved helicase core, composed of two tandemly repeated RecA-like globular domains (DOMAINS 1 and 2) (Fig. 1) that structurally resemble the bacterial recombinase A protein (RecA)[29]. In terms of three-dimensional architecture, Domains 1 and 2 are connected by aflexible linker region and form a conserved structural fold facilitating the motor protein function associated with helicase activ-ity. Within this helicase core, at least 12 characteristic motifs, so called helicase motifs Q, I, Ia, Ib, Ic, II, III, IV, V, Va, Vb and VI are arranged sequen-tially and located at defined positions (Fig. 1). These conserved motifs comprise residues essential for ATP binding and hydrolysis (motifs Q, I, II and VI), RNA binding and translocation (motifs Ia, Ib, Ic, IV, V and Vb) and for coupling ATP hydrolysis to unwinding (motif III)[22,23,25,29, 30]. However, not all motifs are present in each helicase family, whereas the levels of sequence conservation in these motifs are decreasing among different families[25]. Notably, additional domains have been identified in some helicases[31]. These accessory domains may rigidify the relative position of Domains 1 and 2, providing an extensive RNA in-terface and assisting in presenting RNA substrates to the helicase core [30].
The conserved helicase core is surrounded by variable auxiliary domains (Fig. 1). These C- and N-terminal regions are generally not conserved within or between families[25]. These extended C- and N-terminal domains are usually larger than the helicase core and believed to confer functional specificity to individual helicases. For example, these
flanking regions have been shown to modulate enzymatic activity, to facilitate recognition of specific nucleic acid regions or to provide docking sites for protein–protein interactions[22,23,32].
The majority of RNA helicases belong to superfamily 2 (SF2) com-prised offive subfamilies, further distinguished based on the amino acid sequence of the conserved helicase motif II (DEAD, DEAH, DExH and DExD helicases)[25]. Of note, not only these RNA helicases facilitate RNA duplex separation, but also many of them catalyze strand ex-change, strand annealing and RNA assembly/disassembly[22]. Biologi-cally, the human DExD/H RNA helicases have a range of function in nearly every cellular process associated with RNA metabolism, including pre-mRNA processing, ribosome biogenesis, RNA turnover, export, trans-lation, surveillance, storage and decay[32,33]. It is now clear that human DExD/H RNA helicases generally act as components of multi-protein complex with additional ATP-independent roles presumably conferred through their interactions with protein partners. Such complexity and multi-functionality would allow DExD/H RNA helicases to integrate dif-ferent processes in RNA metabolism.
3. DExD/H RNA helicase-mediated regulation of cell survival under stress
As the human DExD/H RNA helicases participate in almost every step of RNA metabolism, it is not surprising that these helicases play a critical role in gene expression regulation during cellular stress re-sponse. Indeed, emerging evidence demonstrates that human DExD/H RNA helicases could act as hubs in the cellular networks to coordinate cellular stress responses to maintain cellular homeostasis for survival or to trigger cell death. In this review, we will attempt to summarize recent advances in understanding of the molecular mechanisms that enable human DExD/H RNA helicases to sustain cell survival, coordinate stress responses, and mediate cell death, especially focusing on microbial pathogen-induced signaling cascades and stress granule formation triggered by various stress stimuli. We apologize for the omission of a large number of references that we could not include here due to space limitations. In addition, we would like to refer the reader to excel-lent reviews that provide more in depth analysis of prokaryotic and plant RNA helicases in stress response[34–36].
3.1. Defense against pathogen infections— cell death induction by DExD/H RNA helicases
Various microbial pathogen infections impose different levels of stress on the infected cells. During infections, pathogens are detected by innate immune cells with the help of various receptors recognizing cer-tain derived conserved structures, known as pathogen-associated molecular patterns (PAMPs)[37]. Pathogen-derived nucleic acids, including their genomic RNA and DNA, as well as replication inter-mediates and double-stranded RNA, have long been recognized as a PAMP and detected by several cellular receptor proteins. These receptors facilitate downstream signaling events to induce antiviral interferon
Fig. 1. Structure of RNA helicase core in SF1 and SF2 proteins. The helicase core region of SF1 and SF2 RNA helicases consists of two RecA (recombinase A)-related domains (domains 1 and 2) containing at least 12 conserved motifs. The motifs are shown with color according to their primary function (red, ATP binding and hydrolysis; yellow, coordination between NTP- and RNA-binding sites; blue, RNA binding). Notably, Motif Ib is not shared in all SF1 and SF2 families. Green ribbons with asterisks indicate insertions of additional motifs. Apart from the con-served functional helicase core, most SF1 and SF2 helicases possess additional variable N- and C-terminal extensions that provide RNA/protein cofactor specificity to individual helicases. The lengths of the motifs and the distance between the conserved domains are not drawn to scale.
(IFN) production and initiate a series of immune responses. Apart from interferon production and immune response, programmed cell death of infected cells induced by these receptors is now being recognized as another line of defense against microbial pathogens[38–42]. Since path-ogen propagation requires a functional host metabolism, death of host cell could interfere the replicative cycle of pathogens, expose the patho-gen to extracellular immune surveillance and promote activation of adap-tive immune system by presenting microbial antigens to T cells[43,44]. Accordingly, to diminish the replicative niche of pathogens, pro-survival defense mechanisms need to befine-tuned with cellular suicide program in response to pathogen infections.
In recent years, it has emerged that several DExD/H-box RNA helicases contribute to anti-pathogen immunity and cell survival regula-tion upon pathogen infecregula-tion. One of the main groups of receptors sensing pathogen-derived nucleic acids is the RIG-like receptors (RLRs), which are part of the DExH-box RNA helicases[45,46]. In addition to RLRs, numer-ous DExD/H-box helicases have recently been implicated in the sensing of pathogen-derived nucleic acids and/or facilitating downstream signal-ing events to induce interferon production, immune responses or cell death[46–49]. Here, we summarize the current knowledge on DExD/H RNA helicases in defense against pathogen infections and their functional role in cell fate decision.Table 1contains a list of known pathogen targets, natural and synthetic ligands of these receptors.
3.1.1. RIG-I-like receptors (RLRs)
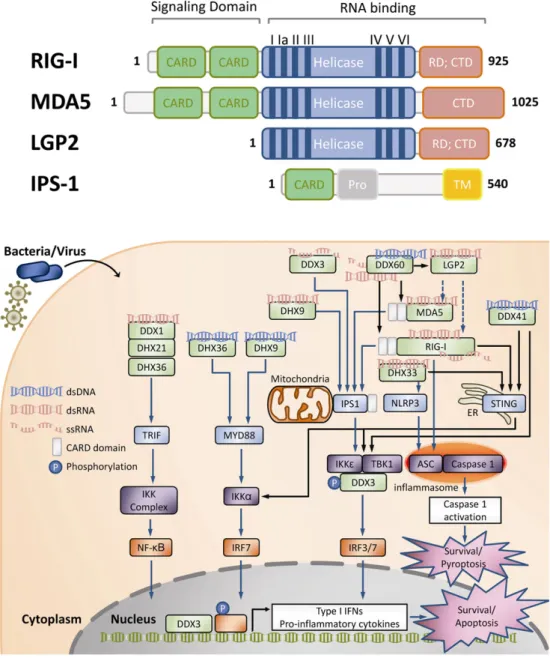
The RIG-I-like receptors (RLRs) are cytosolic proteins recognizing pathogen-derived nucleic acid species. In resting, these proteins are expressed at low levels in most cell types. However, upon stimulation by pathogen-derived nucleic acids, RLRs are induced to high levels. RLRs belong to the family of DExH-box helicases and include three members: RIG-I (retinoic acid-inducible gene I), MDA5 (melanoma differentiation-associated antigen 5) and LGP2 (Laboratory of Genetics and Physiology 2). Structurally, RLRs are related to each other (Fig. 2A). RLRs contain the central helicase core region comprising the conserved two RecA-like domains and a C-terminal regulatory domain (CTD) which mediates spe-cific recognition of nucleic acids[50,51]. Of note, RIG-I and MDA5 harbor CARD (cysteine-aspartic protease (caspase)-recruiting domain) for inter-action with the mitochondrial CARD-domain containing adaptor IPS-1 (IFNB-promoter stimulator 1; also known as MAVS, CARDIF and VISA; Fig. 2A and B). The binding of RLR to RNA ligands results in ATP hydrolysis, along with RLR oligomerization and translocation to mitochondria, pro-moting RLR interaction with IPS-1 (Fig. 2B). These interactions further in-duce phosphorylation of IRF3/7 by the IKK-related kinases TBK1 and IKKε, leading to activation of these transcription factors and the downstream expression of type I IFNs and pro-inflammatory cytokines. Subsequent type I IFN signaling triggers a potent antiviral response defined by the rapid induction of hundreds of IFN-stimulated genes. These genes encode
proteins that confine viral infection through regulation of cellular meta-bolic activity, modulation of inflammation and/or eliciting apoptosis.
3.1.1.1. RIG-I (retinoic acid-inducible gene I; also known as DDX58). RIG-I harbors an ssRNA/dsRNA-binding C-terminal domain (CTD) which functions as a repressor domain (RD) when unbound to RNA ligands (Fig. 2A). The prototypical RIG-I ligand is short RNA with an uncapped 5′ triphosphate end and blunt-ended base pairing. In addition, RIG-I has been shown to bind to various dsRNA and ssRNA ligands[52,53]. Consistent with this notion, RIG-I has been demonstrated to be involved in the recognition of various viruses, including antisense (−)ssRNA viruses or sense (+)ssRNA/dsRNA viruses[54]. Moreover, RIG-I could be indirectly activated by cytosolic viral and bacterial double-stranded DNA (dsDNA), as these pathogen AT-rich dsDNA can be transcribed by RNA polymerase III to generate dsRNA with 5′ triphosphate ends [55,56].
Recent structural studies have provided a better understanding of ligand binding and activation of RIG-I. Specific binding of 5′ppp is medi-ated by the CTD whereas the helicase domain binds the double-stranded part of the RNA. In the inactive state, RIG-I is held in an auto-repressed conformation with unexposed CARD. Ligand binding results in the release of the two repeats of CARD at the N terminus. These conformation chang-es enable RIG-I to interact with CARD-containing adaptor protein IPS-1 located at the cytosolic face of the outer mitochondrial membrane (Fig. 2B). This mitochondrial association is necessary for triggering subse-quent downstream signaling events[57,58]. In addition, RIG-I could in-teract with the adaptor protein ASC, resulting in in flammasome-dependent caspase 1 activation and the subsequent pyroptosis (Fig. 2B) [42]. RIG-I, but not MDA5, was also shown to interact with STING (stim-ulator of IFN genes; also known as MITA, MPYS and ERIS), which is an adaptor protein predominantly found in the endoplasmic reticulum[59, 60]. This interaction potentiates RIG-I-mediated signaling through TBK1 following RNA virus infection (Fig. 2B).
3.1.1.2. MDA5 (melanoma differentiation-associated antigen 5; also known as IFIH1). MDA5 has been implicated in the response to long dsRNA. It has also been suggested that MDA5 recognizes particularly large and branched RNA structures generated during the viral replication[61]. Compared with RIG-I, MDA5 seems to mediate responses to distinct sets of viruses. However, MDA5 has also been shown to be of impor-tance in cooperation with RIG-I during the antiviral response[62]. MDA5 is structurally homologous to RIG-I (Fig. 2A). Similarly to RIG-I, upon binding of long dsRNA fragments, MDA5 exposes its CARD and ini-tiates subsequent cytokine and type I IFN production through interac-tion with IPS-1 (Fig. 2B)[63].
Table 1
Cytoplasmic RNA helicases critical for sensing pathogen-derived nucleic acid.
Nucleic acid sensor Sensed pathogens Natural ligand Synthetic ligand Note Reference RLR
RIG-I/Ddx58 (−)ssRNA virus, (+)ssRNA virus, dsRNA virus, DNA virus, bacteria
Viral ss/dsRNA 5′ppp-dsRNA Positive regulator [62,65,163–173] MDA5/Ifih1/Helicard (−)ssRNA virus, (+)ssRNA virus, dsRNA
virus, DNA virus
Viral long dsRNA Poly(I:C) Positive regulator [62,164,167,168,171,174–176] LGP2/DHX58 (+)ssRNA virus dsRNA Negative/positive regulator [64–66,177]
Non-RLR helicases
DDX1/DDX21/DHX36 (−)ssRNA virus, dsRNA virus Viral dsRNA Poly(I:C) Positive regulator [79] DDX3 (−)ssRNA virus Viral stem-loop RNA Poly(I:C) Positive regulator [72] DHX9 (−)ssRNA virus, dsRNA virus dsDNA, dsRNA Poly(I:C) CpG-B ODNs Positive regulator [77,78]
DHX36 DNA virus dsDNA CpG-A ODNs Positive regulator [78]
DDX41 DNA virus Synthetic AT-rich B DNA Positive regulator [74]
DDX60 (−)ssRNA virus, DNA virus Viral ss/dsRNA; dsDNA Positive regulator [80] DHX33 dsRNA virus, bacteria Viral dsRNA Poly(I:C) Positive regulator [81,82]
5′ppp, 5′ triphosphate end; dsRNA, double-stranded RNA; LGP2, Laboratory of Genetics and Physiology 2; MDA5, melanoma differentiation-associated protein 5; ODN, oligodeoxynucleotide; poly(I:C), polyinosinic–polycytidylic acid; RIG-I, retinoic acid-inducible gene I; ssRNA, single-stranded RNA.
3.1.1.3. LGP2 (Laboratory of Genetics and Physiology 2; also known as DHX58). LGP2 is the third member of the RLRs and less well-characterized. The LGP2 contains a similar helicase core and CTD as RIG-I and MDA5, but lacks the CARD region present in RIG-I and MDA5 (Fig. 2A). As the CARD region is responsible for the association
with IPS-1 and the following signaling events, LGP2 is initially thought to be a negative regulator of RLR signaling via interaction between the RD region of LGP2 and that of RIG-I (Fig. 2A)[64,65]. However, both positive and negative effects have been observed in recent reports, de-pending on the exact nature of the RNA ligand, the virus studied and
Fig. 2. Pathogen-derived nucleic acid-sensing and responsive machinery composed of cytoplasmic RNA helicases. (A) Structure of the RIG-I like receptors (RLRs) and their adaptor IPS-1. RLRs are cytosolic proteins and structurally related to each other. These proteins harbor three key structural domains: (1) the two N-terminal tandem caspase activation and recruitment domains (CARDs), present in RIG-I and MDA5 but absent in LGP2, are required for interaction with IPS-1 and downstream signaling. (2) The central helicase domain involved in RNA binding, translocation/unwinding of RNA and ATP hydrolysis required for RLR function. (3) The unique C-terminal domain (CTD) containing a repressor domain (RD) involved in auto-regulation. MDA5 lacks the RD. The adaptor IPS-1 consists of a transmembrane domain (TM) on its C-terminus, a proline-rich region (Pro) and the CARD for interaction with RIG-I and MDA5. (B) Overview of the nucleic acid-sensing RLRs and non-RLR RNA helicases in cell survival regulation during pathogen infections. In the cytosol, the microbial pathogen-derived nucleic acids could be recognized by various receptors, including the RIG-I-like receptors (RLRs) and some other DExD/H-box helicases. These receptors could induce the expression of pro-inflammatory cytokines/type I IFNs or trigger inflammasome formation to promote a potent antiviral response through activation of various signaling cascades, which may eventually lead to apoptosis or pyroptosis. RLRs are structurally conserved to each other. RIG-I recognizes ss- and ds-viral RNAs, whereas MDA5 responds to highly structured ssRNA and dsRNA species. LGP2 has been proposed as both a positive and a negative regulator of RIG-I and MDA5 signaling. The two N-terminal CARD regions, present in RIG-I and MDA5 but absent in LGP2, are required for interaction with IPS-1 and downstream IPS1-mediated activation of TBK1 and IKKε and subsequent activation of transcription factors IRF3, IRF7 and NFκB. Additionally, RIG-I may interact with the adaptor protein ASC, leading to inflammasome-dependent caspase 1 activation and a following pro-inflammatory form of cell death called pyroptosis. RIG-I was also shown to interact with STING (stimulator of IFN genes), an adaptor protein located in the ER. This interaction potentiates TBK1-mediated RIG-I signaling. DDX60 has been proposed to sensitize the RLRs to viral ssRNA, dsRNA as well as dsDNA ligands through association with RIG-I, MDA5, and LGP2 but not with IPS-1. The proposed cytosolic DNA receptors DDX41 interact with STING and TBK1 to activate TBK1, IKKε and the IKK complex. DHX9 has been proposed to sense dsRNA and trigger an IPS-1-dependent signaling. In addi-tion, DHX36 and DHX9 activate TRIF-dependent and MYD88-dependent signaling in response to CpG-A and CpG-B DNA, respectively. DDX1, DDX21 and DHX36 have been suggested to detect dsRNA and form a TRIF-interacting complex. DHX33 has been proposed to sense viral and bacterial RNA to activate NLRP3-dependent or STING-dependent signaling. DDX3 has been reported to be a signaling adaptor through its interaction with TBK1 and IKKε. DDX3 has also been suggested to act as a sensor of viral RNA in conjunction with the RIG-I and MDA5. Notably, DDX3 was demonstrated to be recruited to the IFN promoter, suggesting its transcriptional regulator role during infections.
the RLR recognizing this virus[66]. Therefore, LGP2 is proposed to be a modulator of the innate immune response, but not a sensor due to its incapability to initiate antiviral gene expression. Interestingly, LGP2 has been shown to enhance MDA5-dependent IFN induction and to bind dsRNA independent of 5′-triphosphates[67]. Given that the affinity of the MDA5 CTD for dsRNA is relatively low, LGP2 might serve as a co-receptor enhancing the ability of MDA5 to sense long dsRNA structures by complexing with MDA5[67]. It will require more investigations to un-derstand how LGP2 modulates RIG-I-dependent responses to different RNA ligands and MDA5-mediated recognition of dsRNA.
3.1.2. Non-RLR helicases
In addition to RLRs, several other DExD/H-box helicases, including DDX3, DDX60, DDX41, DDX1, DDX21, DHX9, DDX33 and DHX36, have recently been implicated as additional receptors for pathogen-derived nucleic acids and to induce downstream signaling and programmed cell death (Fig. 2B). Interestingly, as mentioned above, many of these helicases contain auxiliary variable N- and C-terminal regions conferring their functional specificities, such as the ability to bind specific RNA targets or to induce downstream signaling through protein/protein in-teraction[46–48].
3.1.2.1. DDX3/DDX3X/DBX/CAP-Rf. Human DDX3 is a ubiquitously expressed RNA helicase. DDX3 gene is located on the X chromosome and is highly homologous to DBY, which is present on the Y chromosome and only expressed in the male germ line[68]. The signaling adaptor role of DDX3 in anti-viral innate immune signaling pathways has been pro-posed in two reports[69,70]. DDX3 was identified as an interacting partner and phosphorylation substrate of TBK1 (TANK-binding kinase 1), a central kinase for the induction of interferon in response to patho-gens. DDX3 knockdown impairs interferon production whereas TBK1-mediated phosphorylation of DDX3 is required for DDX3 to synergize with TBK1 to stimulate the IFN promoter, suggesting that DDX3 is a crit-ical TBK1 effector necessary for IFN induction[69](Fig. 2B). Additionally, DDX3 was demonstrated to be recruited to the IFN promoter, highlight-ing its possible transcriptional regulator role after TBK1 activation[69] (Fig. 2B). Another report demonstrated an interaction between DDX3 and IKKε[70](Fig. 2B). Notably, DDX3 enhanced IFN promoter induction by TBK1/IKKε in an ATPase and RNA unwinding activities-independent manner[70]. However, the unique N-terminal fragment of DDX3 was re-quired for this activity[70]. As DDX3 was identified as a host target of the vaccinia virus (VACV) protein K7 in the same work, the authors proposed that K7 forms a complex with DDX3 and antagonizes TBK1/IKK ε-mediated IFN-beta promoter activation to inhibit IFN-beta induction [70]. Recently, the same research group confirmed the direct DDX3/ IKKε interaction and identified the requirement of IKKε-mediated DDX3 phosphorylation at serine 102 for the recruitment of IRF3 into the complex, IRF3 activation as well as IFN promoter induction[71] (Fig. 2B). Therefore, thesefindings propose DDX3 as a scaffolding adaptor coordinating the activation of IKKε and IFN induction.
More recently, Oshiumi et al. demonstrated an interaction between DDX3 and IPS1 and suggested that DDX3 might directly bind to vesicular stomatitis virus (VSV) RNA in conjunction with RIG-I and MDA5 to enhance the IFN-I response, supporting a regulatory role of DDX3 in the RLR pathway[72](Fig. 2B). Also noted, DDX3 is targeted by vaccinia virus protein K7[70]and HCV core protein[73]to abrogate DDX3-enhanced IPS-1 signaling. Collectively, thesefindings underscore the rel-evance of DDX3 in efficient sensing of pathogen-derived nucleic acids. 3.1.2.2. DDX41. DDX41 was shown to bind specifically to DNA rather than RNA. This binding was further mapped to the DEAD domain of DDX41[74]. In addition, DDX41 could directly interact with TBK1 and STING, a crucial adaptor molecule for cytoplasmic DNA receptors, but not IPS1[74](Fig. 2B). Indeed, silencing of DDX41 expression in murine myeloid dendritic cells (mDCs) led to a marked decrease of IFN and pro-inflammatory cytokine production in response to cytosolic DNA[74].
Another recent report confirmed the DNA receptor role for DDX41 during adenovirus infection in the murine macrophage cell line[75]. Collectively, thesefindings identify DDX41 as an additional intracellular STING-dependent DNA sensor for pathogenic DNA. Notably, DDX41 has also been demonstrated to recognize bacterial secondary messenger molecules cyclic di-GMP and cyclic di-AMP (cyclic dinucleotides; CDNs) and form a complex with STING to activate the interferon response, highlighting its importance in innate immune response[76].
3.1.2.3. DHX9/RNA Helicase A (RHA)/Nuclear DNA Helicase II (NDHII). In myeloid dendritic cells (mDCs), DHX9 is required for the production of pro-inflammatory cytokines and IFNα/β in response to influenza virus, reovirus and poly(I:C) (polyinosinic–polycytidylic acid, a sub-strate used as a mimic of viral dsRNA)[77]. Interestingly, DHX9 binds dsRNA via its two N-terminal dsRNA-binding domains, whereas its C-terminus interacts with the CARDs of IPS-1, linking DHX9 into the RLH signaling pathway[77](Fig. 2B). In addition, DHX9 is required for the activation of NF-κB and IFN regulatory factor 3 by dsRNA, suggesting an IPS-1-dependent RNA sensor role for DHX9 in pathogenic RNA-sensing. It is worthy to note that DHX9 is recruited by various viruses [46], suggesting that DHX9, like DDX3, may have a dual function as an essential host cofactor for viral replication and an innate immune medi-ator in the battle between viruses and the host immune system. 3.1.2.4. DHX9 and DHX36 as CpG sensors. In addition to the dsRNA sen-sor role of DHX9 mentioned above, DHX9 has also been identified in plasmacytoid dendritic cells (pDCs) as a sensor for CpG DNA[78]. CpG DNA (or CpG ODN) are oligodeoxynucleotides containing immunostimulatory unmethylated dinucleotide CpG motifs. These motifs are considered pathogen-associated molecular patterns (PAMPs) due to their abundance in microbial genomes and their rarity in vertebrate genomes. DHX9 was shown to bind CpG-B, whereas DHX36 was characterized as a CpG-A binding protein[78]. Interestingly, DHX36 binds CpG-A via the helicase domain, while DHX9 interacts with CpG-B through its C-terminal region[78]. Also noted, DHX9 is associated with TNFα and IL-6 production as well as NFκB activation in re-sponse to CpG-B, but DHX36 is important for IFNα production in re-sponse to CpG-A. As both helicases activate the cytosolic adapter protein MyD88 (myeloid differentiation primary response gene 88) through binding to its TIR (Toll-interleukin receptor) domain, these ob-servations demonstrate that DHX9/DHX36 represent the MyD88-dependent DNA sensors in pDCs (Fig. 2B).
3.1.2.5. DDX1/DDX21/DHX36 complex. In myeloid dendritic cells (mDCs), another viral sensing complex consisting of RNA helicase DDX1, DDX21 and DHX36 has been identified by isolation of poly(I:C)-binding pro-teins[79]. In response to poly(I:C), this DDX1/DDX21/DHX36 complex triggers an antiviral program dependent of the adapter molecule TRIF (TIR-domain containing adapter-inducing interferon-β). Intriguingly, only DDX1 directly bound to poly(I:C) via its helicase domain, whereas both DHX36 and DDX21 bind the TIR domain of TRIF[79](Fig. 2B). This complex was shown to be required for the IFN induction in response to long or short poly(I:C), influenza and reovirus infection[79].
3.1.2.6. DDX60. DDX60 localizes at the cytoplasmic region and its expres-sion could be induced upon viral infection[80]. Intriguingly, DDX60 could bind viral ssRNA, dsRNA as well as dsDNA[80], enhancing the IFN response to RNA/DNA viruses and DNA stimulation through associ-ation with RIG-I, MDA5, and LGP2 but not with IPS-1[80]. Thus, DDX60 is proposed to act as a cofactor promoting RIG-I-like receptor-mediated signaling (Fig. 2B).
3.1.2.7. DHX33. In innate immune responses, the NLRP3 (NLR Pyrin-domain containing 3)-induced inflammasome plays a critical role by ac-tivating caspase-1, resulting in secretion of IL-1β and IL-18. Recently, RNA helicase DHX33 was identified as a cytosolic RNA receptor able to
activate the NLRP3 inflammasome in human macrophages [81] (Fig. 2B). DHX33 is involved in the activation of caspase-1 and secretion of IL-1β/IL-18 induced by cytosolic poly(I:C), reoviral RNA or bacterial RNA[81]. Notably, DHX33 was shown to bind dsRNA and NLPR3 through its helicase C domain and DEAD domain, respectively[81]. In agreement with thesefindings, another recent study confirmed the role of DHX33 in the sensing of cytosolic poly(I:C) and reoviral RNA in myeloid DCs[82]. However, in this report, poly(I:C)-induced activation of NF-κB and IRF3 was demonstrated to be mediated by the interaction between DHX33 and IPS-1, which was mapped to the helicase C domain of DHX33[82].
To ensure viral replicative niche, many viruses have evolved strate-gies for interfering with the potent anti-viral effect of type I interferons. Numerous examples of viral immune evasion proteins and strategies have been reported, including masking of viral RNA ligands and direct inhibition of nuclei acid receptors mentioned above[83]. Most intrigu-ingly, numerous viruses rely greatly on host RNA helicases to mediate RNA remodeling events during their replication cycle. Consistent with this notion, several DExD/H-box helicases have been identified as essen-tial host factors for the viral replication[46]. For example, DDX3 has been shown to be essential for the replication of several viruses, including the most famous HCV and HIV[46,84–86]. Similarly, DDX1 and DHX9 are re-quired for the replication of various viruses[46]. Thus, the host RNA helicases utilized by viruses may also be the very same ones implicated in anti-viral innate immune responses. Future investigation on this ongo-ing co-evolutionary combat between host and viruses would lead to new insights into the host anti-viral immune response and cell death. 3.2. Stress granule formation to suppress apoptosis under stress
3.2.1. Translation silencing, stress granule assembly and cell survival upon stress
Cells have evolved multiple strategies to cope with various environ-mental insults, such as oxidative, genotoxic, hyperosmotic, heat shock and viral infection. The cellular response to these stresses involves a global silencing of protein translation. One hallmark of the stress response re-ported recently is the temporary formation of large cytoplasmic RNP foci, known as stress granules (SGs)[87–91].
3.2.1.1. Translational regulation and stress granule formation. Translational regulation plays a central role in the control of gene expression. Under most circumstances, recruitment of ribosomes to mRNA is the rate-limiting step in translation initiation and a primary target for translational control[92]. This process is facilitated by numerous eukaryotic initiation factors (eIFs). One of these, eIF4G, an essential scaffold protein, is a sub-unit of the hetero-trimeric eIF4F complex associating with the mRNA m7G cap and facilitating ribosome joining to the mRNA. Two other
com-ponents of eIF4F complex are the cap-binding protein eIF4E and the RNA helicase eIF4A. Additionally, eIF4G interacts with the poly(A) binding protein (PABP), which stimulates initiation factor recruitment to mRNA and leads to mRNA circularization[93,94]. The PABP–eIF4G interaction is required for efficient translation, and it stimulates the formation of both 48S and 80S ribosome complexes[93].
In response to environmental stress (e.g., heat, oxidative conditions, UV irradiation, and hypoxia), aggregates of stalled translation initiation complex localize to cytoplasmic foci called stress granules (SGs)[87,90, 95]. SGs have been observed in various species and reported in vivo[87, 88,91]. Although the composition may vary with stress stimuli and cell type[87,89], SGs typically harbor most components of the stalled 48S pre-initiation complex (i.e., small ribosomal subunits, mRNA tran-scripts, eIF3, eIF4A, eIF4E, eIF4G, eIF2, eIF2B and PABP1). These core con-stituents of SGs are universal markers for all SGs. In addition, many other protein components, including RNA helicases, RNA-binding pro-teins, transcription factors, translation regulators and signaling mole-cule have been reported to accumulate in SGs[87–89]. The selective SG recruitment of both stabilizing and destabilizing proteins supports
a model in which these dynamic cytoplasmic foci are sites of mRNA triage during stress.
Apart from stalled translation initiation during a stress response, SG formation could be induced by pharmacological inhibition of translation initiation, knockdown of specific initiation factors, or overexpression of translational repressors[88]. However, not all initiation obstruction in-duces SGs, suggesting that SG assembly occurs upon certain impeded ini-tiation processes. Interestingly, several SG-associated proteins promote SG assembly when overexpressed (e.g., TIA-1 or TIAR, G3BP, CPEB, FAST, FXR1 and FRM), whereas other proteins have no effect or inhibit SG assembly[87]. Thesefindings indicate that ‘primary aggregation’ is probably mediated by these SG nucleators that initiate mRNP aggregation and thus physically nucleate SG assembly.
In addition, SGs are closely linked to another RNP granule, processing body (PB), cytoplasmic mRNA degradation machinery[96]. These find-ings therefore raise a model in which SGs serve as triage centers that sort, remodel, and export specific transcripts for re-initiation, storage, or decay[87–89]. Most notably, SGs are highly dynamic structures and re-cently proposed to act as signaling hubs by intercepting a group of signal-ing molecules to modulate metabolism, growth and survival upon stress [97]. Despite recent progress, a complete list of SG components is still lacking. Thus, a deeper insight into the nature of the mRNP complex with-in SGs would provide a better view of their function.
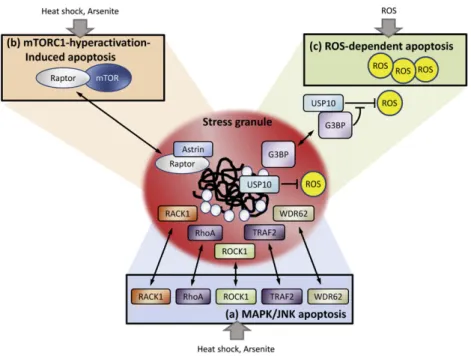
3.2.1.2. Stress granules and apoptosis. In addition to being critical for mRNA regulation during stress, SGs modulate the signaling balancing apoptosis and cell survival. Accumulating evidence reveals that stress granule assembly and apoptotic cell death tend to occur in a mutually exclusive fashion. Interventions preventing stress granule formation, such as knockout of SG proteins, render cells more vulnerable to stress, highlighting the cyto-protective role of SGs against environmental insults[88]. Although the precise mechanism underlying how SGs inhibit apoptosis has not been fully elucidated, recent reports pro-vided evidence of the crosstalk between SG assembly and apoptosis (Fig. 3).
One emerging scenario of the protective effect of SGs is the sequestra-tion of pro-apoptosis factors into SGs to nullify their activities. Thefirst case reported is the TRAF2 (TNF receptor-associated factor 2), a central signaling molecule in TNF-α-mediated NF-κB pro-inflammatory signal-ing. Upon treatment with various stress-inducing agents (heat shock, arsenite, puromycin [polysome destabilizer] and FCCP [a chemical uncou-pler of electron transport and oxidative phosphorylation]), TRAF2 is se-questered in to SGs. Under heat stress, TRAF2 is retained in SGs through the interaction with SG component eIF4G, resulting in the blockage of TRAF2-mediated TNF signaling and downstream pro-inflammatory responses responsible for cell death[98](Fig. 3). Similarly, several key molecules facilitating the MAPK/JNK apoptotic pathway, such as RACK1 (The Receptor for Activated C Kinase 1), ROCK1 (Rho-associated, coiled-coil containing protein kinase 1), small GTPase RhoA (Ras homolog gene family member A) and WDR62 (WD repeat domain 62) were shown to be sequestered into SGs induced by heat shock or arsenite [99–101], thereby protecting cells from apoptosis (Fig. 3). More recently, an essential negative regulator of mTORC1 (mammalian target of rapamycin complex 1) in the cellular stress response, astrin, was identi-fied. Under stress conditions, astrin recruits the mTORC1 component rap-tor to SGs, thus inhibiting mTORC1 association and avoiding mTORC1-hyperactivation-induced apoptosis[102](Fig. 3).
Another cell survival mechanism of SGs is linked to reactive oxygen species (ROS) production. A recent study reported that SGs harbor anti-oxidant activity, partly mediated by two SG components, G3BP1 (GTPase-activating protein SH3 domain binding protein 1) and USP10 (ubiquitin-specific protease 10)[103]. USP10 possesses an antioxidant activity. However, under steady-state conditions, its activity is sup-pressed by excess G3BP1. Upon stress, G3BP1 and USP10 cooperatively induce SGs. Meanwhile, SGs disrupt G3BP1-mediated inhibition against USP10, possibly by altering the conformation of USP10 and/or G3BP1,
thereby uncovering the antioxidant activity of USP10 to reduce ROS production[103]. The authors proposed that SGs may act as rapidly inducible antioxidant machinery protecting cells from ROS-induced apoptosis[103](Fig. 3).
An additional interesting possibility is that SG assembly is required for optimal translation of stress-responsive mRNAs. When canonical cap-dependent translation is compromised during stress, those mRNAs preferentially translated under these conditions tend to initiate translation by non-canonical mechanisms and their translation upon stress largely depends on uORF (upstream open reading frame) and IRES (internal ribosome entry site)[104–108]. Recent estimates suggest that 10–15% of cellular transcripts possess IRES activity, whose transla-tion mayfluctuate depending on the stress condition[106]. Both uORF and IRES rely on various trans-acting factors, such as eIF3[109], hnRNP A1[110]and PCBP2[111]. Interestingly, these factors are critical SG components, supporting that translation initiation of specific mRNAs may require SG formation and even occur in SGs.
Moreover, SGs were reported to be associated with the stability of certain mRNAs, such as ARE-containing mRNAs. The AREs (AU-rich elements), prominent instability cis-acting elements within the 3′ untranslated regions of unstable mRNAs, direct rapid mRNA decay process named‘ARE-mediated mRNA decay (AMD)’ and are responsible for the rapid decay of many short-lived mRNAs including cytokines, growth factors, and proto-oncogenes. In addition to the AREs, the AMD pathway involves trans-acting factors known as ARE-binding pro-teins (ARE-BPs) that positively or negatively affect the AMD process. Most remarkably, numerous ARE-BPs were recruited into SGs upon stress[90,112]. Because SGs contain proteins involved in both degrada-tion and stabilizadegrada-tion of mRNAs, their assembly might be critical for the regulation of mRNA decay during stress[90,112]. Indeed, current knowledge indicates that SGs do affect the stability of an array of stress-responsive mRNAs, including ARE-containing messages p21 mRNA, with consequences on cell survival[113]. Thus, SG-mediated reprogramming of mRNA translation and decay assembly may recon fig-ure the proteome to facilitate cellular adaption to adverse environmental conditions.
Collectively, the role of SGs in controlling apoptosis could be linked to sequestration of apoptosis-promoting factors, suppression of ROS production and reprogramming of mRNA expression upon stress. 3.2.2. Stress granule-associated RNA helicases
Considering the various RNA remodeling processes occurring in SGs, it is unexpected that only a few RNA helicases were found to be associ-ated with these foci (Table 2). And it will be no surprise if the number of RNA helicases associated with SGs is revealed to be much more. Here we summarize the relevance of these SG-associated RNA helicases in SG assembly, reprogramming of gene expression and cellular survival regulation during stressed conditions.
3.2.2.1. DDX1. RNA helicase DDX1 associates with poly(A) RNA[114] and is present in RNA-transporting granules[115]. When subjected to arsenite stress, DDX1 was found to colocalize with YB-1 and MBNL1 (muscleblind-like splicing regulator 1) at cytoplasmic stress granules, suggesting a role for these three proteins in mRNA metabolism in the cytoplasm during stress[116]. In addition, DDX1 was found in the cyto-plasmic PQBP1 (polyglutamine-binding protein 1) protein complex and co-localized with PQBP1 in SGs upon arsenite-induced oxidative stress [117]. As PQBP1 has been linked to several X-linked intellectual disability disorders and progressive neurodegenerative diseases, thesefindings may suggest a role for DDX1 in cellular survival regulation during the pathogenesis of intellectual disability.
Most recently, the interaction between DDX1 and another component of PQBP1 complex, KSRP (K homology Splicing Regulatory Protein), was further characterized[118]. KSRP is a decay-promoting ARE-binding pro-tein and predominately localizes in the nucleus. Previously, KSRP has been reported to be phosphorylated by AKT and this phosphorylation enhances its interaction with 14–3–3ζ, rendering its localization to the nucleus[119]. Under oxidative stress, KSRP accumulates in SGs and colo-calizes with DDX1[117]. Interestingly, down-regulation of DDX1 elevated cytoplasmic levels of KSRP and facilitated ARE-mediated mRNA decay [118]. As DDX1 could compete with 14–3–3 for interaction with KSRP, the competing interactions of DDX1 or 14–3–3 with KSRP may control
Fig. 3. Cell survival mechanisms linked with SG formation. SGs have been shown to suppress apoptosis by various mechanisms: (a) upon heat stress or arsenite treatment, several central molecules facilitating the MAPK/JNK apoptotic pathway, such as RACK1, RhoA, ROCK1, TRAF2 and WDR62 were retained in SGs, and thereby nullify their activities and protect cells from apoptosis. (b) Under stress conditions like heat shock or arsenite, astrin recruits the mTORC1 component raptor to SGs, thus inhibiting mTORC1 association and avoiding mTORC1-hyperactivation-induced apoptosis. (c) SGs have antioxidant activity, partly mediated by two SG components, G3BP1 and USP10. USP10 possesses an antioxidant activity; however, its activity is suppressed by excess G3BP1 under steady-state conditions. Upon ROS stress, SGs disrupt G3BP1-mediated inhibition against USP10 probably through altering the conformation, thereby uncovering the antioxidant activity of USP10 to reduce ROS production and protect cells from ROS-dependent apoptosis.
the cytoplasmic-nuclear shuttling of KSRP, leading to a modulation of its AMD activity[118]. Accordingly, this DDX1-mediated AMD reg-ulation may play an important role during oxidative stress-induced reprogramming of mRNA expression and cell survival decision. 3.2.2.2. DDX2/eIF4A. eIF4A, which has been extensively characterized both biochemically and structurally, is the prototype and founding member of the DEAD box RNA helicase family. eIF4A is the most abundant translation initiation factor and is required for recruitment of ribosomes to cellular, and most viral, mRNAs. eIF4A exists as a free form or as a sub-unit of eIF4F, a heterotrimeric complex composed of eIF4E (the m7GpppN
cap binding protein) and the scaffolding protein eIF4G. The helicase activ-ity of eIF4A could be stimulated through transient association with eIF4B [120]. Notably, eIF4A is one of the core constituents of SGs[88,121]. Arse-nite, hippuristanol and pateamine exposure induced the recruitment of eIF4A to SGs[121,122]. Interestingly, inactivation of eIF4A activities, through either inhibition of its RNA binding or blockage of eIF4F assembly, could promote SG assembly[121,122]. As numerous stress-responsive mRNA possesses a long and structured 5′ un-translated region (UTR) facilitating its translation by a‘shunting’mechanism that does not require eIF4A-dependent scanning[107,123,124], these mRNAs might be-come preferred candidates for translation under stress when eIF4A isinactivated. Thus, the modulation of eIF4A activity during SG assembly may contribute to the stress-induced reprogramming of mRNA expres-sion and cell adaption.
3.2.2.3. DDX3/DDX3X/DBX/CAP-Rf. DDX3, as mentioned above, is a ubiqui-tously expressed DEAD box RNA helicase and a nucleo-cytoplasmic shut-tling protein possessing RNA dependent ATPase/helicase activity[86, 125]. Recent studies have revealed the multi-facets of DDX3 in transla-tional control[126–133]. Our previous work demonstrated that DDX3 functions as an eIF4E inhibitory protein to specifically repress cap-dependent translation through its N-terminal eIF4E-binding consensus
38YIPPHLR44[130]. DDX3 could thereby block the formation of
pre-initiation complex eIF4F and translation pre-initiation[130].
Consistently, DDX3 was previously reported to localize into cyto-plasmic SGs while overexpressed or after arsenite treatment[128,134, 135]. Our subsequent study described coordinative roles for DDX3 and its interactions with eIF4E/PABP1 in SG assembly and stress response [135]. A wide variety of stress stimuli, including heat shock, UV irradia-tion, ER, oxidative and/or osmotic stress, direct DDX3 with eIF4E and PABP1 into SGs[135]. Interestingly, DDX3 knockdown interfered with SG assembly, led to nuclear accumulation of PABP1 and reduced cell vi-ability following stress. Conversely, supplementation with DDX3 could restore these defects[135]. Notably, the SG-inducing capacity of DDX3 is independent of its ATPase and helicase activities, but mapped to the eIF4E-binding region of DDX3[135]. Moreover, the eIF4E-binding defective mutant DDX3 was impaired in its SG-inducing ability and
protective effect on cell survival under adverse conditions. These obser-vations characterize DDX3 as a pivotal SG nucleating factor and illus-trate coordinative roles for DDX3, eIF4E and PABP1 in integrating environmental signal, mRNA translation and cellular survival under ad-verse conditions.
3.2.2.4. DDX6 (RCK/p54/Dhh1/Me31B/Cgh-1). The human DEAD-box RNA helicase, DDX6, is an abundant protein found bound to non-translating mRNA in the cytoplasm and acts as a translational repressor[136]. DDX6 is highly expressed in malignant cell lines, and its expression is linked to the regulation of differentiation and cell growth of cancer cells. DDX6 directly interacts with AGO1 and AGO2 in RNA-induced silencing complex (RISC), facilitates P-bodies formation, and plays a role in Dicer-independent miRNA processing as well as ARE-mediated mRNA decay. Notably, DDX6 is a prototypic component of processing bodies and localizes to SGs upon arsenite or heat shock treatment [136–138].
Interestingly, mutagenesis of conserved DDX6 helicase motifs relieves its suppression on translation, reduces accumulation of DDX6 in P-bodies, inhibits its capacity to assemble P-bodies and impairs its interaction with oocyte repressor complex components[139]. More sur-prisingly, although C-terminal D2 domain of DDX6 alone is deficient for P-body assembly, this region alone is sufficient for translational repres-sion and complete accumulation in P-bodies[139]. Thus, the D2 domain may act as a protein binding platform, whereas the ATPase/helicase ac-tivity allows protein complex remodeling. In addition, two of DDX6-interacting factors, Ataxin-2 and Ataxin-2 like, are shown to regulate P-bodies and SG assembly, supporting the importance of DDX6 in the biogenesis of these two RNA granules[138,140]. Also noted, DDX6 was recently shown to interact with the VEGF mRNA 5′-UTR and regulate VEGF IRES-mediated translation under hypoxia[141]. Since SGs sequester and inhibit VEGF transcripts during hypoxia to regulate tumor cell surviv-al after irradiation via an unknown mechanism[142], these observations provide evidence for the DDX6-mediated modulation of VEGF mRNA translation and its functional consequences under hypoxic conditions. 3.2.2.5. DHX36/RHAU (RNA Helicase Associated with AU-rich element). RHAU is a DEAH box helicase (DHX36) associated with AU-rich element (ARE) of urokinase-type plasminogen activator (uPA) mRNA[143]. RHAU is a nucleocytoplasmic shuttling protein predominantly found in the nucleus. ATPase activity is necessary for RHAU function in the decay of uPA mRNA and for its nuclear localization[143,144]. RHAU was reported to associate with SGs upon treatments with various stress inducers, including arsenite, heat shock, hippuristanol and CCCP[145]. In the same study, RHAU was shown to physically interact with RNA through a unique N-terminal RNA-binding domain composed of a G-rich region and an RHAU-specific motif that is highly conserved be-tween RHAU orthologs. The same RNA-binding domain is necessary and
Table 2
Stress granule-associated RNA helicases.
RNA helicases Present in Relevant binding partners SG-inducing condition Note Refs DDX1 SG YB-1, MBNL1, KSRP, PQBP1 Arsenite • DDX1 moves from nucleus to cytoplasmic SGs
upon arsenite stress
[116–118]
DDX2/eIF4A SG eIF4B Arsenite,
Hippuristanol, Pateamine,
• Reduction of eIF4A levels and activity induces SG formation
[121,122]
DDX3 SG, PB eIF4E, PABP1, eIF4A, eIF4G, TDRD3 Arsenite, Heat shock, DTT, UV,
Sorbitol
• Nucleates SGs
• Knockdown of DDX3 impairs SG formation • N-terminal domain is essential for SG localization
[128,134,135,178,179]
DDX6(RCK/p54) SG, PB Ataxin 2, Ataxin 2-like Arsenite, Heat shock
• Reduced levels of Ataxin 2 or Ataxin 2-like lead to impaired SG assembly [137,138,140,179–181] DHX36/RHAU SG Arsenite, Heat shock, Hippuristanol, CCCP
• RHAU knockdown had no effect on SG formation • N-terminal RNA-binding domain is essential for
RHAU localization in SGs
sufficient for RHAU recruitment to SGs, whereas the ATPase activity of RHAU is involved in the RNA interaction and in the dynamic regulation of RHAU shuttling into and out of SGs[145]. As RHAU knockdown had no effect on arsenite-induced SG formation, it seems that RHAU does not participate actively in SG assembly[145]. However, considering that RHAU is a cis-acting factor involved in ARE-mediated decay of uPA mRNA and that its association with SG is regulated by RNA interac-tion, the significance of RHAU for SGs is proposed to associate with mRNA turnover regulation during stress.
3.3. Multifaceted function of DExD/H RNA helicases in stress survival regulation
As RNA remodeling enzymes, DExD/H RNA helicases have been im-plicated in an intriguing array of cellular functions associated with RNA metabolism. In this review, although we concentrate mainly on two kinds of cellular stress responses (microbial pathogen-induced signaling cascades and stress granule formation triggered by various stress stim-uli), it is important to note that there are additional ways in which DExD/H RNA helicases can exert their effect to regulate cellular survival during stress.
For example, significant attention has been focused on the quadruplex-resolving activity of DHX36/RHAU. DHX36 has been described to recognize and remove so-called quadruplex knots (G4-loops) present in gene promoter regions where they block transcription. The specificity of DHX36 for G4-loops is mediated by its N-terminal RNA-binding domain rather than its helicase core region[146]. Indeed, DHX36 has been suggested to resolve G4-loops present in the promoter of the transcriptional regulator YY1 and to thus facilitate its transcrip-tion[147]. Since YY1 has recently been shown to negatively regulate
the ifnb promoter, thesefindings may provide additional functional link between DHX36 and anti-viral immunity[148].
In addition, roles of transcriptional co-regulator have been shown for DDX3, DDX5 (also known as p68), DDX17 (also known as p72) and DDX20 (also known as gemin 3), suggesting their possible involvement in transcriptional regulation during stress response. Indeed, DDX5/p68, an established co-activator of the p53 tumor suppressor, has recently been shown to be required for the induction of p53-dependent p21 ex-pression and cell cycle arrest after DNA damage, highlighting a novel function of p68 as a modulator of the decision between p53-mediated growth arrest and apoptosis[149].
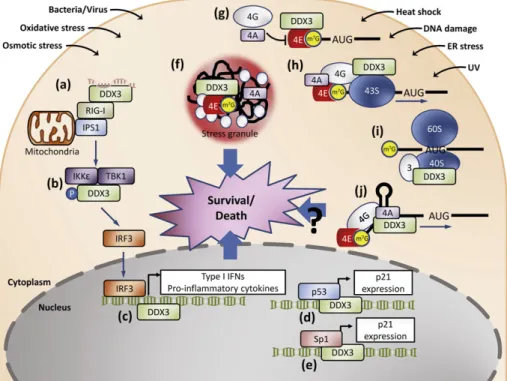
Moreover, like DHX36, one single RNA helicase may participate in stress response and survival regulation at multiple different levels. Over the past few years, DDX3 has been reported to participate in vari-ous mRNA biogenesis steps and may serve as a typical example to illus-trate multifaceted function of RNA helicases in stress survival regulation (Fig. 4). DDX3 wasfirst identified as an interacting partner of HCV (Hep-atitis C Virus) core protein[125,150,151]. As a member of DEAD box RNA helicase family, DDX3 is a nucleo-cytoplasmic shuttling protein and possesses RNA dependent ATPase/helicase activity[86,125]. Nucle-ar export of DDX3 can be mediated not only by the CRM1-dependent export pathway[86], but may also in part through the TAP-mediated pathway recently[128]. Over the past few years, an array of divergent cellular functions for DDX3 has been reported, including mRNA splicing, export, transport, ribosome biogenesis, as well as transcriptional and translational regulation[115,131,152–155]. As mentioned above, during microbial pathogen infection, DDX3 could act as a sensor for pathogen-derived nucleic acids in conjunction with the RLRs (Fig. 4)[72]. Addition-ally, DDX3 serves as a signaling intermediate through its interaction with TBK1 and IKKε (Fig. 4)[69,70]. Furthermore, DDX3 was recruited to the
Fig. 4. The multifaceted role of RNA helicase DDX3 in stress survival regulation. RNA helicase DDX3 has been proposed to have multifaceted function in response to various environmental insults. During microbial pathogen infection, DDX3 has been suggested to act as a sensor for pathogen-derived nucleic acids in conjunction with the RLRs (a). In addition, DDX3 could serve as a signaling intermediate through its association with TBK1 and IKKε (b). DDX3 has been also demonstrated to be recruited to the IFN promoter during infections, suggesting its tran-scriptional modulator role (c). After DNA damage, DDX3 could promote the retention and accumulation of p53 in the nucleus probably through its interaction with p53, and thereby reg-ulate apoptotic signaling (d). Notably, DDX3 could interact and cooperate with Sp1 to up-regreg-ulate the promoter activity of p21 in a p53-independent manner to exert its tumor suppressor function (e). Upon stimuli of various stresses, such as heat shock, UV irradiation, ER, oxidative or osmotic stress, DDX3 is required for the formation of cytoplasmic stress granules, which could suppress apoptosis (f). Different models proposed by recent studies for the role of DDX3 in translation initiation may confer optimal translation of stress-responsive mRNAs: DDX3 could sequester eIF4E from its binding to eIF4G resulting in the ubiquitous blockage of cap-dependent translation (g); DDX3 could promote ribosomal scanning of selected mRNAs harboring a highly structured 5′-UTR (h); DDX3 could promote 80S ribosome assembly through its association with eIF3 and the 40S ribosomal subunit (i); DDX3 is required for translation of certain mRNAs containing structures near the m7
GTP cap moiety (j). Considering the different effects exerted by DDX3 on the translation of mRNA with various structural features, these models may account for how cells reconfigure the proteome and determine their fate and survival during adverse conditions (see text).
IFN promoter, suggesting its transcriptional regulator role after TBK1 activation (Fig. 4)[69]. Notably, DDX3 associates with p53, increases its accumulation, and thereby positively regulates apoptotic signaling following DNA damage (Fig. 4)[156]. However, DDX3 could interact and cooperate with Sp1 to up-regulate the promoter activity of p21 in a p53-independent manner to exert its tumor suppressor function (Fig. 4)[152]. Upon stimuli of various stresses, such as heat shock, UV irradiation, ER, oxidative and/or osmotic stress, DDX3 is required for SG assembly, which could suppress stress-induced apoptosis (Fig. 4)[135].
Recently, multiple functional roles for DDX3 in translational regula-tion proposed by recent evidence may confer optimal translaregula-tion of stress-responsive mRNAs (Fig. 4): DDX3 could function as an eIF4E in-hibitory protein to specifically repress cap-dependent translation through its eIF4E-binding consensus motif eIF4E, and thereby block the formation of pre-initiation complex eIF4F and translation initiation [130]. In addition, DDX3 may preferentially promote the translation ini-tiation of structured 5′ untranslated regions upon its RNA helicase activ-ity[128,129]. Interestingly, DDX3 could promote 80S ribosome assembly through its association with eIF3 and the 40S ribosomal subunit[127]. Also noted, DDX3 is required for translation of certain mRNAs containing structures near the m7GTP cap moiety[133]. It is worthy to note that
numerous stress-responsive transcripts preferentially translated under adverse conditions tend to initiate translation by non-canonical mechanisms largely dependent on uORF and highly structured IRES [104–108]. Since both uORF and IRES rely heavily on various trans-acting factors, such as eIF3[109], the diverse scenarios proposed for the role of DDX3 in translation initiation of different mRNAs may sug-gest the relevance of DDX3 in the specific and optimal translation of stress-responsive mRNAs and subsequent survival decision during stress. Other than DDX3, nucleic acid receptor DHX9 has been proposed to have an additional role in regulating transcription of IFN-stimulated genes or subsequent processing of these mRNAs[157]. The multifaceted function is likely to reflect the fundamental roles that RNA helicases play in response to stress. Undoubtedly, future studies will explore the involvement and underlying mechanisms of more RNA helicases in stress response and survival regulation.
4. Conclusions and future perspectives
The cellular mechanisms responsible for the cell survival regulation under stress represent a complex and diverse program. Deciphering molecules and pathways involved infine-tuning of cell demise and cyto-protective processes upon stress stimuli is of utmost meaning in biomedicine, since its deregulation is tightly linked to the pathogenesis of numerous diseases, such as pathogen infections, diabetes, neurode-generative diseases and cancer[158]. Actually, recent development clearly demonstrates that our capability to manipulate these endoge-nous survival programs will lead to functional treatment approaches [158].
Notably, the requirements of precise gene regulation during stress response must be achieved at different levels. Considering the impor-tance of RNA processing as critical steps in gene expression, RNA helicases are relevant and potential regulators of cellular stress re-sponses. Indeed, as summarized in this review, these RNA remodeling enzymes have emerged as central players orchestrating the multilayered cellular stress responses. Due to their involvement in lots of regulatory pathways that have direct implications in human health, RNA helicases have gained much attention during the last few years. Mutations in RNA helicases or alterations of their expression levels have been associ-ated with infections, neurological disorders, cancer and aging processes [159,160]. Several RNA helicases are also host factors required for the replication of human pathogenic viruses[46]. Not surprisingly, these en-zymes have been proposed as potential cellular targets for alternative therapies whereas specific inhibitors targeting their catalytic activities have already been developed [161]. According to recent findings outlined here, it is quite evident that RNA helicases are capable of
performing more than one, non-overlapping functions under normal or stress conditions. Nevertheless, there also appear to be a variety of path-ways by which stress-associated RNA helicases can exert their influence. Undeniably, examples of RNA helicases involved in stress responses will continue to expand. Although identification of RNA helicases intimately associated with stress response program has just emerged, numerous fundamental questions remain unanswered. On the mechanistic side, focused structure–function studies are needed to verify interacting part-ners of RNA helicases. In addition, the study of RNA helicases in authentic RNP complexes and their rearrangement activities during stress re-sponse will probably become central theme. With regard to the cellular function of RNA helicases in stress response, focus will undoubtedly be on identification of specific RNA targets, on elucidating pathways by which environmental stress regulates RNA helicase activity or gene ex-pression, on clarifying means by which these proteins are recruited to their sites of action, and on devising mechanisms by which the physio-logical and biochemical functions of RNA helicases are integrated. Given that several RNA helicases are reported to undergo posttranslation-al modifications which provide the opportunity to directly link helicase activity with environmental sensing-signal cascades[162], it will be rele-vant to investigate the functional impact of post-translational modi fica-tions on these events.
In this respect, alterations in post-translational modification of RNA helicases could also influence their localization, their associations with partners and their impact on different cellular processes, adding more complexity. Thus a given RNA helicase may have a pro-survival role in some contexts whereas a death-promoting role in others. This context dependence would obviously have paramount implications for the consideration of RNA helicases as possible biomarkers or therapeutic targets. Therefore, the huge spectrum of RNA helicase functions sug-gests the requirement of more than ever a comprehensive understand-ing of its biology before any medical application. Clearly, there is much research to be performed in this area. In a near future, the fast-growing field of molecular designs and RNA technology should yield new in-sights into the regulation exerted by RNA helicases in all aspects of stress responses and survival program.
Acknowledgements
This work isfinancially supported by the National Science Council, Taiwan, Republic of China [grant numbers NSC100-2320-B-009-007-MY3, NSC101-2320-B-009-001-MY3 and NSC102-2911-I-009-101 (to Y.-H.W.L.); grant numbers NSC101-2917-I-564-022, NSC102-2811-B-038-006 and NSC102-2321-B-038-008 (to J.-W.S.)] and the Ministry of Education, Taiwan, Republic of China for a grant of‘Aiming for the Top University Program’ to the National Chiao Tung University.
References
[1]Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 2010;40:238–52.
[2]Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell 2010;40:253–66.
[3]Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010;40:280–93.
[4]Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell 2010;40:228–37.
[5]Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol 2010;2010:214074.
[6] Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007;35:495–516.
[7]Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011;13:184–90.
[8]He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009;43:67–93.
[9]Bialik S, Zalckvar E, Ber Y, Rubinstein AD, Kimchi A. Systems biology analysis of pro-grammed cell death. Trends Biochem Sci 2010;35:556–64.
[10]Ouyang L, Shi Z, Zhao S, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 2012;45:487–98. [11]Gonzalez-Polo RA, Boya P, Pauleau AL, et al. The apoptosis/autophagy paradox: