Choice of Optimal Shift Parameter for the Intruder State Removal
Techniques in Multireference Perturbation Theory
Shu-Wei Chang and Henryk A. Witek*
Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung University, Hsinchu, Taiwan
*
S Supporting InformationABSTRACT: An extensive critical evaluation of intruder state removal techniques (aka shift techniques) applicable to
multireference perturbation theory (MRPT) shows that the magnitude of the shift parameter σ does not influence the spectroscopic parameters of diatomics to a significant degree, provided that the shift is chosen to be sufficiently large. In such case, typical variation of spectroscopic parameters over a wide range of shift parameters is smaller than 0.005 Å for equilibrium distances, 30 cm−1for harmonic vibrational frequencies, and 0.1 eV for dissociation energies. It is found that large values ofσ not only remove intruder states but they also bring the MRPT energies and properties closer to experimental values. The presented analysis allows us to determine optimal values of the shift parameters to be used in conjunction with various versions of MRPT; these values are recommended to replace the ad hoc values of σ suggested in MRPT manuals in calculations for diatomics. Transferability of the optimal shift parameters to larger molecular systems and to other basis sets than aug-cc-pVTZ is anticipated but remains to be formally established.
1. INTRODUCTION
Multireference perturbation theory (MRPT) is a collection of robust quantum chemical techniques that can be used on a routine basis to calculations on ground and excited states of small and medium size molecules, determination of potential energy surfaces and reaction paths, explication of reaction mechanisms, analysis of open-shell systems, and many other applications which require more than one Slater determinant to describe properly the character of the wave function.1−3 At present, MRPT can be considered as the only feasible and commercially available way of treating systems whose physically realistic description calls for using more than 12 active orbitals in the description of the zeroth-order wave functions, such as transition metal clusters, multicenter organometallic systems, high-lying excited molecular states, etc. MRPT is not only a well-established computational tool; it also constitutes afield of vigorous methodological development aiming, besides others, at enhancing its accuracy,4 finding optimal partitioning schemes5−9leading to better accuracy and better perturbation series convergence,10−14 designing alternative algorithmic schemes,15−23 and eliminating its various unwelcomed draw-backs.24−31
The most serious drawback of MRPT is the existence of intruder states,32which are defined as some of the intermediate states k in the perturbation expansion with zeroth-order energies Ek(0) degenerated (or quasidegenerated) with the
zeroth-order energy Eα(0)of the reference state α. It is easy to
see that such quasidegeneracies result in exceedingly small perturbation energy denominators possibly leading to com-pletely unphysical, very large energy corrections already at low orders of perturbation theory. This problem was first recognized in MRPT by Andersson and Roos in their work on the chromium dimer,33 for which they obtained a discontinuous potential energy surface plagued with local singularities. The problem of intruder states was tackled by
Andersson and Roos by introducing a downward energy shift applied to the zeroth-order energy of the reference state,33 which can be mathematically justified as a repartitioning of the original Hamiltonian into new zeroth-order Hamiltonian and perturbation operators. It is intuitively obvious that such a downward energy shift (i.e., transferring a fraction of the zeroth-order energy of the reference state to the perturbation operator) would remove the existing quasidegeneracies, but it is entirely possible that it would introduce instead other degeneracies or quasidegeneracies. Such behavior was indeed observed. For many systems, a small downward shift (up to 0.1 au) was found to be too small to guarantee physically meaningful characteristics of MRPT energies and wave functions, which could be obtained only with substantially larger repartitionings. This situation is well illustrated by our recent work34on the ground state potential energy surface of the manganese dimer, for which we have determined the zeroth-order energies Eα(0) of the ground state along the
internuclear coordinate and plotted it against the zeroth-order energy spectrum of the singly and doubly excited intermediate states. It turned out that for two out of eight possible classes of excitations, i.e., for the one- and two-electron active→ virtual excitations, Eα(0)is completely embedded in the lower portion of the dense spectrum of the zeroth-order Hamiltonian of the intermediate states k. A downward energy shift allows for bringing the reference stateα below the dense spectrum, but in some situations the corresponding shift magnitude has to be quite substantial.
The downward energy shift technique described above is usually referred to as the level shift technique or as the real shift (RS) technique.33 A viable alternative was developed by Forsberg and Malmqvist,35 who added an imaginary shift to Received: October 6, 2011
Published: September 10, 2012
the zeroth-order energy of the reference state. This resulted in a complex value of Eα(0), which thus became well separated from the purely real zeroth-order spectrum of the intermediate states. Consequently, all the degeneracies and quasidegeneracies were completely removed at the cost of additional imaginary component in the resulting MRPT energy, Eα(0+1+2). This
obstacle was solved by neglecting the imaginary component and retaining only the real part of the MRPT energy as thefinal answer. This approach is usually referred to as the imaginary shift (IS) technique.35Yet another approach, usually referred to as the intruder state avoidance (ISA) technique,36 was developed by Witek and collaborators. It was based on the observation that a modification of Eα(0)affects the contribution
to the perturbation series from all possible intermediate states, while it would be desirable to introduce the modification only for the intruding intermediate states. Following this observa-tion, it was suggested to modify the zeroth-order energies of the intermediate states instead of modifying Eα(0); the corresponding
shift magnitude was made proportional to the intruding power of a given intermediate state computed as the inverse of the zeroth-order energies difference, Eα(0)− Ek(0). It is important to
highlight here that all the intruder state removal shift techniques discussed above are not simply ad hoc corrections to the second-order perturbation energy but constitute well-defined perturbation schemes differing from the original versions of MRPT by slightly different partitioning of the original Hamiltonian into the zeroth-order operator and the perturbation. It was explicitly demonstrated for ISA that the repartitioning does not alter the infinite-order PT solutions, provided that both series, MRPT and ISA-MRPT, are convergent.36
The intruder state removal shift techniques were imple-mented in quantum chemistry packages37−39 and are available for popular variants of MRPT. The complete active space second-order perturbation theory (CASPT2) implementation40 in the MOLPRO package37can be used in conjunction with the RS technique.33 Both RS33and IS35are available as the shift techniques for the CASPT2 implementation41,42 in the MOLCAS package.38 The ISA technique36 can be used in conjunction with the multirefence Møller−Plesset (MRMP) perturbation theory of Hirao43 and the multiconfigurational quasi-degenerate perturbation theory (MCQDPT) of Nakano44 as implemented in the GAMESS quantum chemistry package.39 In most of the recent applications of MRPT to chemical problems, the intruder state removal techniques are used automatically with a small value of the intruder state shift parameter.
It is thus quite surprising how little research effort34,45−47was devoted so far to verifying the accuracy of the shifted MRPT calculations and their dependence on the magnitude of the shift parameterσ. It is generally believed that the magnitude of the shift parameter does not have a strong influence on the computed MRPT results. This conviction, however, does not have very strong scientific grounds.45 Rather opposite, the legitimacy of shift techniques has been recently questioned by showing that the use of MRPT together with intruder state shift techniques for weakly bound species may actually lead to serious conceptual difficulties. Second-order MRPT computa-tional studies46,48performed with RS-CASPT2/MOLPRO, IS-CASPT2/MOLCAS, and ISA-MRMP/GAMESS for the ground and excited states of transition metal dimers (Mn2
and Sc2) showed that their spectroscopic parameters depend on the magnitude of the employed shift parameterσ. Equilibrium
bond lengths, vibrational frequencies, and dissociation energies underwent noticeable changes when the shift parameter was made larger; these changes were particularly pronounced when the studied electronic state had a weakly bound character. The most unexpected finding associated with using the shift techniques in MRPT was probably the prediction of a false ground state for the scandium dimer.46,47,49MRPT calculations performed with the (12o,6e) active space and small values of the shift parameter predicted 3Σu− as the ground state, while
larger values of the shift parameter indicated that the ground state is 5Σ
u
−. Analogous calculations, performed with a larger
active space (18o,6e) and independently with the second- and third-order n-electron valence perturbation theory (NEVPT), were free of the intruder state problem and unequivocally showed that5Σ
u
− is located a few kilocalories per mole lower
than 3Σ u −.50
Note in passing that NEVPT19,20,51 is the only version of the commercially available37 popular variants of MRPT that seems to be completely free of the intruder state problem owing to an enhanced, partially bielectronic definition of the zeroth-order Hamiltonian developed by Dyall,52 while the other methods (CASPT2, MRMP, and MCQDPT) use a multireference generalization of a purely one-electron Møller− Plesset operator to determine the zeroth-order Hamiltonian and the corresponding perturbation operator.
In the present manuscript, we use a statistical approach to analyze in detail the dependence of various intruder-state removal MRPT techniques on the magnitude of the shift parameter. Our work is motivated by the above-mentioned apparent discord between the conceptual problems associated with using small values ofσ and a common approach of MRPT practitioners, who, following manual suggestions, choose σ as small as possible. We aim, in particular, at resolving two important issues: (i) How big can the change be in spectroscopic parameters induced by varying σ? (ii) What is the optimal value ofσ? Both issues require some explanation. It is clear that using infinite shift in MRPT would result in vanishing low-order PT correction energies, leading to exactly the same energies and spectroscopic parameters (for bound states) as in CASSCF. Therefore, the variation in spectroscopic parameters induced by changing σ cannot be too large, as CASSCF usually provides a sensible approximation to MRPT. Intuitively, it is natural to expect that the largest variation should be observed for small values ofσ; for larger values, slow convergence toward CASSCF is anticipated (again, for bound states). As discussed above, using small values ofσ should be avoided, as it leads to serious conceptual problems for weakly bound electronic states. We are destined then to use a rather large value ofσ. We are able to show in the present study that for diatomic molecules larger values ofσ not only remove the intruder states but also bring the shifted MRPT results closer to experimental values. Thisfinding is not difficult to understand. It is a well-known characteristic of second-order perturbation theory that it overestimates the correlation energy; using larger values of σ cancels partially this effect, resulting in better correspondence of the computed spectroscopic parameters to experimental values. Thisfinding gives us an easily quantified base for determination of an optimal value of the shift parameter to be used in conjunction with each of the available shift techniques. The optimal values of shift parameters determined in this fashion should be more robust than the developed ad hoc values ofσ given in MRPT manuals and used every year in numerous MRPT applications,45,53−76 both with respect to the accuracy of the results and with respect to their
credibility. Note, however, that the optimal shift parameters are determined in the current study only for bound states of diatomic molecules in conjunction with a single basis set (aug-cc-pVTZ); transferability of these results to other molecular systems as well as to other basis sets yet remains to be established.
2. COMPUTATIONAL DETAILS
In order to find optimal values of the shift parameters σ, we study potential energy curves of 65 low-lying electronic states of 15 diatomic molecules consisting of thefirst- and second-row elements. The electronic states used in our analysis are listed in Table 1. Each potential curve was determined using five different MRPT approaches: (i) CASPT2 implemented in MOLPRO together with the RS shift technique, (ii) MRMP (or its multistate variant, MCQDPT) implemented in GAMESS together with the ISA shift technique, (iii) CASPT2 implemented in MOLCAS together with the IS shift technique and no IPEA shift, (iv) CASPT2 implemented in MOLCAS together with the IS shift technique and the default IPEA shift of 0.25 hartree,77 and (v) CASPT2 implemented in MOLCAS together with the IS shift technique, no IPEA shift, and the g1 modification of the zeroth-order
Hamiltonian.78For brevity, these approaches will be referred to in the current study as RS-CASPT2/MOLPRO, ISA-MRMP/ GAMESS, IS-CASPT2/MOLCAS, IS-IPEA-CASPT2/MOL-CAS, and IS-g1-CASPT2/MOLCAS, respectively. For every
approach, we used a number of applicable shift parameters: 0.0−1.0 with spacing of 0.1 for RS; 0.0, 0.0001, 0.0005, 0.001, 0.002, 0.005, 0.01, 0.03, 0.05, 0.07, 0.1, 0.2, 0.3, 0.4, and 0.5 for ISA; and 0.0−0.7 with spacing of 0.1 and 0.9−1.5 with spacing of 0.2 for IS. Each potential energy curve was determined by computing more than 150 single point energies for geometries located between 1.2 and 20.0 bohr. The equilibrium distance re
for each potential energy curve was determined by choosing 11 points located symmetrically around the lowest calculated single point energy; the spacing between two adjacent points was selected as 0.01 bohr. These points were subsequently fitted to a fourth-order polynomial using the GRACE program,79 yielding directly the equilibrium distance as one of thefitting constants. The harmonic vibrational frequency ωe was determined in a similar fashion using the quadratic term of thefit. The dissociation energy Defor a given electronic state was determined as the difference between the fitted energy at the equilibrium distance and the computed MRPT energy at large internuclear separation. The spectroscopic parameters determined in this way were compared against the correspond-Table 1. List of Electronic States Used in the Current Study Together with the Compilation of Available Experimental Spectroscopic Parametersa state re ωe De state re ωe De BH X1Σ+ 1.232 2367 3.52 FO X2Π 1.326 1029 1.67 a3Π 1.201 B 2 X3∑g− 1.590 1051 3.10 A1Π 1.219 2251 0.66 C 2 X1∑g+ 1.243 1855 6.35 b3Σ− 1.227 1Δ g(1) CH X2Π 1.120 2859 3.65 1∑ g +(2) a4Σ− 1.085 3145 2.92 a3Π u 1.312 1641 6.26 A2Δ 1.102 2931 2.03 b3∑ g − 1.369 1470 5.56 NH X3Σ− 1.036 3282 3.67 A1Π u 1.318 1608 5.31 a1Δ 1.034 3188 4.50 c3∑ g + 1.230 1962 4.70 b1Σ+ 1.036 3352 4.62 d3Π g 1.266 1788 3.87 A3Π 1.037 3231 2.36 e3Π g 1.535 1107 2.56 OH X2Π 0.970 3738 4.62 C1Π g 1.255 1809 2.11 A2Σ+ 1.012 3179 2.53 N 2 X1∑g+ 1.098 2359 9.91 FH X1Σ+ 0.917 4138 6.11 A3∑ u + 1.287 1461 3.68 BF X1Σ+ 1.263 1402 7.96 B3Π g 1.213 1733 4.90 A1Π 1.308 1324 4.34 W3Δ u 1501 4.87 a3Π 1.304 1265 1.61 B′3∑ u − 1.278 1517 5.26 CN A2Π 1.233 1813 6.55 a′1∑ u − 1.276 1530 6.22 CO X1Σ+ 1.128 2170 11.24 a1Π g 1.220 1694 6.08 A1Π 1.235 1518 3.18 w1Δ u 1.268 1559 5.73 I1Σ− 1.391 1092 3.17 C3Π u 1.149 2047 1.24 D1Δ 1.399 1094 3.07 O 2 X1∑g− 1.208 1580 5.21 a′3Σ+ 1.352 1229 4.32 a1Δ g 1.216 1484 4.23 d3Δ 1.370 1172 3.67 b1∑ g + 1.227 1433 3.58 a3Π 1.206 1743 5.21 c1∑ u − 1.517 794 1.11 e3Σ− 1.384 1118 3.28 A′3Δ u 1.480 850 0.91 5Π(1) A3∑ u + 1.522 799 0.82 NO X2Π 1.151 1904 6.61 F 2 X1∑g+ 1.412 917 1.66 B2Π 1.417 1037 3.30 1∑ g −(1) 2Φ(1) 1Π g(1) a4Π 1017 1.84 1Π u(1) b4Σ− 1206 2.54 3Π u(1) B′2Δ 1.302 1217 1.50 aEquilibrium distances r
ing experimental values taken from the compilation of Herzberg and Huber and available online from the homepage of NIST.80 The experimental values of De were determined from the available D0 constants81 by adding half of the
experimental harmonic frequency and a quarter of the first anharmonicity (if available). The compilation of reference experimental data obtained in this way is given in Table 1. Note that for the 65 selected electronic states, not all the desired experimental values are actually available; we use only 54 equilibrium distances, 55 harmonic vibrational frequencies, and 54 dissociation energies in the statistical analysis of the computed MRPT spectroscopic constants.
The MRPT calculations were performed with the complete active space self-consistent field (CASSCF) reference wave functions using the augmented correlation-consistent polarized valence triple-ζ (aug-cc-pVTZ) basis sets of Dunning.82,83 For all studied molecules, we used full valence active space consisting of the 1s atomic orbital for hydrogen and the 2s and 2p atomic orbitals for the remaining elements. The inner shell molecular orbitals were correlated in the MRPT calculations. The CASSCF optimization for HF, CN, FO, and B2was performed state-specifically, while for other systems, we
applied a state-averaged CASSCF scheme including all explicitly considered here electronic states (see Table 1). All CASSCF calculations were performed with MOLPRO,37which provides a very convenient way of identification of the Λ quantum number of linear molecules via the LQUANT option. The resulting optimized CASSCF molecular orbitals were then transformed into the MOLCAS and GAMESS formats and used in the CASCI calculations for determination of appropriate reference wave functions for the subsequent MRPT calculations. The validity of the orbital transformation process was confirmed by comparing the CASSCF energies from MOLPRO and the CASSCI energies from MOLCAS and GAMESS. The validation was particularly important for the GAMESS program, as the orbital transformation from the spherical harmonics basis to the Cartesian basis required a number of error-prone steps. Using an identical set of molecular orbitals for all three quantum chemistry packages was very important from the computational point of view, assuring that the differences between the MRMP/GAMESS, CASPT2/MOLPRO, and CASPT2/MOLCAS results originate solely from different characteristics of these three flavors of MRPT. The calculations were performed with the MOLPRO (version 2009), MOLCAS (version 7.4), and GAMESS (version 2009) quantum chemistry packages.
3. RESULTS
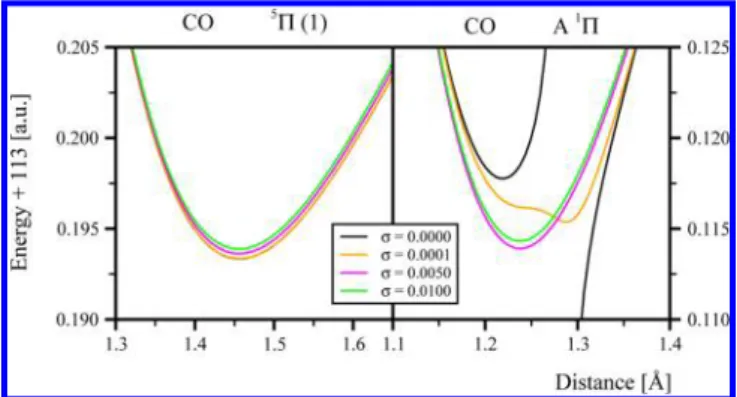
Before giving a statistical analysis of the results, let us shortly illustrate the general influence of the shift techniques on the MRPT results. A typical answer of the computed MRPT potential energy curves to the intruder state removal shift techniques is presented in the left panel of Figure 1. We show there the equilibrium region of the PES of the lowest5Π state
of carbon monoxide computed with the ISA-MRMP technique with four values of theσ parameter: 0, 0.0001, 0.005, and 0.01. It is clear that the original MRMP curve (σ = 0.0) is not affected by intruder states. An application of the intruder state removal technique elevates the curve slightly, to a degree proportional to the shift magnitude. The observed elevation directly corresponds to the decrease in the amount of recovered dynamical electron correlation. However, the answer of the computed PESs to shift techniques can be by far much less
regular. An example of such a behavior is presented in the right panel of Figure 1, which gives the equilibrium region of the ISA-MRMP potential energy curves for the lowest1Π state of
CO again computed using four values of σ.84 The original MRMP curve (σ = 0) shows a discontinuity located at approximately 1.275 Å, with one branch of the potential energy curve diverging toward infinity and the other one, toward negative infinity. An application of a very small shift (σ = 0.0001) removes the discontinuity. However, the shape of the curve still displays a “memory effect” of the removed discontinuity. Clearly, determination of equilibrium distance or vibrational frequency from such a curve would be subject to large numerical errors. An application of a larger shift (σ = 0.005 andσ = 0.01) removes effectively the memory effect and produces potential energy curves displaying only slight dependence on the shift parameter, analogous to that discussed previously for the5Π state of CO. The memory effect behavior
of the1Π curve around equilibrium can be directly linked to the previously discovered prediction of a false ground state (3Σ
u −
instead of5Σu−) of the scandium dimer when too small a value
of the shift parameter was used.
The statistical analysis presented here is based on a large number of single point MRPT calculations performed with various values of the shift parameterσ, which were performed to construct over 3600 potential energy curves for the 65 analyzed here electronic states of diatomic molecules (for details, see Table 1). For each of the curves, we extracted its equilibrium distance re, harmonic vibrational frequencyωe, and
dissociation energy De (details given in section 2). The dependence of the resulting spectroscopic parameters on the magnitude of the shift parameterσ is presented graphically in the Supporting Information in Figures SB1−SB65. As explained in the previous paragraph, it was not possible to extract reliable spectroscopic parameters from all the calculated potential energy curves, particularly due to the lack of convergence in the CASPT2 procedure, PES discontinuities around the equili-brium, or deformed or distorted shape of the resulting PESs. Such situations were highlighted using yellow regions in Figures SB1−SB65. Note that these yellow regions appear only for small values ofσ and that their right border corresponds to the minimal value of the shift parameter allowing for complete removal of the memory effect of the intruding degeneracies or quasidegeneracies.
It is not possible (and probably not desirable) to discuss here all 65 resulting dependence schemes (Figures SB1−SB65). Instead, we present in Figure 2 one of the resulting dependence
Figure 1.Fragment of PES for the lowest5Π (left panel) and1Π (right
panel) states of CO computed using MRMP without (σ = 0) and with (σ = 0.0001, 0.005, and 0.01) the intruder state removal ISA technique.84
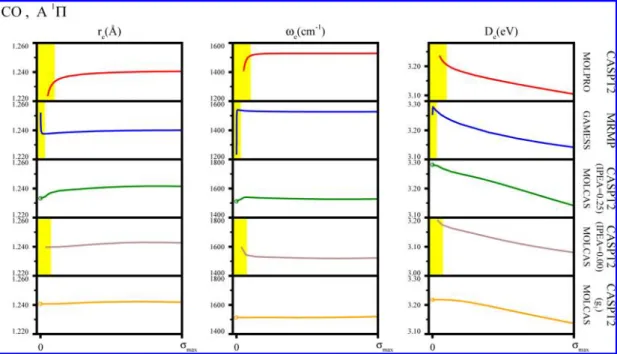
schemes obtained for the lowest1Π state of CO to exemplify its main features. The left panel of Figure 2 shows the dependence of the equilibrium distance on the shift parameterσ, while the middle and right panels show analogous dependence of the harmonic vibrational frequencies and dissociation energies. Five rows in each of the columns correspond to the various five MRPT approaches studied here. Three out of five studied shifted MRPT approaches suffer from numerical instabilities observed as rapid changes of spectroscopic parameters for small values ofσ; the instability regions are depicted in yellow in the plots. An analysis of all 65 sets of spectroscopic parameters shows that the MRPT approach most vulnerable to the intruder state problem is MRMP with yellow regions in 21 graphs. The original CASPT2 methods implemented in MOLPRO and MOLCAS are less vulnerable with 16 and 13 yellow regions, respectively. Using an IPEA shift of 0.25 hartree helps considerably to immunize the CASPT2/MOLCAS method against the intruder state problem, giving the yellow region warnings only in two cases. The most effective in solving the intruder state problem is the g1redefinition of the zeroth-order Hamiltonian, for which no numerical instabilities were detected for small values of the shift parameter. This aspect of our analysis gives a lower bound of the shift parameterσ for each of the intruder state removal shift techniques. The minimal values of the shift parameters that should be used in conjunction with each studied shifted MRPT techniques are 0.3 for RS-CASPT2/MOLPRO, 0.03 for ISA-MRMP/GAMESS, 0.2 for IS-CASPT2/MOLCAS, 0.1 for IS-IPEA-CASPT2/MOLCAS, and 0 for IS-g1-CASPT2/MOLCAS. For future use, we will
refer to these values asσmin.
Further analysis of the graphs presented in Figure 2 shows that outside the yellow regions, reandωechange only slightly,
while De diminishes linearly. An inspection of the remaining
dependence schemes (Figures SB1−SB65) shows that the actual dependence patterns are quite complicated and can be analyzed only using a statistical approach. For each plot, we
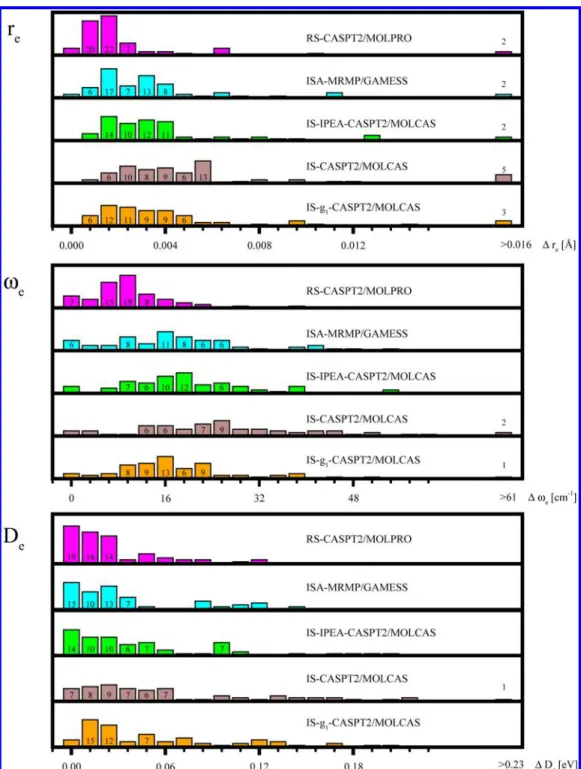
thus determine the difference between the largest and the smallest values of a given spectroscopic constant determined with the shift parameter between σmin and σmax. Histograms made of such values are shown in Figure 3. The most striking observation concerning the plotted histograms is a surprisingly small change in the spectroscopic parameters induced by variation of the shift parameter. The majority of equilibrium distances are varied in that way by less than 0.005 Å. A similar observation can be made for the harmonic vibrational frequencies and dissociation energies: almost all changes in ωeare smaller than 30 cm−1, and almost all changes in Deare
smaller than 0.1 eV. These values are smaller or comparable to the anticipated accuracy of the second-order multireference perturbation theory. These findings suggest that the actual value of the shift parameter σ used in the shifted MRPT calculations does not play a crucial role in the computational process, provided that σ is larger from the minimal allowed valueσmin. Such behavior can be understood from a theoretical
viewpoint quite easily. It signifies that all analyzed σ repartitionings of the original Hamiltonian into a zeroth-order Hamiltonian and a perturbation yield perturbation theories giving similar second-order perturbation correction, provided thatσ is large enough to remove the quasidegeneracies and the memory effects associated with them.
The above discussed mild dependence of the computed potential energy surfaces on the magnitude of the shift parameter can be treated as an advantage of the shifted MRPT approaches. We can use it in a practical manner for choosing an optimal value of the shift parameterσoptyielding best agreement with experimental values. Such a treatment can be compared in its spirit to the spin-component scaled MP2 method introduced by Grimme,85,86 where adjustable scaling factors were used to ensure best agreement with available experimental data. To determine σopt for each of the MRPT approaches studied here, we have compared the computed spectroscopic constant with available experimental data and
Figure 2.Dependence of spectroscopic parameters of the lowest1Π state of CO on the magnitude of the intruder state removal shift parameter σ
computed using various version of multireference perturbation theory. Analogous results for other states are given in the Supporting Information in Figures SB1−SB65. Yellow regions highlight the values of σ with large variance in the spectroscopic parameters. The maximal value of the shift parameter,σmax, is 1.0 for RS-CASPT2/MOLPRO, 0.5 for ISA-MRMP/GAMESS, and 1.5 for IS-CASPT2/MOLCAS.
determined the corresponding overall mean absolute deviations (MAD) of re, ωe, and De for each studied MRPT approach.
Detailed results of such investigation are given in the Supporting Information as Figures SC1−SC18. The computed mean absolute deviations are then used for determination of a combined errorε, which is defined here as
ε= MAD( )r + −ω + D 0.001Å MAD( ) 1cm MAD( ) 0.01eV e e 1 e (1)
where 0.001 Å, 1 cm−1, and 0.01 eV were used as desired accuracy units for the equilibrium frequency, vibrational frequencies, and dissociation energies, respectively. In Figure 4, we present the dependence of the combined errorε on the
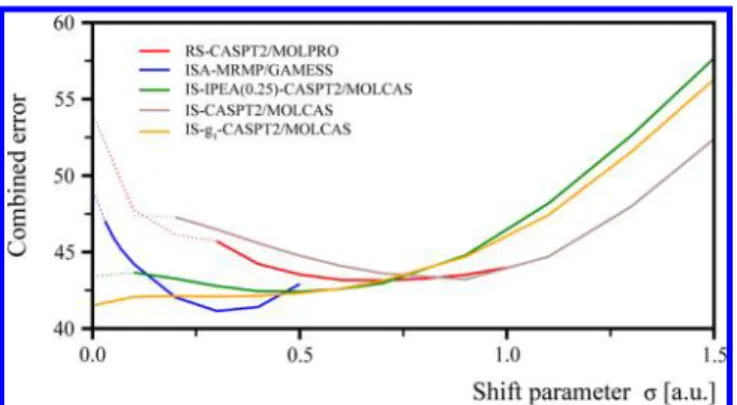
employed shift parameter σ. The optimal values of the shift parameter σopt that minimize the combined error can be
characterized as follows. The ISA-MRMP/GAMESS approach shows a parabola-like dependence ofε on σ with a minimum at approximately 0.3. Similar behavior is observed for the original IS-CASPT2 implementation in MOLCAS (with no IPEA shift), where the optimal value of the shiftσ can be characterized as 0.9, and for the RS-CASPT2/MOLPRO approach with an optimal shift of 0.7. The modified IS-CASPT2 approaches, both with g1 and IPEA = 0.25, show quite flat dependence of the
combined errorε on σ, suggesting that the optimal shift can be chosen quite freely in the range 0.1−0.5. Note that the optimal values of the shift parameter σ determined here are sizably
Figure 3.Distribution of maximal changes in spectroscopic parameters of the 65 studied PESs induced by varying the shift parameter fromσminto
larger than the values suggested in the MRPT manuals and bear certain resemblance to large denominator shifts defined in the context of forced valence orbital degeneracy (FD) partitioning in the variant of MRPT developed by Freed and co-workers.87−100
One should keep in mind that the optimal values given here are determined with the second-order MRPT for three specific spectroscopic parameters of diatomic molecules and corre-spond to a single basis set, aug-cc-pVTZ. Transferability of these optimal parameters to higher orders of perturbation theory, other basis sets, to larger molecular systems, and to other molecular properties (e.g., vertical excitation energies) remains to be established. We anticipate that the observed regularities, i.e., large variability of MRPT molecular properties with the shift for small values of σ, mild dependence of the properties on σ for larger values of the shift, and optimal agreement with experimental values for relatively large values of σ, are general phenomena. We also anticipate that the actual value of the optimal shift parameter will vary slightly with the studied property and with the size of the molecular system and the basis set, which should not constitute a serious practical problem, as we have shown here that the combined error does not change much provided that the shift parameter is chosen to be larger than σmin. We also expect that the values of the optimal shift parameters determined with higher orders of MRPT may be quite different, as can be justified by the following argument. The third-order MRPT is known, in contrast to the second-order MRPT, to underestimate the correlation energy. Therefore, the optimal shift parameter is expected to be as small as possible to avoid additional removal of dynamical correlation. An interesting insight into the variability of the optimal shift parameter can be obtained by replacing the reference data obtained from the experiment by the reference data obtained from analogous multireference configuration interaction (MRCI) calculations; this approach minimizes the effect of the basis set, since both sets of quantities are obtained from calculations using the same set of atomic orbitals. To this end, we have computed the deviation from MRCI of re,ωe, and Decomputed using the RS-CASPT2/
MOLPRO approach; the dependence of the mean absolute deviation of these three spectroscopic parameters on the value of the shift parameterσ is shown in the Supporting Information in Figures SC19−SC21. The optimal value of the shift parameter determined from the analogous plot of the combined error (Figure SC22 in Supporting Information) is 1.2, which is
to be compared with the value of 0.7 obtained with experimental data. Both values are quite large, but the discord between them is quite substantial. We are planning to address the signalized issues in our next publications. We hope that the results and discussion presented here will also motivate other groups to investigate these topics.
4. CONCLUSIONS
The dependence of various versions of MRPT on the magnitude of the intruder state removal shift parameter is studied using potential energy curves of 65 electronic states of 15 diatomic molecules. An analysis of the resulting spectro-scopic parameters (re,ωe, and De) yields a number of important observations:
1. Small values of the employed shift parameterσ in many cases can lead to large numerical errors in the calculated spectroscopic parameters. The errors originate from anomalous shapes of the resulting potential energy curves due to the memory effect of the discontinuities caused by intruder states. Such artifacts can be avoided if a given intruder state removal shift technique is used with sufficiently large shift parameter. The minimal values of the shift parameterσminrequired for such an effect are 0.3
for RS-CASPT2/MOLPRO, 0.03 for ISA-MRMP/ GAMESS, 0.2 for CASPT2/MOLCAS, 0.1 for IS-IPEA-CASPT2/MOLCAS, and 0.0 for IS-g1-CASPT2/
MOLCAS.
2. If the employed shift parameterσ is larger than σmin, the
resulting spectroscopic parameters depend only weakly on the actual magnitude of the shift parameter. We have found that forσ in the range [σmin,σmax], whereσmax is 1.0 for the real shift technique, 0.5 for the intruder state avoidance technique, and 1.5 for the imaginary shift technique, typical variation of spectroscopic constants is smaller than 0.005 Å for equilibrium distances, 30 cm−1 for harmonic vibrational frequencies, and 0.1 eV for dissociation energies.
3. The best agreement with experimental results can be achieved if the shift parameter takes on a specific value σopt. Statistical analysis of the data and a comparison with
available experimental data allowed us to determine the optimal values of σopt, which are 0.7 for RS-CASPT2/
MOLPRO, 0.3 for ISA-MRMP/GAMESS, and 0.9 for IS-CASPT2/MOLCAS. For the IS-IPEA(0.25)-CASPT2/ MOLCAS and IS-g1-CASPT2/MOLCAS approaches, the
optimal shift can be chosen quite freely in the range 0.1− 0.5. The optimized values should be a sensible replacement for the ad hoc values of 0.1, 0.2, and 0.3 for RS and IS and 0.02 for ISA quoted in the MOLPRO, MOLCAS, and GAMESS manuals. The presented optimal values were determined for standard chemical molecules consisting of elements from the first and second rows of the periodic table. Therefore,σoptgiven
here should be used in calculations for standard chemical diatomic structures consisting of such elements in conjunction with the aug-cc-pVTZ basis set. Note that the transferability to other systems, e.g, to transition metals, to large organic molecules, or to heavy metals, requires additional checks and verifications of the dependence of the calculated results on the magnitude of the employed shift parameter. We are planning to investigate these issues in a forthcoming publication.
Figure 4.Dependence of the combined error ε (see eq 1) on the magnitude of the shift parameter σ. Minima of the plotted curves correspond to the choice of the optimal shift parameterσoptfor each
4. It is found that the accuracy of ISA-MRMP/GAMESS, RS-CASPT2/MOLPRO, CASPT2/MOLCAS, IS-IPEA(0.25)-CASPT2/MOLCAS, and IS-g1-CASPT2/
MOLCAS used with the optimal shift parameter is comparable.
■
ASSOCIATED CONTENT*
S Supporting InformationDetailed graphical analysis of the dependence of the resulting spectroscopic parameters on the magnitude of the shift parameterσ for all 65 studied electronic states of 15 diatomic molecules (Figures SB1−SB65) and statistical analysis of their mean absolute deviations with respect to experimental results (Figures SC1−SC18) and with respect to MRCI (Figures SC19−SC22). This material is available free of charge via the Internet at http://pubs.acs.org.
■
AUTHOR INFORMATIONCorresponding Author
*E-mail: hwitek@mail.nctu.edu.tw. Phone: +886 (3)5712121 ext. 56583. Fax: +886 (3)5723764.
Notes
The authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSNational Science Council (grant NSC99-2113-M-009-011-MY3) and Ministry of Education of Taiwan (MOE-ATU project) are acknowledged forfinancial support. We are grateful to the National Center for High-Performance Computing for computer time and facilities.
■
REFERENCES(1) Roos, B. O.; Andersson, K.; Fülscher, M. P.; Malmqvist, P.-Å.; Serrano-Andrés, L.; Pierloot, K.; Merchán, M. In Multiconfigurational Perturbation Theory: Applications in Electronic Spectroscopy; Wiley: New York, 2007; pp 219−331.
(2) Nakano, H.; Hirao, K. Bull. Korean Chem. Soc. 2003, 24, 812. (3) Nakano, H.; Yamanishi, M.; Hirao, K. Trends Chem. Phys. 1997, 6, 167−214.
(4) Szabados, A.; Nagy, P. J. Phys. Chem. A 2011, 115, 523. (5) Witek, H. A.; Nakano, H.; Hirao, K. J. Chem. Phys. 2003, 118, 8197.
(6) Witek, H. A.; Nakano, H.; Hirao, K. J. Comput. Chem. 2003, 24, 1390.
(7) Rosta, E.; Surjan, P. R. J. Chem. Phys. 2001, 116, 878. (8) Chen, F. W. J. Chem. Theory Comput. 2009, 5, 931.
(9) Surjan, P. R.; Rolik, Z.; Szabados, A.; Kohalmi, D. Ann. Phys. 2004, 13, 223.
(10) Surjan, P. R.; Szabados, A. Int. J. Quantum Chem. 2002, 90, 20. (11) Alexandrov, V. I.; Zaitsevskii, A. V.; Dementev, A. I. Chem. Phys. Lett. 1994, 218, 206−211.
(12) Herman, M. S.; Hagedorn, G. A. Int. J. Quantum Chem. 2009, 109, 210.
(13) Fink, R. F. Chem. Phys. 2009, 356, 39.
(14) Surjan, P. R.; Szabados, A. Collect. Czech. Chem. Commun. 2004, 69, 105.
(15) Finley, J. P. J. Chem. Phys. 1998, 108, 1081. (16) Finley, J. P. J. Chem. Phys. 1998, 109, 7725.
(17) Choe, Y.-K.; Finley, J. P.; Nakano, H.; Hirao, K. J. Chem. Phys. 2000, 113, 7773.
(18) Finley, J. P.; Witek, H. A. J. Chem. Phys. 2000, 112, 3958. (19) Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. J. Chem. Phys. 2001, 114, 10252.
(20) Angeli, C.; Bories, B.; Cavallini, A.; Cimiraglia, R. J. Chem. Phys. 2006, 124, 054108.
(21) Granovsky, A. A. J. Chem. Phys. 2011, 134, 214113.
(22) Kobayashi, M.; Szabados, A.; Nakai, H.; Surjan, P. R. J. Chem. Theory Comput. 2010, 6, 2024.
(23) Khait, Y. G.; Jiang, W.; Hoffmann, M. R. Int. J. Quantum Chem. 2009, 109, 1855.
(24) Khait, Y. G.; Song, J.; Hoffmann, M. R. J. Chem. Phys. 1998, 108, 8317.
(25) Khait, Y. G.; Song, J.; Hoffmann, M. R. J. Chem. Phys. 2002, 117, 4133.
(26) Robinson, D.; McDouall, J. J. W. J. Phys. Chem. A 2007, 111, 9815.
(27) Robinson, D.; McDouall, J. J. W. J. Chem. Theory Comput. 2007, 3, 1306.
(28) Robinson, D.; McDouall, J. J. W. Mol. Phys. 2006, 104, 681. (29) Nicolaides, C. A. Int. J. Quantum Chem. 2005, 102, 250. (30) Szabados, A.; Rolik, Z.; Toth, G.; Surjan, P. R. J. Chem. Phys. 2005, 122, 114104.
(31) Roskop, L.; Gordon, M. S. J. Chem. Phys. 2011, 135, 044101. (32) Choe, Y.-K.; Witek, H. A.; Finley, J. P.; Hirao, K. J. Chem. Phys. 2001, 114, 3913.
(33) Roos, B. O.; Andersson, K. Chem. Phys. Lett. 1995, 245, 215. (34) Camacho, C.; Witek, H. A.; Yamamoto, S. J. Comput. Chem. 2009, 30, 468.
(35) Forsberg, N.; Malmqvist, P. Å. Chem. Phys. Lett. 1997, 274, 196. (36) Witek, H. A.; Choe, Y.-K.; Finley, J. P.; Hirao, K. J. Comput. Chem. 2002, 23, 957.
(37) Werner, H. J.; Knowles, P. J.; Knizia, G.; Manby, F. R.; Schütz, M.; Celani, P.; Korona, T.; Lindh, R.; Mitrushenkov, A.; Rauhut, G.; Shamasundar, K. R.; Adler, T. B.; Amos, R. D.; Bernhardsson, A.; Berning, A.; Cooper, D. L.; Deegan, M. J. O.; Dobbyn, A. J.; Eckert, F.; Goll, E.; Hampel, C.; Hesselmann, A.; Hetzer, G.; Hrenar, T.; Jansen, G.; Köppl, C.; Liu, Y.; Lloyd, A. W.; Mata, R. A.; May, A. J.; McNicholas, S. J.; Meyer, W.; Mura, M. E.; Nicklass, A.; O’Neill, D. P.; Palmieri, P.; Pflüger, K.; Pitzer, R.; Reiher, M.; Shiozaki, T.; Stoll, H.; Stone, A. J.; Tarroni, R.; Thorsteinsson, T.; Wang, M.; Wolf, A. MOLPRO, version 2010.1; Cardiff University: Cardiff, U. K.; Universität Stuttgart: Stuttgart, Germany, 2010. http://www.molpro. net (accessed Sept. 2012).
(38) Aquilante, F.; De Vico, L.; Ferré, N.; Ghigo, G.; Malmqvist, P. Å.; Neogrády, P.; Pedersen, T. B.; Piton̆ák, M.; Reiher, M.; Roos, B. O.; Serrano-Andrés, L.; Urban, M.; Veryazov, V.; Lindh, R. J. Comput. Chem. 2010, 31, 224.
(39) Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.; Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, N.; Nguyen, K. A.; Su, S.; Windus, T. L.; Dupuis, M.; Montgomery, J. A., Jr. J. Comput. Chem. 1993, 14, 1347.
(40) Celani, P.; Werner, H.-J. J. Chem. Phys. 2000, 112, 5546. (41) Andersson, K.; Malmqvist, P. Å.; Roos, B. O.; Sadlej, A. J.; Wolinski, K. J. Phys. Chem. 1990, 94, 5483.
(42) Andersson, K.; Malmqvist, P. Å.; Roos, B. O. J. Chem. Phys. 1992, 96, 1218.
(43) Hirao, K. Chem. Phys. Lett. 1992, 196, 397. (44) Nakano, H. J. Chem. Phys. 1993, 99, 7983.
(45) Roos, B. O.; Andersson, K.; Fulscher, M. P.; Serrano-Andrés, L.; Pierloot, K.; Merchán, M.; Molina, V. J. Mol. Struct. (THEOCHEM) 1996, 388, 257−276.
(46) Camacho, C.; Cimiraglia, R.; Witek, H. A. Phys. Chem. Chem. Phys. 2010, 12, 5058.
(47) Camacho, C.; Cimiraglia, R.; Witek, H. A. Phys. Chem. Chem. Phys. 2011, 13, 7232.
(48) Camacho, C.; Yamamoto, S.; Witek, H. A. Phys. Chem. Chem. Phys. 2008, 10, 5128.
(49) Soto, J.; Avila, F.; Otero, J. C.; Arenas, J. F. Phys. Chem. Chem. Phys. 2011, 13, 7230.
(50) Camacho, C.; Witek, H. A.; Cimiraglia, R. J. Chem. Phys. 2010, 132, 244306.
(51) Angeli, C.; Borini, S.; Cestari, M.; Cimiraglia, R. J. Chem. Phys. 2004, 121, 4043.
(53) Chen, H.; Li, S. J. Chem. Phys. 2006, 124, 154315.
(54) Pierloot, K.; van Besien, E. J. Chem. Phys. 2005, 123, 204309. (55) de Graaf, C.; Hozoi, L.; Broer, R. J. Chem. Phys. 2004, 120, 961. (56) Pierloot, K.; Delabie, A.; Groothaert, M. H.; Schoonheydt, R. A. Phys. Chem. Chem. Phys. 2001, 3, 2174.
(57) Rubio, M.; Roos, B. O.; Serrano-Andrés, L.; Merchán, M. J. Chem. Phys. 1999, 110, 7202.
(58) Yu, X. F.; Yamazaki, S.; Taketsugu, T. J. Chem. Theory Comput. 2011, 7, 1006.
(59) Assmann, M.; Perez-Hernandez, G.; Gonzalez, L. J. Phys. Chem. A 2010, 114, 9342.
(60) Bokareva, O. S.; Bataev, V. A.; Godunov, I. A. J. Mol. Struct. (THEOCHEM) 2009, 913, 254.
(61) Doyle, R. J.; Campo, R. D.; Taylor, P. R.; Mackenzie, S. R. J. Chem. Phys. 2004, 121, 835.
(62) Pierloot, K.; Vancoillie, S. J. Chem. Phys. 2006, 125, 124303. (63) Oliver, T. A. A.; Taylor, P. R.; Doyle, R. J.; Mackenzie, S. R. J. Chem. Phys. 2007, 127, 024301.
(64) Olaso-González, G.; Roca-Sanjuán, D.; Serrano-Andrés, L.; Merchán, M. J. Chem. Phys. 2006, 125, 231102.
(65) Zou, W.; Liu, W. J. Chem. Phys. 2006, 124, 154312.
(66) de Oliveira-Filho, A. G. S.; Aoto, Y. A.; Ornellas, F. R. J. Chem. Phys. 2011, 135, 044308.
(67) Pierloot, K.; Zhao, H.; Vancoillie, S. Inorg. Chem. 2010, 49, 10316.
(68) Gomez-Jimenez, M. D.; Pou-Amerigo, R.; Orti, E. J. Chem. Phys. 2009, 131, 244105.
(69) Fedorov, D. G.; Nakajima, T.; Hirao, K. J. Chem. Phys. 2003, 118, 4970.
(70) Iuchi, S.; Morita, A.; Kato, S. J. Chem. Phys. 2004, 121, 8446. (71) Patchkovskii, S. Phys. Chem. Chem. Phys. 2006, 8, 926. (72) Sliznev, V. V.; Vogt, N.; Vogt, J. J. Mol. Struct. 2006, 780−781, 247.
(73) Gopakumar, G.; Lievens, P.; Nguyen, M. T. J. Chem. Phys. 2006, 124, 214312.
(74) Shirai, S.; Iwata, S.; Tani, T.; Inagaki, S. J. Phys. Chem. A 2011, 115, 7687.
(75) Sliznev, V. V.; Belova, N. V.; Girichev, G. V. J. Struct. Chem. 2010, 51, 622.
(76) Zeng, T.; Fedorov, D. G.; Klobukowski, M. J. Chem. Phys. 2009, 131, 124109.
(77) Ghigo, G.; Roos, B. O.; Malmqvist, P. Å. Chem. Phys. Lett. 2004, 396, 142.
(78) Andersson, K. Theor. Chem. Acc. 1995, 91, 31.
(79) Grace. http://plasma-gate.weizmann.ac.il/Grace (accessed Sept. 2012).
(80) Huber, K. P.; Herzberg, G. Constants of Diatomic Molecules (data prepared by Gallagher, J. W.; Johnson, R. D., III). In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P. J., Mallard, W. G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD. http://webbook.nist.gov (ac-cessed March 15, 2011).
(81) Darwent, B. Bond Dissociation Energies in Simple Molecules; U.S. National Bureau of Standards: Washington, DC, 1970.
(82) Dunning, T., Jr. J. Chem. Phys. 1989, 90, 1007.
(83) Kendall, R. A.; Dunning, T. H., Jr.; Harrison, R. J. J. Chem. Phys. 1992, 96, 6796.
(84) Note that the strong intruder state around the equilibrium geometry of the lowest singletΠ state of carbon monoxide is not a particular feature of MRMP. Analogous calculations performed with RS-CASPT2/MOLPRO and IS-CASPT2/MOLCAS show very similar behavior to that discussed here (for details, see Figure SA1 in the Supporting Information).
(85) Grimme, S. J. Chem. Phys. 2003, 118, 9095. (86) Fink, R. F. J. Chem. Phys. 2010, 133, 174113.
(87) Sun, H.; Sheppard, M. G.; Freed, K. F. J. Chem. Phys. 1981, 74, 6842.
(88) Takada, T.; Sheppard, M. G. J. Chem. Phys. 1983, 79, 325. (89) Lee, Y. S.; Freed, K. F. J. Chem. Phys. 1982, 77, 1984.
(90) Wang, X.-C.; Freed, K. F. J. Chem. Phys. 1987, 86, 2899. (91) Wang, X.-C.; Freed, K. F. J. Chem. Phys. 1989, 91, 3002. (92) Wang, X.-C.; Freed, K. F. J. Chem. Phys. 1989, 91, 1142. (93) Graham, R. L.; Freed, K. F. J. Chem. Phys. 1992, 96, 1304. (94) Stevens, J.; Graham, R. L.; Freed, K. F. J. Chem. Phys. 1994, 101, 4832.
(95) Martin, C. H.; Graham, R. L.; Freed, K. F. J. Chem. Phys. 1993, 99, 7833.
(96) Finley, J. P.; Freed, K. F. J. Chem. Phys. 1995, 102, 1306. (97) Finley, J. P.; Chaudhuri, R. K.; Freed, K. F. J. Chem. Phys. 1995, 103, 4990.
(98) Chaudhuri, R. K.; Finley, J. P.; Freed, K. F. J. Chem. Phys. 1997, 106, 4067.
(99) Finley, J. P.; Chaudhuri, R. K.; Freed, K. F. Phys. Rev. A 1996, 54, 343.