MJ-29 Inhibits Tubulin Polymerization, Induces Mitotic Arrest, and
Triggers Apoptosis via Cyclin-Dependent Kinase 1-Mediated
Bcl-2 Phosphorylation in Human Leukemia U937 Cells
□
SJai-Sing Yang, Mann-Jen Hour, Wen-Wen Huang, Kuei-Li Lin, Sheng-Chu Kuo, and
Jing-Gung Chung
Department of Pharmacology (J.-S.Y.), School of Pharmacy (M.-J.H.), Department of Biological Science and Technology (W.-W.H., J.-G.C.), Graduate Institute of Pharmaceutical Chemistry (S.-C.K.), China Medical University, Taichung, Taiwan; Department of Radiation Oncology, Chi Mei Medical Center, Tainan, Taiwan (K.-L.L.); and Department of Biotechnology, Asia University, Wufeng, Taichung, Taiwan (J.-G.C.)
Received December 31, 2009; accepted May 11, 2010
ABSTRACT
We investigated the signaling pathways associated with microtubule interaction and apoptosis in U937 cells in vitro and in the U937 xenograft model in vivo by using 6-pyrrolidinyl-2-(2-hydroxyphenyl)-4-quinazolinone (MJ-29). MJ-29 induced growth inhibition and cell death of leukemia cell lines (U937, HL-60, K562, and KG-1) in a dose-and time-dependent manner but did not obviously impair the viability of normal cells (peripheral blood mononuclear cells and human um-bilical vein endothelial cells). MJ-29 interacted with␣- and -tubulin, inhibited tubulin polymerization both in vitro and in vivo, and dis-rupted microtubule organization. MJ-29 caused mitotic arrest by activating cyclin-dependent kinase 1 (CDK1)/cyclin B complex activ-ity. MJ-29-induced growth inhibition and activation of CDK1 activity were significantly attenuated by roscovitine (CDK inhibitor) and CDK1 small interfering RNA (siRNA). Furthermore, MJ-29-induced Bcl-2
phosphorylation was also significantly attenuated by CDK1 siRNA. MJ-29 caused an increase in the protein levels of cytosolic cyto-chrome c, apoptotic protease-activating factor-1, procaspase-9, and apoptosis-inducing factor. MJ-29-promoted activation of caspase-9 and caspase-3 during apoptosis was significantly attenuated by caspase-9 and caspase-3 inhibitors. It is noteworthy that in BALB/ cnu/numice bearing U937 xenograft tumors MJ-29 inhibited tumor
growth in vivo. The terminal deoxynucleotidyl transferase-mediated d-UTP nick end-labeling-positive apoptotic cells of tumor sections significantly increased in MJ-29-treated mice compared with the control group. In conclusion, our results suggest that MJ-29 induces mitotic arrest and apoptosis in U937 cells via CDK1-mediated Bcl-2 phosphorylation and inhibits the in vivo tumor growth of U937 xeno-graft mice.
Leukemia is one of the most common hematologic malig-nancies in humans. Approximately 3.8 of every 100,000 peo-ple die each year of leukemia, and it is the 12th most common malignancy in Taiwan based on a report from the Depart-ment of Health, Executive Yuan of Taiwan in 2008
(Depart-ment of Health, Taiwan, http://www.doh.gov.tw/EN2006/ index_EN.aspx). In the clinical therapy of leukemia patients, bone marrow transplant, radiotherapy, and chemotherapy are applied (Nau and Lewis, 2008; Fotoohi et al., 2009). Microtubule-targeting agents (MTAs) are primarily used, and they are the most effective drugs in leukemia treatment (Perez, 2009). However, clinically used MTAs remain highly toxic to normal tissues (Tallman, 1996; Sornsuvit et al., 2008; Itzykson et al., 2009; Sitaresmi et al., 2009). Therefore, dis-covering a new antileukemia agent that is more effective and less toxic for leukemia patients is necessary.
The anticancer activities of MTAs, which can be classically subdivided into microtubule-stabilizing (e.g., taxanes, epothi-This work was supported by the National Science Council of Taiwan [Grants
NSC 94-2320-B-264-002, NSC94-2320-B-039-013] and China Medical Univer-sity, Taichung, Taiwan [Grant CMU98-S-12].
S.-C.K. and J.-G.C. contributed equally to this work.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.109.165415.
□S The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
ABBREVIATIONS: MJ-29, 6-pyrrolidinyl-2-(2-hydroxyphenyl)-4-quinazolinone; AIF, apoptosis-inducing factor; Apaf-1, apoptotic protease-acti-vating factor-1; CDK, cyclin-dependent kinase; CAK, CDK-actiprotease-acti-vating kinase; DCFH-DA, 2⬘-7⬘-dichlorfluorescein-diacetate; DiOC6, 3,3 ⬘-dihexyl-oxacarbocyanine iodide; DMSO, dimethyl sulfoxide; ECL, enzyme chemiluminescence; FADD, Fas-associated protein with death domain; FasL, Fas ligand; HRP, horseradish peroxidase; MTA, microtubule-targeting agent; NAC, N-acetyl-cysteine; PBS, phosphate-buffered saline; PI, propidium iodide; PMSF, phenylmethane sulfonyl fluoride; HUVEC, human umbilical vein endothelial cell; PBMC, peripheral blood mononuclear cell; TNF, tumor necrosis factor; TNF-R1, TNF receptor 1; TRAIL-R1, TNF-related apoptosis-inducing ligand receptor 1; TRAIL-R2, TNF-related apoptosis-inducing ligand receptor 2; TUNEL, terminal deoxynucleotidyl transferase-mediated d-UTP nick end-labeling; siRNA, small interfering RNA; RT-PCR, reverse transcription-polymerase chain reaction; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; DTT, dithiothreitol; ANOVA, analysis of variance;⌬⌿m, mitochondrial membrane potential; ROS, reactive oxygen species.
THEJOURNAL OFPHARMACOLOGY ANDEXPERIMENTALTHERAPEUTICS Vol. 334, No. 2 Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics 165415/3607556
JPET 334:477–488, 2010 Printed in U.S.A.
lones, and discodermolide) and microtubule-depolymerizing agents (e.g., vinca alkaloids), were demonstrated many years ago in in vitro and in vivo studies (Perez, 2009). MTAs act as inhibitors in the mitotic phase because they might disrupt G2–M transition and induce cell-cycle arrest and apoptosis in
tumor cells. Cyclin-dependent protein kinase 1 (CDK1; p34cdc2) and cyclin B play regulatory key roles in the G
2–M
transition (Allan and Clarke, 2007). The CDK1/cyclin B pro-gression from G2 to the M phase can be affected by the
phosphorylation of CDK1. The phosphorylation of Thr161 by Cdk-activating kinase (CAK; a heterodimer of cyclin H and Cdk7) is strictly required for CDK1 to be activated (Kaldis, 1999; Lolli and Johnson, 2005). CDK1 plays a proapoptotic role rather than an antiapoptotic function in the mechanism of anticancer drugs. An increase in Cdk1 activity has been found in numerous apoptotic conditions. MTAs, such as pac-litaxel and vinca alkaloids, are able to increase CDK1 activ-ity and induce apoptosis in leukemia cells (Ibrado et al., 1998). Mitochondria play a central role in the signaling path-way, and CDK1 can trigger mitochondrial membrane perme-abilization by targeting Bcl-2 family proteins and subse-quently inducing cell apoptosis (Debatin et al., 2002). Cancer cells exposed to MTAs can stimulate Bcl-2 phosphorylation and lost antiapoptotic function. CDK1 can phosphorylate Bcl-2 on Ser70 and suppress its antiapoptotic function (Wang et al., 1999).
Several plants containing alkaloids with 4-quinazolinone nuclei have been reported in antimalarial, anti-inflamma-tory, antibacterial, and anticancer activities (Hour et al., 2007). In recent years, we have designed and synthesized a series of 2-phenyl 6-pyrrolidinyl-4-quinazolinone derivatives as new antimitotic agents. We found that many of these compounds exhibited potent cytotoxicity against leukemia cell lines. 6-Pyrrolidinyl-2-(2-hydroxyphenyl)-4-quinazolin-one) (MJ-29) (Supplementary Fig. 1A) is the most potent compound against leukemia cells (Hour et al., 2007). How-ever, the cytotoxic effects of MJ-29 on leukemia cells and normal cells or the molecular mechanisms underlying its anticancer activity have not been revealed. In our pilot study, we observed significant growth of inhibition activity of MJ-29 on leukemia cells. We also demonstrated that MJ-29 inhibits the polymerization of microtubules and induces mitotic ar-rest and apoptosis in U937 cells. Thus, we suggest that MJ-29 induces mitotic arrest and apoptosis through CDK1-mediated Bcl-2 phosphorylation.
Materials and Methods
Reagents and Antibodies. Propidium iodide (PI), RNase A, Tri-ton X-100, proteinase K, and N-acetyl-cysteine (NAC) were pur-chased from Sigma-Aldrich (St. Louis, MO) and dissolved in H2O. z-Leu-Glu-His-Asp-fluoromethyl ketone (caspase-9 inhibitor), z-Ile-Glu-Thr-Asp-fluoromethyl ketone (caspase-8 inhibitor), and z-Asp-Met-Gln-Asp-fluoromethyl ketone (caspase-3 inhibitor) were pur-chased from R&D Systems (Minneapolis, MN) and dissolved in DMSO. Roscovitine (CDK1 inhibitor) was purchased from Calbio-chem (La Jolla, CA) and dissolved in DMSO. Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA). Polyclonal antibodies specific for phospho-CDK1 (Thr161), phospho-Bcl-2 (Ser70), phos-pho-Histon H3, Apaf-1, caspase-9, and caspase-3 were obtained from Cell Signaling Technology (Danvers, MA), and monoclonal antibod-ies specific for-tubulin, cyclin B, CDK1, GAPDH, Bcl-2, Bax, cyclin A, Cdc25c, Weel-1, CDK7, cytochrome c, FADD, TNF-R1, TRAIL-R1
(DR4), TRAIL-R2 (DR5), and -tubulin-fluorescein isothiocyarate, and all peroxidase-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Monoclonal antibodies specific for AIF, complex IV, Fas/CD95, and FasL were purchased from Abcam Inc. (Cambridge, MA). Enhanced chemilumi-nescence (ECL), a Western blot detection reagent, was purchased from Pierce Chemical (Rockford, IL). MJ-29 was designed and syn-thesized by M.-J.H. and S.-C.K.
Cell Lines. The leukemia cell lines, HL-60 (human promyelo-cytic leukemia), U937 (human lymphoma cancer cell), K562 (hu-man chronic myelogenous leukemia), KG-1 (hu(hu-man acute myelog-enous leukemia), and human umbilical vein endothelial cells (HUVECs) were purchased from the Culture Collection and Re-search Center, Food Industry ReRe-search and Development Insti-tute (Hsinchu, Taiwan) and were originally from the American Type Culture Collection (Manassas, VA). Cells were cultured in RPMI medium 1640 (Invitrogen) and supplemented with 10% heat-inactivated fetal calf serum (HyClone Laboratories, Logan, UT), 100 units/ml penicillin, 100g/ml streptomycin, and 2 mM
L-glutamine at 37°C in a 5% CO2humidified incubator. The pe-ripheral blood mononuclear cells (PBMCs) were isolated from the heparinized whole blood of healthy volunteers by using Ficoll-Paque Plus (GE Healthcare, Little Chalfont, Buckinghamshire, UK). HUVECs were passaged at preconfluent densities by using a solution containing 0.05% trypsin and 0.5 mM EDTA (Invitrogen). Cell Viability Assay. The cell viabilities of six cell lines after exposure to various concentrations of MJ-29 were measured by using a PI exclusion method. Cells (2.5⫻ 105/well) were seeded into each well of a 24-well plate and incubated with 0, 0.5, 1, 5, and 10M MJ-29 for 24, 48, and 72 h. For incubation with inhibitors, cells were seeded into 24-well plates and pretreated with NAC (10 mM), rosco-vitine (20M), caspase-3 inhibitor (z-Asp-Met-Gln-Asp-fluoromethyl ketone), caspase-9 inhibitor (z-Leu-Glu-His-Asp-fluoromethyl tone), and caspase-8 inhibitor (z-Ile-Glu-Thr-Asp-fluoromethyl ke-tone) for 1 h, followed by treatment with or without 1M MJ-29. Cells were harvested, washed, resuspended with phosphate-buffered saline (PBS) containing 4g/ml PI, and then analyzed with a flow cytometer (FACSCalibur; BD Biosciences, San Jose, CA) equipped with a laser at 488-nm wavelength. The percentage of cell viability was calculated as a ratio of the number of drug-treated cells to that of 0.1% DMSO vehicle-control cells. Viability assays were performed in triplicate for three independent experiments. The concentration of MJ-29 inhibiting 50% of cells (IC50) was calculated by using the software Dose-Effect Analysis with Microcomputers of SPSS soft-ware (version 13.5 for Windows; SPSS Inc., Chicago, IL) (Yang et al., 2004; Ji et al., 2009).
Flow Cytometric Analysis for Apoptosis by TUNEL Assay. TUNEL staining was performed according to the manufacturer’s protocols (in situ cell death detection kit; Roche Diagnostics, Mann-heim, Germany). U937 cells, PBMCs, and HUVECs (1⫻ 106/well) were individually plated into six-well plates and exposed to 1M MJ-29, paclitaxel, and vincristine for 24 h. After treatment, cells were collected, fixed in 70% ethanol overnight, washed in PBS twice, and incubated in the dark for 30 min at 37°C in 100l of terminal deoxynucleotidyl transferase-containing solution. After TUNEL staining, all samples were washed once and resuspended in 0.5 ml of PBS containing PI (10g/ml) and DNase free-RNase A (200 g/ml). TUNEL-positive cells were analyzed by flow cytometry. The median fluorescence intensity was quantified with CellQuest software (BD Biosciences). TUNEL assays were performed in triplicate for three independent experiments (Chung et al., 2007).
Flow Cytometric Analysis for DNA Content and Apoptosis. U937 cells (2.5⫻ 105/well) were seeded into each well of 24-well plates, and cells were incubated with 1M MJ-29 for 0, 4, 8, and 12 h. Then cells were collected, fixed in 70% ethanol overnight, washed in PBS once, and resuspended in 40l of 192 mM Na2HPO4, 4 mM citric acid, pH 7.8 at 25°C for 30 min. The cells were stained with 0.5 ml of PBS containing 1 mg/ml RNase and 10 g/ml PI,
incubated for 30 min in the dark at 25°C, and then analyzed by flow cytometry. The DNA content and apoptotic cells were analyzed by using the ModFit DNA analysis program (Verity Software House, Topsham, ME). Cell cycle analysis was performed in triplicate for three independent experiments (Chen et al., 2009).
Molecular Modeling of the MJ-29 Protein Complex. The crystal structure of the tubulin-colchicine-stathmin-like domain complex (Protein Data Bank code 1SA0) was retrieved from the Protein Data Bank (http://www.rcsb.org/pdb). Automated docking was then carried out. LigandFit within the software package Discov-ery Studio 2.1 (Accelrys, San Diego, CA) was used to evaluate and predict the in silico binding free energy of the inhibitors within the macromolecules. First, water molecules and the phosphate group of the receptor molecule were removed, hydrogen was added under a condition of pH 7.4, and the alternative conformations were cor-rected. The Protein Data Bank files were energy-minimized by 500 iterations of the steepest descent and 500 iterations of conjugate gradient optimization and subjected to molecular dynamics simula-tions at 300 K. The backbone was initially condensed, and then it was set free. A binding pocket of the native colchicine ligand was selected as the binding site for the study. After typing the receptor model with the CHARMm force field, the binding site was identified by the LigandFit flood-filling algorithm. This docking protocol used total ligand flexibility whereby the final ligand conformations were determined by the Monte Carlo conformation search method set to a variable number of trial runs. The docked ligands were further refined by in situ ligand minimization with the Smart Minimizer algorithm. Each minimization was carried out in two steps, first using the steepest descent minimization for 200 cycles and then using conjugate gradient minimization, until the average gradient fell below 0.01 kcal/mol. All atoms within 6.0 Å of the inhibitor were allowed to relax during the minimization, whereas those atoms be-yond 6.0 Å were held rigid. Dock score was used to estimate the binding free energies of the ligands. Among the docked conforma-tions, the pose with the highest value of dock score was selected for the calculation of binding free energy (⌬Gb) and inhibition constant (Ki) (Hong et al., 2009).
In Vitro Tubulin Turbidity Assay. Tubulin polymerization
as-say was performed according to the manufacturer’s protocols (tubu-lin polymerization assay kit; Cytoskeleton Inc., Denver, CO). In brief, tubulin proteins (⬎99% purity, included in this kit) were sus-pended in G-PEM buffer containing 80 mM PIPES, 2 mM MgCl2, 0.5 mM EDTA, and 1.0 mM GTP (pH 6.9) and 5% glycerol with or without paclitaxel (1M), vincristine (1 M), and MJ-29 (0, 0.5, and 1M) in a 96-well plate, and the absorbance was measured at 340 nm from 0 to 30 min (SpectraMAX Plus; Molecular Devices, Sunny-vale, CA). In vitro tubulin turbidity assay was performed in three independent experiments (Chang et al., 2009).
Immunofluorescence Staining. U937 cells (1⫻ 104/well) were seeded into each well of the eight-well chamber slides and treated with MJ-29 (1M), paclitaxel (1 M), and vincristine (1 M) for 12 h. Cells were collected fixed in 100% methanol at⫺20°C overnight and then incubated in 1% bovine serum albumin containing 0.1% Triton X-100 at 37°C for 30 min. The cells were washed twice with PBS three times, stained with-tubulin primary antibodies at 37°C for 1 h, and then stained with fluorescein isothiocyanate-conjugated secondary antibody at 37°C for 1 h. The cells were analyzed with a confocal laser microscopic system (TCS SP2; Leica, Wetzlar, Ger-many) (Chung et al., 2007).
In Vivo Microtubule Assembly Assay. We used an established
method to measure soluble (depolymerized) and assembled (polymer-ized) tubulin (Wang et al., 2008). U937, HL-60, K562, and KG-1 cells (5⫻ 107/flask) were seeded into the 75-T flask. Cells were exposed to paclitaxel (1M), vincristine (1 M), colchicine (1 M), and MJ-29 (1 M) for 12 h. After treatment, cells were collected and washed twice with ice-cold PBS then lysed at 37°C for 5 min with 50l of hypotonic buffer (1 mM MgCl2, 2 mM EGTA, 0.5% NP-40, 2 mM PMSF, 200 units/ml aprotinin, 5 mM amino caproic acid, 1 mM benzamidine,
and 20 mM Tris-HCl, pH 6.8). The cell lysates were centrifuged at 13,000 rpm for 10 min at 25°C. The supernatants containing soluble (cytosolic) tubulin were separated from the pellets containing poly-merized (cytoskeletal) tubulin. The pellets were resuspended in 100 l of hypotonic buffer, sonicated on ice, mixed with 5⫻ sample buffer, and heated for 5 min at 100°C. Equal amounts of the two fractions were partitioned by SDS-polyacrylamide gel electrophoresis. Immu-noblots were probed with-tubulin monoclonal antibody and second-ary HRP-conjugated antibody. The blots were developed by using an ECL kit and Kodak Bio-MAX MR film (Eastman Kodak, Rochester, NY). All results are from three independent experiments.
CDK1 Activity Assay. CDK1 activity assay was performed
ac-cording to the manufacturer’s protocols (CycLex Cdc2-Cyclin B Ki-nase Assay Kit; MBL International, Nagoya, Japan). U937, HL-60, K562, and KG-1 cells (1⫻ 107/dish) were seeded into a 10-cm dish and exposed to paclitaxel (1M) and MJ-29 (1 M) or with/without roscovitine (10mol/l) for 0, 4, 8, and 12 h. Then cells were harvested and washed twice with ice-cold PBS. The cell pellet was resuspended with 500l of extraction buffer (20 mM Tris-HCl, pH 8.5, 150 mM NaCl, 0.2% NP-40, 1 mM DTT, 1 mM EDTA, 1 mM EGTA, 0.2 mM PMSF, 1g/ml pepstatin, 0.5 g/ml leupeptin, 5 mM -glycerophos-phate, 5 mM NaF, 1 mM Na3VO4, and 5 mM-mercaptoethanol), and the resuspended cells were lysed by using three cycles of freezing and thawing. Cell extracts were diluted 1:5 with Q-buffer (20 mM Tris-HCl, pH 8.5, 0.2 mM EDTA, 1 mM EGTA, 1g/ml pepstatin, 0.5 g/ml leupeptin, 0.2 mM Na3VO4, and 5 mM-mercaptoethanol). They were allowed to stand on ice for 15 min, and 100l of sample was added to the wells and then incubated for 30 min at 30°C. The wells were washed, and 100 l of HRP-conjugated antiphospho-specific antibody was added and then incubated for 1 h at room temperature. The cells were washed, and 100l of substrate reagent and 100l of stop solution were added. CDK1 activity was measured by absorbance using an enzyme-linked immunosorbent assay reader at OD450. All results are from three independent experiments (Chou et al., 2009).
siRNA Transfection. U937 cells (1⫻ 105/well) were transfected with siRNA oligonucleotides in six-well plates by using Lipo-fectamine 2000 according to the manufacturer’s instructions (In-vitrogen). Cells were transfected with scramble LO GC Duplex Stealth RNAi Negative Control (100 nM) (Invitrogen) or CDK1 siRNA (100 nM) (Invitrogen). After 24-h transfection, CDK1 mRNA was rechecked by reverse transcription-polymerase chain reaction (RT-PCR). Transfected cells were treated with MJ-29 (1 M) or paclitaxel (1M) for 24 h. Cell viability was analyzed by the exclu-sion method, and the level of phosphor-Bcl-2 protein was determined by Western blot analysis (Kuo et al., 2009).
RT-PCR. To check CDK1 mRNA levels, cells were analyzed by
RT-PCR after 24-h transfection. Total cellular RNA was extracted with a Qiagen RNeasy Mini kit (QIAGEN, Valencia, CA). The RNA was quantified by determining absorbance at 260 nm. After reverse tran-scription of 3g RNA, the cDNA product was amplified by PCR using the Amp PCR System 2700 (Applied Biosystems, Foster City, CA) in a final reaction volume of 20 l for 35 cycles by using the One-Step RT-PCR kit (QIAGEN). Amplification of CDK1 and GAPDH was per-formed in one reaction. Primers used for amplification were: CDK1, sense, 5⬘-GGGGATTCAGAAATTGATCA-3⬘; antisense, 5⬘-TGTCA-GAAAGCTACATCTTC-3⬘; and GAPDH, sense, 5⬘-ACGGATTTG-GTCGTATTGGGCG-3⬘; antisense, 5⬘-CTCCTGGAAGATGGTGATGG-30. The cycling conditions were as follows: 5 min at 95°C, followed by 35 cycles at 95°C for 1 min, 55°C for 1 min, and 72°C for 1 min. The amplified products were separated by 2% agarose gels, stained with 0.1 g/ml ethidium bromide, and photographed under ultraviolet light (Kuo et al., 2009).
Reactive Oxygen Species and Mitochondrial Membrane Po-tential Assays. The levels of reactive oxygen species (ROS) and
mitochondrial membrane potential (⌬⌿m) were determined by flow
cytometry using DCFH-DA and DiOC6as fluorescent probes. U937 cells (2.5⫻ 105/well) were seeded into each well of 24-well plates and
treated with MJ-29 (1M) for 0, 6, 12, and 24 h. Then cells were harvested, washed with PBS, resuspended in 1 ml of PBS, and incubated with 5M DCFH-DA and 40 nM DiOC6for 30 min. ROS and ⌬⌿m were analyzed for fluorescence intensity by using flow cytometry. The median fluorescence intensity was quantified by CellQuest software (Kuo et al., 2009). All results are from three independent experiments.
Western Blot. Cytosolic and mitochondrial fractions and total protein were prepared and determined as described previously (Chung et al., 2007). U937, HL-60, K562, and KG-1 cells (5⫻ 107/ flask) were seeded into a 75-T flask and exposed to MJ-29 (1M) for 0, 4, 6, 8, 12, 18, and 24 h. The harvested cells were suspended in five times volume of buffer A (20 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA, 1 mM DTT, 1g/ml aprotinin, 100 g/ml PMSF, and 250 mM sucrose). Cells were homogenized for 40 strokes and centrifuged at 1200 rpm for 10 min at 4°C. The supernatant was collected and further cen-trifuged at 60,000 rpm for 60 min at 4°C to isolate cytosolic and mitochondrial fractions. The protein concentration was measured by using a BCA assay kit (Pierce Chemical, Rockford, IL). Equal amounts of cell lysate were run on 10 to 12% SDS-polyacrylamide gel electrophoresis and electrotransferred to a nitrocellulose membrane by using the iBot Dry Blotting System (Invitrogen). The transferred membranes were blocked for 1 h in 5% nonfat dry milk in Tris-buffered saline/Tween 20 and incubated with primary antibodies at 4°C overnight. Membranes were washed three times with Tris-buff-ered saline/Tween 20 for 10 min and incubated with secondary HRP-conjugated antibody. The blots were developed by using an ECL kit and Kodak Bio-MAX MR film (Eastman Kodak). All results are from three independent experiments (Chung et al., 2007).
Caspase Activities Assay. Caspase-3, caspase-8, and caspase-9 activities were performed according to the manufacturer’s protocols (caspase-3, caspase-8, and caspase-9 Colorimetric Assay Kit; R&D Systems). U937, HL-60, K562, and KG-1 cells (1⫻ 107/dish) were seeded into a 10-cm dish and incubated with MJ-29 (1M) for 0, 12, 16, 20, and 24 h. Then cells were harvested and lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 10 mM EGTA, 10 mM digitonin, and 2 mM DTT). The resulting cell lysates (50 g of protein) were incubated with caspase-3-, caspase-8-, and caspase-9-specific substrates (Ac-DEVD-pNA, Ac-IETD-pNA, and Ac-LEHD-pNA. respectively) (R&D Systems) for 1 h at 37°C. Caspase activity and absorbance were measured with an enzyme-linked immunosor-bent assay reader at OD405. All results are from three independent experiments (Chung et al., 2007; Yang et al., 2009).
Xenograft Model and in Vivo Antitumor Activity Assay. Twenty-eight male BALB/cnu/nu mice (4 – 6 weeks of age) were ob-tained from the Laboratory Animal Center of National Applied Re-search Laboratories (Taipei, Taiwan). All mice were fed a commercial diet and water. U937 cells (total 1⫻ 106cells) were resuspended in serum-free RPMI medium 1640 with Matrigel basement membrane matrix at a 1:1 ratio (total volume 100l). U937 cells were subcu-taneously injected into the flanks of mice. Tumor mass was mea-sured every 3 days. When tumors reached an approximate volume of 100 mm3, mice were selected and distributed for drug studies (day 0). Animals with tumors were randomly assigned to four treatment groups. Animals (seven mice/group) were given vehicle control (PBS/ Tween 80 1:1), MJ-29 (5 and 25 mg/kg), or vincristine (10 mg/kg) by intraperitoneal injection for twice-daily treatment. Body weight and tumors volume were measured every 3 days with a caliper. Tumor volumes were determined by measuring the length (l) and the width (w), and the volumes were calculated as V⫽ lw2/2. The mice were sacrificed when the tumor burden was more than 1800 mm3(day 45). All experiments were conducted according to the Institutional Ani-mal Care and Use Committee, China Medical University, Taichung, Taiwan (Ho et al., 2009; Ji et al., 2009).
Apoptosis Detection by TUNEL in Tumor Tissue Sections. Apoptosis was detected by using a TUNEL in situ apoptosis detection kit (Roche Diagnostics). Tumor tissue samples were fixed in 4%
buffered formaldehyde and embedded in paraffin wax. The paraffin sections were mounted on glass slides and treated with xylene for 5 min and then 100, 95, or 75% ethanol. The deparaffinized tumor samples were incubated with proteinase K (2 mg/ml) at 25°C for 15 min. After PBS washing, endogenous peroxidase was blocked by the addition of 3% H2O2. Tissues were then treated with terminal de-oxynucleotidyl transferase and biotinylated dUTP. After stopping the reaction with TB buffer (30 mM sodium chloride, 30 mM sodium citrate), the samples were stained with 3,3⬘-diaminobenzidine. He-matoxylin counterstaining was also done to identify the cell types that were positive by TUNEL. The percentage of TUNEL-positive cells relative to total cells was calculated for each sample, counting at least 200 cells. All results are from three independent experiments (Giatromanolaki et al., 2001).
Statistical Analyses. Data are presented as the mean⫾ S.E.M. for the indicated number of separate experiments. Statistical anal-yses of data were done by one-way ANOVA, and P ⬍ 0.05 was considered significant.
Results
MJ-29 Induces Growth Inhibition and Apoptosis via G2–M Phase Arrest in Leukemia Cells. We investigated the growth inhibition effect of MJ-29 on U937 cells. As shown in Fig. 1A, MJ-29 inhibited cell growth of U937 cells in a dose- and time-dependent manner. MJ-29 also reduced the cell growth of HL-60, K562, and KG-1 cells in a dose-depen-dent manner (Supplemental Fig. 1B). The half-maximal in-hibitory concentrations (IC50s) for 24-h treatment of MJ-29
in U937, KG-1, K562, and HL-60 cell lines were 1.06⫾ 0.54, 16.32⫾ 2.25, 11.23 ⫾ 1.41, and 5.32 ⫾ 1.29 M, respectively. In comparing the growth inhibition effect of MJ-29 on U937 cells and normal PBMCs and HUVECs, we found that MJ-29 has a much less cytotoxic effect on PBMCs and HUVECs than on U937 cells (Fig. 1B). On the other hand, apoptotic evidence in MJ-29 (1M)-treated U937 cells was less appar-ent in normal PBMCs and HUVECs in a TUNEL assay (TUNEL-positive cells, U937: 38.65⫾ 6.28%; PBMCs: 6.15 ⫾ 3.24%; HUVECs: 4.95⫾ 2.51%). However, paclitaxel (1 M) and vincristine (1 M) induced significant apoptotic cell death in normal PBMCs and HUVECs (paclitaxel treatment: TUNEL-positive cells, U937, 42.25⫾ 6.32%; PBMCs, 84.23 ⫾ 4.69%; HUVECs, 87.26 ⫾ 6.28%; vincristine treatment: TUNEL-positive cells, U937, 43.62⫾ 4.26%; PBMCs, 58.26 ⫾ 7.99%; HUVECs, 43.25⫾ 8.01%) (Fig. 1C.).
Next, we assessed the effect of MJ-29 on cell cycle distri-bution in U937 cells. As shown in Fig. 1A, MJ-29 induced the accumulation of G2–M phase and losses in G0–G1phase with
a maximum effect observed at 12 h (Fig. 1D). The accumula-tion of cells with G2–M DNA content was followed by an
increase in the number of hypodiploid cells (sub-G1
popula-tion) at 12 and 24 h, which are indicated as apoptotic cells (Table 1). Our data suggest that MJ-29 induced G2–M phase
arrest of the cell cycle followed by apoptosis in U937 cells. MJ-29 induced selective cytotoxicity in human leukemia cells but had a less cytotoxic effect on PBMCs and HUVECs. MJ-29 might have fewer side effects than paclitaxel and vincristine.
MJ-29 Inhibits the Polymerization of Microtubules. To predict the major target site of MJ-29, the docking simulation of MJ-29 and tubulin was carried out with the program Discovery Studio modeling 2.1 (Accelrys). The three-dimensional crystal structure of tubulin-colchicine was downloaded from the Pro-tein Data Bank web site (ProPro-tein Data Bank code1SA0). The
computational modeling of the MJ-29-tubulin interaction indi-cated that MJ-29 can bind to the active site of␣- and -tubulin. As shown in Fig. 2A, MJ-29 formed three H-bonds between N1, C8-H, and⫺OH atoms of quinazoline and Asn-␣101, Asn-258, and Glu-␣183, respectively. In addition, MJ-29 formed several hydrophobic interactions with Ala-␣180, Glu-␣183, Leu-255, Leu-248, and Asn-258. These interactions made MJ-29 bind readily to␣- and -tubulin with low potential energy.
Because MJ-29 caused G2–M phase arrest, we investigated
whether MJ-29 affects microtubule organization. As shown in Fig. 2B, in an in vitro microtubule assembly assay MJ-29 inhibited tubulin polymerization in a dose-dependent man-ner. Our results also show that tubulin polymerization was significantly promoted by paclitaxel (1M) and completely inhibited by vincristine (1M). We further investigated the effect of MJ-29 on tubulin polymerization in U937 cells in vivo. Immunofluorescence staining results are shown in Fig. 2C. Paclitaxel promoted microtubule polymerization with an increase in the density of cellular microtubules. Treatment with MJ-29 (1 M) and vincristine (1 M) resulted in the inhibition of microtubule polymerization and the appearance of short microtubule fragments in the cytoplasm. The protein levels of soluble tubulin (S) and polymerize tubulin (P) were detected on MJ-29-, paclitaxel-, colchicines-, and
vincristine-treated U937 cells. As shown in Fig. 2D, U937 cells were treated with MJ-29, colchicine, and vincristine for 12 h, caus-ing inhibition of microtubule assembly. However, paclitaxel significantly induced promotion of tubulin polymerization. In addition, HL-60, K562, and KG-1 cells were treated with MJ-29 for 12 h, causing inhibition of microtubule assembly (Supplemental Fig. 2A). Our results indicate that MJ-29 induced mitotic arrest and inhibited the polymerization of microtubules in leukemia cells.
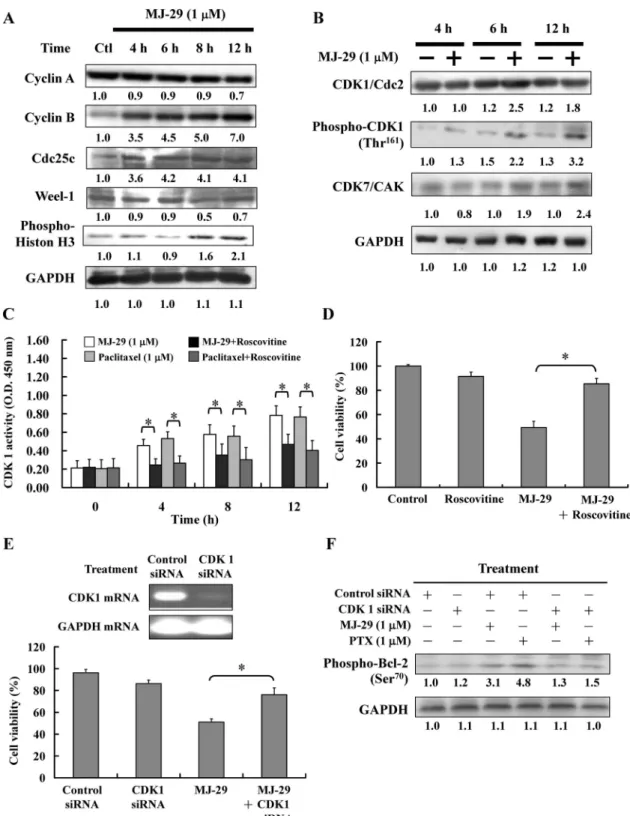
MJ-29 Induces Mitotic Arrest and Apoptosis via CDK1-Mediated Bcl-2 Phosphorylation. We investigated the protein levels in the mitotic phase of the cell cycle. As shown in Fig. 3A, MJ-29 caused an increase in the protein levels of cyclin B, Cdc25c, and phospho-histone H3 (a marker for mitotic progression) and caused a decrease in the protein level of Weel-1 in U937 cells (Rodriguez-Collazo et al., 2008). MJ-29 did not change the level of cyclin A. As shown in Fig. 3B, MJ-29 and paclitaxel caused an increase in the protein levels of CDK1, phospho-CDK1 (Thr161), and CDK7 in U937 cells. In addition, MJ-29 increased the protein levels of cyclin B, CDK1, and phospho-CDK1 (Thr161) in HL-60, K562, and KG-1 cells (Supplemental Fig. 2B).
We investigated CDK1 activity in the mitotic phase. As shown in Fig. 3C, MJ-29 and paclitaxel caused a significant
Fig. 1. Effects of MJ-29 on cell viability, cell cycle distribution, and apoptosis in leukemia cell lines. A, U937 cells were treated with MJ-29 (0, 0.5, 1, 5 and 10 M) for 24, 48 and 72 h. B, U937 cells, PBMCs, and HUVECs were exposed to MJ-29 (0, 0.5, 1, 5, and 10M) for 24 h. Cell viability was determined by the PI exclusion method and analyzed by flow cytometry. Mean⫾ S.E.M. of three inde-pendent experiments.ⴱ, P ⬍ 0.05, signif-icantly different compared with the con-trol group by one-way ANOVA. C, U937 cells, PBMCs, and HUVECs were exposed to MJ-29 (1M), paclitaxel (1 M), and vincristine (1 M) for 24 h. Cells were stained with TUNEL and analyzed by flow cytometry. Mean⫾ S.E.M. of three independent experiments. ⴱ, P ⬍ 0.05, significantly different compared with the untreated control group by one-way ANOVA. D, U937 cells after MJ-29 (1 M) treatment for the indicated time pe-riods were fixed and stained with PI for analysis of DNA content cell cycle distri-bution by flow cytometry.
TABLE 1
Cell cycle distribution (%) in U937 cells after exposure to MJ-29
U937 cells (2.5⫻ 105cells/ml/well) were treated with 1M MJ-29 for 0, 4, 8, 12, and 24 h. The cells were collected, and the cell cycle and sub-G
1phase were examined by
flow cytometric assay as described under Materials and Methods. Each point is mean⫾ S.E.M. of three experiments.
Time Sub-G1 G0–G1 S G2–M h % 0 0.11⫾ 0.01 47.27⫾ 0.62 45.01⫾ 1.04 7.61⫾ 1.01 4 2.10⫾ 0.16* 31.87⫾ 0.41* 41.37⫾ 2.36* 24.66⫾ 3.01* 8 11.80⫾ 1.04* 21.76⫾ 0.80* 34.99⫾ 2.45* 31.45⫾ 2.03* 12 27.25⫾ 4.24* 1.57⫾ 0.71* 6.70⫾ 4.11* 64.48.⫾ 3.04* 24 50.33⫾ 3.14* 0.55⫾ 1.08* 0.50⫾ 3.12* 48.62⫾ 3.02*
increase in CDK1 activity from 4 to 12 h. Cells were pre-treated with roscovitine (10mol/l) and then incubated with MJ-29 for 12 h, significantly abolishing MJ-29-induced CDK1 activity and viability inhibition (Fig. 3D). MJ-29 also caused an increase in CDK1 activity at 12-h treatment in HL-60, K562, and KG-1 cells (Supplemental Fig. 2C).
Next, we showed that the CDK1 mRNA level was knocked down at 24 h after treatment with CDK1 siRNA but not after treatment with control siRNA (Fig. 3E, top). It is noteworthy that knockdown of CDK1 by siRNA led to the significant abolishment of MJ-29-induced cell death (Fig. 3E, bottom). To test the hypothesis that MJ-29 induces mitotic arrest and apoptosis via CDK1-mediated Bcl-2 phosphorylation. Bcl-2 phosphorylation was detected on MJ-29- and paclitaxel-treated CDK1 knockdown U937 cells by Western blot. As shown in Fig. 3F, MJ-29 and paclitaxel caused an increase in the protein level of phospho-Bcl-2 (Ser70) in U937 cells. How-ever, both agents prevented the protein level of phospho-Bcl-2 (Ser70) in CDK1 knockdown U937 cells. Our results suggest that MJ-29 is able to increase CDK1 activity and induce Bcl-2 phosphorylation in U937 cells.
MJ-29 Induces the Mitochondria-Dependent Apopto-tic Signaling Pathway. Mitochondria plays an important role in apoptosis, and CDK1 can trigger mitochondrial mem-brane permeabilization by targeting Bcl-2 family proteins and subsequently inducing cell apoptosis (Debatin et al., 2002). We examined the effects of MJ-29 on ROS produc-tion and loss of⌬⌿m. A remarkable decrease in⌬⌿mwas
observed, and an increase of ROS production was evident after treatment with MJ-29 for 6 to 24 h in U937 cells (Table 2). It is well known that caspases can be activated in two major apoptotic pathways, the death receptor- and mitochondria-mediated signaling pathways (Ashkenazi, 2008; Fulda, 2009). We determined whether both of these pathways contribute to MJ-29-induced apoptosis. As shown in Fig. 4A, MJ-29 caused decreases in mitochondrial protein levels of cytochrome c, Apaf-1, procaspase-9, and AIF in U937 cells (Fig. 4A, left). MJ-29 caused increases in the cytosolic protein levels of cytochrome c, Apaf-1, pro-caspase-9, and AIF in U937 cells (Fig. 4A, right). We examined the protein levels of Bcl-2 family proteins. As shown in Fig. 4B, the Bax protein level was increased and the Bcl-2 protein level was decreased in MJ-29-treated U937 cells. In addition, the phospho-Bcl-2 (Ser70) protein level was increased in a time-dependent manner in MJ-29-treated U937 cells (Fig. 4B). MJ-29 also caused decreases in the protein levels of Bcl-2 and increased protein levels of Bax and phospho-Bcl-2 (Ser70) in HL-60, K562, and KG-1 cells (Supplementary Fig. 3A). Our results revealed the levels of Fas, FasL, TNF-R1, TRAIL-R1 (DR4), TRAIL-R2 (DR5), and protein have no significant influence on MJ-29-treated U937 cells (Fig. 4C) and suggest that the mitochon-drial signaling pathway is mediated by MJ-29-induced apoptotic responses.
MJ-29 Induces Caspase-9 and Caspase-3 Activities. We investigated caspase-9, caspase-8, and caspase-3
activ-Fig. 2. MJ-29 affected microtubule assembly in vitro and in vivo. A, molecular modeling of the interaction of MJ-29 with tubulin. The image shows binding models of MJ-29 with␣- and -tubulin. MJ-29 formed three H-bonds between N1, C8-H, and ⫺OH atoms of quinazoline and Asn-␣101, Asn-258, and Glu-␣183, respectively, and several hydrophobic interactions with Ala-␣180, Glu-␣183, Leu-255, Leu-248, and Asn-258. B, MJ-29 altered microtubule assembly in vitro. In vitro tubulin polymerization assays were performed for 30 min at 37°C in the presence of 0.5% DMSO (the negative control), paclitaxel (1M), vincristine (1 M) (the positive control), and MJ-29 (0.5 and 1 M). The kinetics of tubulin polymerization was monitored by measuring optical density at 340 nm every 2 min. Data are means⫾ S.E.M. from three independent experiments. C, immunofluorescence staining of microtubules in MJ-29-treated U937 cells. Cells were treated for 12 h with 0.5% DMSO as control, paclitaxel (1M), vincristine (1 M), and MJ-29 (1 M), and then stained with -tubulin antibody (green fluorescence). D, after 12-h treatment with vehicle or paclitaxel (1M), vincristine (1 M), colchicine (1 M), and MJ-29 (1 M), cytosolic (S, soluble) and cytoskeletal (P, polymerized tubulin) tubulin fractions were separated and immunoblotted with antibody against-tubulin.
Fig. 3. MJ-29 altered the levels of mitotic-regulated proteins, CDK1 kinase activity, and the phospho-Bcl-2 protein level in U937 cells. A, U937 cells were treated with MJ-29 (1M) for 4, 6, 8, and 12 h. Cells were then harvested and lysed for the detection of levels of cyclin A, cyclin B, Cdc25c, Weel-1, and phosphor-Histon H3 proteins by Western blot analysis. B, U937 cells were treated with MJ-29 (1M) for 4, 6, and 12 h. Cells were then harvested and lysed for the detection of the levels of CDK1/Cdc2, phospho-Thr161-CDK1, CDK7, and GAPDH by Western blot analysis. C, U937 cells were exposed to MJ-29 (1M) or paclitaxel (1 M) for 12 h in the absence or presence of roscovitine (20 M), and CDK1 kinase activity was measured. CDK1 kinase activity was measured in cellular extracts for the ability to phosphorylate MV peptide, a CDK1 kinase-specific substrate, according to the manufacturer (MBL International) of the CycLex Cdc2-Cyclin B Kinase Assay Kit. D, U937 cells were treated with MJ-29 (1M) for 24 h in the absence or presence of roscovitine (20M) for 24 h. The cell viability was determined by PI exclusion and analyzed by flow cytometry. ⴱ, P ⬍ 0.05, significantly different compared with the MJ-29 treatment group by one-way ANOVA. E, U937 cells were transfected with 100 nM CDK1 siRNA or control siRNA for 24 h, and the level of CDK1 mRNA was detected by RT-PCR (top). CDK1 siRNA or control siRNA-transfected U937 cells were treated with MJ-29 (1M) for 24 h, and cell viability was determined by PI exclusion and analyzed by flow cytometry (bottom). Mean ⫾ S.E.M. of three independent experiments.ⴱ, P ⬍ 0.05, significantly different compared with the MJ-29 treatment group by one-way ANOVA. F, U937 cells were transfected with 100 nM CDK1 siRNA or control siRNA. Phosphor-Bcl-2 protein expression was analyzed by Western blot in CDK1 siRNA or control siRNA-transfected U937 cells after 24-h treatment with MJ-29 or paclitaxel.
ities in MJ-29-treated U937 cells. As shown in Fig. 5, MJ-29 caused an increase of caspase-9 (Fig. 5A) and caspase-3 (Fig. 5B) activities in a time-dependent manner, whereas the activity of caspase-8 was not affected (Fig. 5C). MJ-29 also caused an increase of caspase-9 and caspase-3 activities in HL-60, K562, and KG-1 cells (Sup-plemental Fig. 3, B and C). MJ-29-induced caspase-9 and caspase-3 activation was also significantly attenuated by specific inhibitors of caspase-9 or caspase-3. Preincubation
with inhibitors of caspases-9 and caspase-3 or ROS scav-enger (NAC) significantly reduced MJ-29-induced apopto-tic cells. Our results suggest that caspase-9 and caspase-3 activation might be involved in MJ-29-induced apoptotic cell death.
DNA Microarray Analysis for MJ-29-Induced Apoptosis in U937 Cells. DNA microarray analysis was performed to examine the gene expression profile in MJ-29-treated U937 cells. U937 cells were treated with MJ-29 for 6 and 12 h. After exposure to MJ-29 for 6 h, the microarray analysis showed that 16 genes (10 genes, up-regulated; 6 genes, down-regulated) were expressed at least 10-fold compared with the untreated control cells. We observed that RHOC, AKAP13, CHEK1, CDKL2, ZNF610, CDKL2, LOC500409, CCNB2, ANXA2, and ADCY4 mRNA were up-regulated, and CORO2B, CDC14B, PPARGC1B, BCL2L1, MAPK3, and NLN mRNA were down-regulated in the MJ-29-treated cells (Supplemental Table 1). After treatment with MJ-29 for 12 h, 16 genes (nine genes, up-regulated; seven genes, down-regulated) were ex-pressed at least 10-fold compared with the untreated con-trol cells. We also observed that CAPN2, CASP9, CASP3, GPR109A, Clu (clusterin), ZNF238, CASP7, BAX, and
Fig. 4. MJ-29 stimulated the levels of apoptotic relative proteins in U937 cells. Cells were exposed to MJ-29 (1M), and then incubated for 12, 16, 20, and 24 h. The levels of mitochondrial and cytosolic fractions in cytochrome c, Apaf-1, procaspase-9, AIF, complex IV, and GAPDH (A), Bcl-2, phospho-Bcl-2, Bax, and GAPDH (B), and Fas/CD95, FasL, TNF-R1, TRAIL-R1 (DR4), TRAIL-R2 (DR5), FADD, and GAPDH (C) in MJ-29-treated U937 cells were deter-mined. Mitochondrial and cytosolic proteins or whole-cell lysates were prepared and subjected to Western blot anal-ysis, and GADPH served as the loading control.
TABLE 2
Flow cytometric analysis of intracellular ROS and⌬⌿mlevels in U937
cells after MJ-29 treatment
U937 cells (2.5⫻ 105cells/ml/well) were treated with 1M MJ-29. The 0 h was
defined as control. To determine ROS production and ⌬⌿mlevels cells were stained with DCFH-DA and DiOC6, respectively. The stained cells were examined
by flow cytometry as described under Materials and Methods. Values are mean⫾ S.E.M. (n⫽ 3).
Time ROS Production ⌬⌿mLevel
h %
0 3.26⫾ 0.57 98.21⫾ 0.06 6 25.06⫾ 3.26* 90.54⫾ 2.19* 12 38.06⫾ 5.29* 72.33⫾ 5.16* 24 74.25⫾ 4.19* 51.26⫾ 4.29*
CPE mRNA were up-regulated, and CDC14B, CORO2A, BCL2L1, MTFMT, MAP3K3, CCNE1, and TUBGCP3 mRNA were down-regulated in MJ-29-treated cells. These genes also might be involved in the cytotoxic effect on mitotic arrest and apoptosis, inducing the effect of MJ-29 on U937 cells (Supplemental Table 2).
Antitumor Effects of MJ-29 on U937 Cells in a Xeno-graft Model. Based on our in vitro studies, we examined the in vivo antitumor activities of MJ-29 in BALB/cnu/numouse
U937 xenograft models. As seen in Fig. 5A, MJ-29 (5 and 25 mg/kg) and vincristine (10 mg/kg) reduced tumor volume compared with control after treatment from days 24 to 45. Representative tumor weights in the U937 xenograft mice treated with or without MJ-29 are shown in Fig. 6B. MJ-29 and vincristine significantly (P⬍ 0.05) decreased the tumor weight by 40% compared with control.
Fig. 6C shows that the body weights of the xenograft mice were not significantly different after treatment with MJ-29 (5 and 25 mg/kg) and vincristine (10 mg/kg) from days 0 to 45. Treatment with both MJ-29 and vincristine significantly prevented the loss of body weight compared with the control group. We determined apoptotic cells in transplantation tumors from a paraffin section of tumor tissue by TUNEL assay. Our data showed that TUNEL-positive apoptotic cells of tumor sections significantly increased in MJ-29-treated (5 and 25 mg/kg) and vincristine-treated (10 mg/kg) U937 xenograft mice compared with the control group (TUNEL-positive cells: control 3.54⫾ 1.02%; MJ-29 treatment (5 mg/kg): 41.26⫾ 7.26%; MJ-29 treatment (10 mg/kg): 65.33 ⫾ 12.25%; vincristine treatment: 68.29⫾ 8.25%) (Fig. 6D). Our results sug-gest that MJ-29 causes antitumor activity and induces apoptosis in the U937 xenograft animal model.
Discussion
Interference with microtubule assembly/disassembly agents provides a novel approach toward cancer therapy agents (Cheng et al., 2008; Perez, 2009). Although vinca alkaloids and taxanes have antitumor actions in clinical treatment, there are
limitations to their use, because of toxic side effects and drug resistance (Perez, 2009). MJ-29 was designed and synthesized for a promising antileukemia compound in our laboratory (Hour et al., 2007). In this study, we first used molecular modeling (Fig. 2A), and in vitro and in vivo tubulin polymerization assays (Fig. 2, B–D) confirmed that MJ-29 is a MTA. MJ-29 signifi-cantly inhibited the growth of U937, HL-60, K562, and KG-1 cells (Fig. 1B and Supplemental Fig. 1B). It is noteworthy that MJ-29 has much less cytotoxic effect on PBMCs and HUVECs than on U937 cells (Fig. 1B). MJ-29 can be used to develop a safer agent with fewer side effects than can paclitaxel and vincristine based on these observations: 1) The doubling times of PBMCs and HUVECs are approximately 96 and 27 h (Bagley et al., 2003; Macallan et al., 2003). Compared with U937 cells (doubling time: 24 h (Chia et al., 2003), the two normal cell lines need a long time for intermitosis. 2) Paclitaxel and vincristine have lower IC50s than MJ-29 in normal PBMCs and HUVECs.
The IC50s for 24-h treatment of paclitaxel and vincristine in
HUVECs were 0.01 and 5.32⫾ 1.29 M and in PBMCs they were 0.01 and 3.2 nM, respectively (Hirano et al., 1996; Hayot et al., 2002; Grant et al., 2003).
Our results showed that the activity of MJ-29 in U937 and K562 cell lines was more potent than that of paclitaxel. The IC50of paclitaxel for U937 and K562 cells is higher than 1
M (Hirano et al., 1996; Al-alami et al., 1998; Hayot et al., 2002; Grant et al., 2003), so the margin of safety dose is narrower. Moreover, paclitaxel at 0.01 nM can suppress 50% of the growth of normal PBMC leukocytes, but 42.69 times the IC50of MJ-29 is required to inhibit 50% of the growth of
normal PBMC leukocytes. Furthermore, paclitaxel at 0.01 nM can suppress 50% of the growth of normal HUVECs, but 50.24 times higher than the IC50 of MJ-29 is required to
inhibit 50% of the growth of normal PBMC leukocytes. As a result, MJ-29 induces more selective cytotoxicity in human leukemia U937 cells but less in PBMCs. Based on these characteristics, MJ-29 can be used in leukemia patients with decreasing side effects.
Fig. 5. Effects of MJ-29 on caspase-3, caspase-8, and caspase-9 activities in U937 cells. A–C, cells were exposed to MJ-29 (1M) for 12, 16, 20, and 24 h or pretreated with or without specific in-hibitors of caspase-3, caspase-8, and caspase-9 for 1 h, and the whole-cell lysates were determined for caspase-9 (A), caspase-3 (B), and caspase-8 (C) ac-tivities assay. Total cell extracts were incubated with caspase-3-, caspase-9-, and caspase-8-specific substrates (DEVD-pNA, LEHD-pNA, and Ac-IETD-pNA, respectively). The release of pNA was measured at 405 nm by a spec-trophotometer. D, pretreatment with inhibitors of caspase-3, caspase-9, and NAC determined cell viability by PI ex-clusion and flow cytometry. Mean ⫾ S.E.M. of three independent experi-ments.ⴱ, P ⬍ 0.05, significantly differ-ent compared with the 0-h treatmdiffer-ent group by one-way ANOVA.
We have investigated that MJ-29-induced apoptosis in U937 cells is involved in the same apoptotic signaling pathway as in other types of leukemia cell lines. The reasons for the differences in sensitivities of IC50might be:
1) There are different stages of differentiation in leukemia cells. HL-60, K562, and KG-1 cell lines are promyelocytic, chronic, and acute leukemia cells, respectively. However, the U937 cell line is monocyte lymphoma leukemia. Pacli-taxel induces apoptosis in the G2–M–late S phase in both
HL-60 and U937 cells but not in K562 human leukemia cells (Dumontet et al., 2004). 2) The distinct abilities of MJ-29 in inducing apoptosis of U937, HL-60, K562, and KG-1 cells might be caused by differential gene expression in different cell types. It is reported that the 5-lipooxyge-nase inhibitor induced differentiation activity in specific leukemia cell lines. This may be caused by the differences in the gene expression of HL-60, U937, and K562 cell lines (Jing et al., 1999). Other reports showed that etoposide
cannot induce apoptosis in overexpression of the BCR-ABL gene of K562 cells (Stiewe et al., 2000). Moreover, the vital p53 protein is mediated in G2–M-phase arrest and
apopto-sis induction. In our study, HL-60, K652, and KG-1 cells are p53 null, but U937 is p53-mutated (Danova et al., 1990).
We selected the closest concentration to IC50of MJ-29 and
determined antileukemia activity in an in vitro study. Re-sults can be summarized as follows: 1) HL-60, K562, and KG-1 cells were treated with MJ-29 for 12 h, causing inhibi-tion of microtubule assembly (Supplemental Fig. 2A). 2) MJ-29 increased protein levels of cyclin B, CDK1, and phospho-CDK1 (Thr161) in HL-60, K562, and KG-1 cells (Supplemental Fig. 2B). 3) MJ-29 caused an increase in CDK1 activity after 12-h treatment in HL-60, K562, and KG-1 cells (Supplemental Fig. 2C). 4) MJ-29 caused a de-crease in the protein level of Bcl-2 and inde-creased protein levels of Bax and phospho-Bcl-2 (Ser70) in HL-60, K562, and
Fig. 6. MJ-29 inhibited tumor growth and induced apoptosis in the xenograft animal model. Twenty-eight athymic BALB/cnu/nu mice were subcutaneously
implanted with 1⫻ 106U937 cells When
tumors reached an approximate volume of 100 mm3, the mice were randomly
divided into four groups (seven mice/ group). Group 1 was treated with con-trol vehicle (PBS/Tween 80 1:1) intra-peritoneally twice daily; group 2 was treated with 5 mg/kg MJ-29 intraperito-neally twice daily; group 3 was treated with 25 mg/kg MJ-29 intraperitoneally twice daily; and group 4 was treated with 10 mg/kg vincristine intraperitone-ally twice daily. At day 45, all animals were sacrificed. A–C, solid tumor vol-ume (A), tumor weight, (B) and body weight (C) for representative animals are shown. D, apoptotic cells (TUNEL-positive cells) in transplantation tumors from paraffin section of tumor tissue by TUNEL assay are shown. Arrows indi-cate shrinkage and rounding during cell apoptosis. Data are presented as the mean⫾ S.E.M. at days 0 to 45 after tumor implantation. The tumor volumes observed in all groups were compared and analyzed by one-way ANOVA. Data represent the mean⫾ S.E.M. of seven animals. ⴱ, P ⬍ 0.05, significantly different compared with control by one-way ANOVA. E, a proposed model illustrates the molecular mechanism and the overall possible signaling pathways for MJ-29-induced mitotic arrest and apoptosis in U937 leukemia cells.
KG-1 cells (Supplemental Fig. 3A). 5) MJ-29 caused an in-crease in the caspase-9 and caspase-3 activities of HL-60, K562, and KG-1 cells (Supplemental Fig. 3, B and C). Based on these observations, we suggest that MJ-29 induced apoptotic cell death through signaling pathways in HL-60, K562, and KG-1 cells.
Our results demonstrated that MJ-29 induced phospho-histone H3 and induced mitotic arrest in U937 cells (Fig. 3A). Results revealed that MJ-29 increased protein levels of cyclin B and CDK1, increased phosphorylation at the Thr161 site of CDK1 and CDK7, and significantly increased CDK1 kinase activity (Fig. 3 and Supplemental Fig. 2). CAK is a complex of CDK7 and cyclin H, and this complex is involved in cell cycle regulation by phosphorylating an activating residue in the T-loop domain of CDKS (Fisher and Morgan, 1994). We also observed that Chk1, CDK1, and cyclin B mRNA were up-regulated in MJ-29-treated cells (Supplemental Table 1). Previous studies indicated that MTAs promoted activating Thr161 phosphorylation of CDK1 by CAK and reduced inhib-itory phosphorylation at Tyr15 (Yu et al., 1998). Conse-quently, MJ-29 induces mitotic arrest, and CDK1 phosphor-ylation via CAK regulation causes increased CDK1 activity. Induction of apoptosis associated with Bcl-2 phosphoryla-tion by MTAs has been linked with altering a variety of cellular signaling pathways, such as Ras/Raf, protein kinase C, protein kinase A, mitogen-activated protein kinase, c-Jun N-terminal kinase, and CDK1 (Wang et al., 1999; Van-tieghem et al., 2002). CDK1 phosphorylates Bcl-2 on Ser70 and suppresses its antiapoptotic function (Wang et al., 1999). Our results demonstrated that cells treated with MJ-29 and roscovitine (CDK inhibitor) or CDK1 siRNA significantly abolished MJ-29-induced apoptosis (Fig. 3, C–E). We found that knockdown of CDK1 by siRNA led to significant abol-ishment of MJ-29 or paclitaxel-induced Bcl-2 phosphoryla-tion on Ser70 (Fig. 3F). Therefore, we suggest that MJ-29-induced CDK1 activity resulted in apoptosis caused by Ser70 phosphorylation of Bcl-2, then started apoptosis via the in-trinsic pathway.
MJ-29 induced the activation of caspase-9 and caspase-3 in a time-dependent manner (Fig. 4, A and B), suggesting that MJ-29 could trigger intrinsic signaling pathways. When mi-tochondria perceive apoptotic signaling, the outer membrane of mitochondria becomes permeabilized, and then cyto-chrome c, Apaf-1, procaspase-9, and AIF are released into the cytosol before the activation of caspase-3 by caspase-9, lead-ing to apoptotic cell death (Burz et al., 2009). We demon-strated the alteration of⌬⌿m and ROS after MJ-29 treat-ment for 12 h (Table 2), which led to the release of cytochrome c, Apaf-1, procaspase-9, and AIF proteins from mitochondria into the cytosol (Fig. 4A). We discovered that 16 genes changed their expression in the MJ-29-treated U937 cells with a minimal 10-fold change at 12 h after treatment by DNA microarray assay (Supplemental Table 2). We also observed that caspase-9, caspase-3, and Bax mRNA were up-regulated, and Bcl-2 and mitochondrial methionyl-tRNA formyltransferase were down-regulated in MJ-29-treated cells. Our results in Figs. 4C and 5C reveal that the protein levels of Fas, FasL, TNF-R1, TRAIL-R1 (DR4), TRAIL-R2 (DR5), and FADD did not significantly increase. In addition, the level of caspase-8 activity in U937 cells had no significant increase. Based on these results, we can rule out that MJ-29-stimulated apoptotic cell death is involved in the death
receptor apoptotic pathway. These results indicate that MJ-29 might trigger the intrinsic signaling pathways leading to apoptotic cell death.
In our in vitro studies, mitotic arrest and apoptosis induction by MJ-29 were observed. However, these effects are also operative in vivo, and in vivo study is required to establish a potential for drug discovery. We examined the antitumor activities of MJ-29 in a BALB/cnu/nu mouse
U937 xenograft model. MJ-29 and vincristine reduced tu-mor volume compared with control after treatment from 24 to 45 days (Fig. 6A). MJ-29 and vincristine also signifi-cantly decreased tumor weight by 40% compared with control (Fig. 6B). The body weights of the U937 xenograft mice were not significantly different after MJ-29 and vin-cristine treatment from days 0 to 45, but both agents significantly prevented the loss of body weight compared with the control group (Fig. 6C). In support of the apopto-sis mechanism in vitro, we next examined TUNEL staining in tumor specimens from control and MJ-29- or vincristine-treated animals. The increase of TUNEL-positive cells was detected in the MJ-29- or vincristine-treated animals com-pared with control group. Our results support apoptotic evidence in MJ-29-treated U937 xenograft mice.
In conclusion, MJ-29 showed significant cytotoxicity against leukemia cells. However, MJ-29 is less toxic to PBMCs and normal HUVECs. In Fig. 6C, we have outlined the molecular mechanism and the overall possible signal-ing pathways for MJ-29-induced apoptosis in U937 cells. The mechanism of action involves an interaction with tu-bulin, leading to dysregulation of mitotic spindles and induction of mitotic arrest and cell apoptosis. MJ-29 also induced CDK1 activation, phosphorylation of Bcl-2 on Ser70, activation of caspase-9 and caspase-3, and release of AIF and triggered the intrinsic signaling apoptosis path-ways in U937 human leukemia cells.
References
Al-alami O, Sammons J, Martin JH, and Hassan HT (1998) Divergent effect of taxol on proliferation, apoptosis and nitric oxide production in MHH225 CD34 positive and U937 CD34 negative human leukaemia cells. Leuk Res 22:939 –945. Allan LA and Clarke PR (2007) Phosphorylation of caspase-9 by CDK1/cyclin B1
protects mitotic cells against apoptosis. Mol Cell 26:301–310.
Ashkenazi A (2008) Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev 19:325–331.
Bagley RG, Walter-Yohrling J, Cao X, Weber W, Simons B, Cook BP, Chartrand SD, Wang C, Madden SL, and Teicher BA (2003) Endothelial precursor cells as a model of tumor endothelium: characterization and comparison with mature endothelial cells. Cancer Res 63:5866 –5873.
Burz C, Berindan-Neagoe I, Balacescu O, and Irimie A (2009) Apoptosis in cancer: key molecular signaling pathways and therapy targets. Acta Oncol 48:811– 821. Chang YH, Yang JS, Kuo SC, and Chung JG (2009) Induction of mitotic arrest and
apoptosis by a novel synthetic quinolone analogue, CWC-8, via intrinsic and extrinsic apoptotic pathways in human osteogenic sarcoma U-2 OS cells. Antican-cer Res 29:3139 –3148.
Chen JC, Lu KW, Tsai ML, Hsu SC, Kuo CL, Yang JS, Hsia TC, Yu CS, Chou ST, Kao MC, et al. (2009) Gypenosides induced G0/G1 arrest via CHk2 and apoptosis through endoplasmic reticulum stress and mitochondria-dependent pathways in human tongue cancer SCC-4 cells. Oral Oncol 45:273–283.
Cheng KL, Bradley T, and Budman DR (2008) Novel microtubule-targeting agents– the epothilones. Biologics 2:789 – 811.
Chia S, Qadan M, Newton R, Ludlam CA, Fox KA, and Newby DE (2003) Intra-arterial tumor necrosis factor-␣ impairs endothelium-dependent vasodilatation and stimulates local tissue plasminogen activator release in humans. Arterioscler Thromb Vasc Biol 23:695–701.
Chou LC, Yang JS, Huang LJ, Wu HC, Lu CC, Chiang JH, Chen KT, Kuo SC, and Chung JG (2009) The synthesized 2-(2-fluorophenyl)-6,7-methylenedioxyquinolin-4-one (CHM-1) promoted G2/M arrest through inhibition of CDK1 and induced apoptosis through the mitochondrial-dependent pathway in CT-26 murine colorec-tal adenocarcinoma cells. J Gastroenterol 44:1055–1063.
Chung JG, Yang JS, Huang LJ, Lee FY, Teng CM, Tsai SC, Lin KL, Wang SF, and Kuo SC (2007) Proteomic approach to studying the cytotoxicity of YC-1 on U937 leukemia cells and antileukemia activity in orthotopic model of leukemia mice. Proteomics 7:3305–3317.
Danova M, Giordano M, Mazzini G, and Riccardi A (1990) Expression of p53 protein during the cell cycle measured by flow cytometry in human leukemia. Leuk Res 14:417– 422.
Debatin KM, Poncet D, and Kroemer G (2002) Chemotherapy: targeting the mito-chondrial cell death pathway. Oncogene 21:8786 – 8803.
Dumontet C, Jaffrezou JP, Tsuchiya E, Duran GE, Chen G, Derry WB, Wilson L, Jordan MA, and Sikic BI (2004) Resistance to microtubule-targeted cytotoxins in a K562 leukemia cell variant associated with altered tubulin expression and polymerization. Bull Cancer 91:E81–E112.
Fisher RP and Morgan DO (1994) A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 78:713–724.
Fotoohi AK, Assaraf YG, Moshfegh A, Hashemi J, Jansen G, Peters GJ, Larsson C, and Albertioni F (2009) Gene expression profiling of leukemia T-cells resistant to methotrexate and 7-hydroxymethotrexate reveals alterations that preserve intra-cellular levels of folate and nucleotide biosynthesis. Biochem Pharmacol 77:1410 – 1417.
Fulda S (2009) Caspase-8 in cancer biology and therapy. Cancer Lett 281:128 –133. Giatromanolaki A, Sivridis E, Athanassou N, Zois E, Thorpe PE, Brekken RA, Gatter KC, Harris AL, Koukourakis IM, and Koukourakis MI (2001) The angiogenic pathway “vascular endothelial growth factor/flk-1(KDR)-receptor” in rheumatoid arthritis and osteoarthritis. J Pathol 194:101–108.
Grant DS, Williams TL, Zahaczewsky M, and Dicker AP (2003) Comparison of antiangiogenic activities using paclitaxel (taxol) and docetaxel (taxotere). Int J Cancer 104:121–129.
Hayot C, Farinelle S, De Decker R, Decaestecker C, Darro F, Kiss R, and Van Damme M (2002) In vitro pharmacological characterizations of the anti-angiogenic and anti-tumor cell migration properties mediated by microtubule-affecting drugs, with special emphasis on the organization of the actin cytoskeleton. Int J Oncol 21:417– 425.
Hirano T, Oka K, Mimaki Y, Kuroda M, and Sashida Y (1996) Potent growth inhibitory activity of a novel Ornithogalum cholestane glycoside on human cells: induction of apoptosis in promyelocytic leukemia HL-60 cells. Life Sci 58:789 –798. Ho YT, Yang JS, Lu CC, Chiang JH, Li TC, Lin JJ, Lai KC, Liao CL, Lin JG, and Chung JG (2009) Berberine inhibits human tongue squamous carcinoma cancer tumor growth in a murine xenograft model. Phytomedicine 16:887– 890. Hong YB, Kang HJ, Kim HJ, Rosen EM, Dakshanamurthy S, Rondanin R,
Baruch-ello R, Grisolia G, Daniele S, and Bae I (2009) Inhibition of cell proliferation by a resveratrol analog in human pancreatic and breast cancer cells. Exp Mol Med 41:151–160.
Hour MJ, Yang JS, Lien JC, Kuo SC, and Huang LJ (2007) Synthesis and cytotox-icity of 6-pyrrolidinyl-2-(2-substituted phenyl)-4-quinazolinones. J Chin Chem Soc 54:785–790.
Ibrado AM, Kim CN, and Bhalla K (1998) Temporal relationship of CDK1 activation and mitotic arrest to cytosolic accumulation of cytochrome C and caspase-3 activity during Taxol-induced apoptosis of human AML HL-60 cells. Leukemia 12:1930 – 1936.
Itzykson R, Ayari S, Vassilief D, Berger E, Slama B, Vey N, Suarez F, Beyne-Rauzy O, Guerci A, Cheze S, et al. (2009) Is there a role for all-trans retinoic acid in combination with recombinant erythropoetin in myelodysplastic syndromes? A report on 59 cases. Leukemia 23:673– 678.
Ji BC, Hsu WH, Yang JS, Hsia TC, Lu CC, Chiang JH, Yang JL, Lin CH, Lin JJ, Suen LJ, et al. (2009) Gallic acid induces apoptosis via caspase-3 and mitochon-drion-dependent pathways in vitro and suppresses lung xenograft tumor growth in vivo. J Agric Food Chem 57:7596 –7604.
Jing Y, Nakajo S, Xia L, Nakaya K, Fang Q, Waxman S, and Han R (1999) Boswellic acid acetate induces differentiation and apoptosis in leukemia cell lines. Leuk Res 23:43–50.
Kaldis P (1999) The cdk-activating kinase (CAK): from yeast to mammals. Cell Mol Life Sci 55:284 –296.
Kuo TC, Yang JS, Lin MW, Hsu SC, Lin JJ, Lin HJ, Hsia TC, Liao CL, Yang MD, Fan MJ, et al. (2009) Emodin has cytotoxic and protective effects in rat C6 glioma cells: roles of Mdr1a and nuclear factorB in cell survival. J Pharmacol Exp Ther 330:736 –744.
Lolli G and Johnson LN (2005) CAK-cyclin-dependent activating kinase: a key kinase in cell cycle control and a target for drugs? Cell Cycle 4:572–577. Macallan DC, Asquith B, Irvine AJ, Wallace DL, Worth A, Ghattas H, Zhang Y,
Griffin GE, Tough DF, and Beverley PC (2003) Measurement and modeling of human T cell kinetics. Eur J Immunol 33:2316 –2326.
Nau KC and Lewis WD (2008) Multiple myeloma: diagnosis and treatment. Am Fam Physician 78:853– 859.
Perez EA (2009) Microtubule inhibitors: differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol Cancer Ther 8:2086 –2095.
Rodriguez-Collazo P, Snyder SK, Chiffer RC, Bressler EA, Voss TC, Anderson EP, Genieser HG, and Smith CL (2008) cAMP signaling regulates histone H3 phos-phorylation and mitotic entry through a disruption of G2 progression. Exp Cell Res 314:2855–2869.
Sitaresmi MN, Mostert S, Purwanto I, Gundy CM, Sutaryo, and Veerman AJ (2009) Chemotherapy-related side effects in childhood acute lymphoblastic leukemia in Indonesia: parental perceptions. J Pediatr Oncol Nurs 26:198 –207.
Sornsuvit C, Komindr S, Chuncharunee S, Wanikiat P, Archararit N, and Santa-nirand P (2008) Pilot study: effects of parenteral glutamine dipeptide supplemen-tation on neutrophil functions and prevention of chemotherapy-induced side-effects in acute myeloid leukaemia patients. J Int Med Res 36:1383–1391. Stiewe T, Parssanedjad K, Esche H, Opalka B, and Pu¨tzer BM (2000) E1A overcomes
the apoptosis block in BCR-ABL⫹ leukemia cells and renders cells susceptible to induction of apoptosis by chemotherapeutic agents. Cancer Res 60:3957–3964. Tallman MS (1996) Differentiating therapy in acute myeloid leukemia. Leukemia
10:1262–1268.
Vantieghem A, Xu Y, Assefa Z, Piette J, Vandenheede JR, Merlevede W, De Witte PA, and Agostinis P (2002) Phosphorylation of Bcl-2 in G2/M phase-arrested cells following photodynamic therapy with hypericin involves a CDK1-mediated signal and delays the onset of apoptosis. J Biol Chem 277:37718 –37731.
Wang LG, Liu XM, Kreis W, and Budman DR (1999) The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer Chemother Pharmacol 44:355–361.
Wang SW, Pan SL, Huang YC, Guh JH, Chiang PC, Huang DY, Kuo SC, Lee KH, and Teng CM (2008) CHM-1, a novel synthetic quinolone with potent and selective antimitotic antitumor activity against human hepatocellular carcinoma in vitro and in vivo. Mol Cancer Ther 7:350 –360.
Yang JS, Chen GW, Hsia TC, Ho HC, Ho CC, Lin MW, Lin SS, Yeh RD, Ip SW, Lu HF, et al. (2009) Diallyl disulfide induces apoptosis in human colon cancer cell line (COLO 205) through the induction of reactive oxygen species, endoplasmic retic-ulum stress, caspases casade and mitochondrial-dependent pathways. Food Chem Toxicol 47:171–179.
Yang JS, Hour MJ, Kuo SC, Huang LJ, and Lee MR (2004) Selective induction of G2/M arrest and apoptosis in HL-60 by a potent anticancer agent, HMJ-38. Anticancer Res 24:1769 –1778.
Yu D, Jing T, Liu B, Yao J, Tan M, McDonnell TJ, and Hung MC (1998) Overex-pression of ErbB2 blocks Taxol-induced apoptosis by up-regulation of p21Cip1, which inhibits p34Cdc2 kinase. Mol Cell 2:581–591.
Address correspondence to: Jing-Gung Chung, Department of Biological Science and Technology, College of Life Sciences, China Medical University, 91, Hsueh-Shih Road, Taichung 404, Taiwan. E-mail: [email protected]