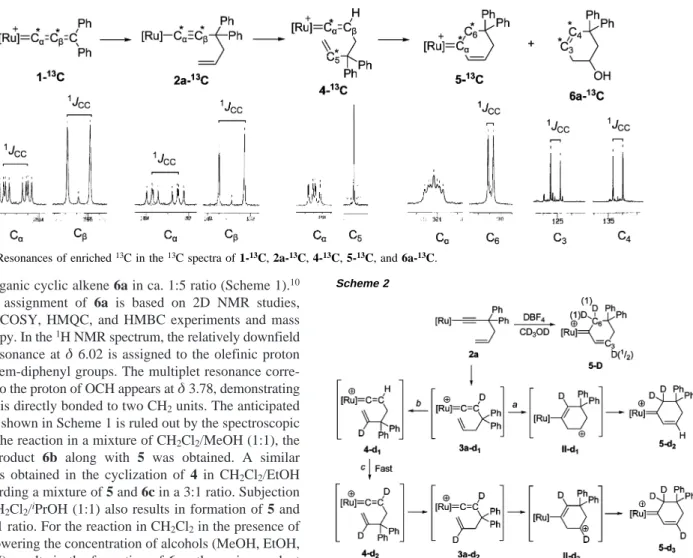
Intramolecular Cyclization of Ruthenium Vinylidene
Complexes with a Tethering Vinyl Group: Facile Cleavage
and Reconstruction of the C
-
C Double Bond
Cheng-Wei Cheng, Yi-Chun Kuo, Shu-Hao Chang, Ying-Chih Lin,* Yi-Hong Liu, and Yu Wang
Contribution from the Department of Chemistry, National Taiwan UniVersity, Taipei, Taiwan 106, Republic of China
Received July 5, 2007; E-mail: yclin@ntu.edu.tw
Abstract:Protonation of ruthenium acetylide complexes [M]-*Ct*CCPh2CH2CHdCH2(2a, [M] )(η5
-C5H5)(P(OPh)3)(PPh3)Ru; 2a′, [M])(η5-C5H5)(dppp)Ru; *C)13C-labeled carbon atom) with HBF4in ether
produces [[M]d*CdCHCH2CPh2*CHdCH2][BF4] (4, 4′) exclusively via a metathesis process of the terminal
vinyl group with the *Cd*C of the resulting vinylidene group. For 4 in methanol, bond reconstruction of the
two labeled *C atoms readily takes place via a retro-metathesis process followed by a cyclization of the resulting vinylidene ligand giving the cyclic carbene complex 5, which is fully characterized by single-crystal X-ray diffraction analysis. The protonation of 2a in MeOH is followed by a cyclization, also giving 5.
Deuterium-labeling study indicates that the C-C bond formation of this cyclization proceeds simultaneously
with the formation of 4 consistent with facile cleavage and reconstruction of CdC bonds. For comparison,
complex 4 in alcohol yields, besides 5, the corresponding alkoxycyclohexene 6. Formation of 6 from 4 also
involves a skeletal rearrangement with reconstruction of the CdC bond. Interestingly, [[Ru′]d*Cd
C(Me)-CH2CPh2*CHdCH2][BF4] (8′) originally from a complex with two connected labeled carbon atoms also
undergoes reestablishment of the *Cd*C bond yielding the cyclic allenyl complex 9′.13C-labeling studies
clearly reveal the reestablishment of two CdC double bonds in the transformation of both 4 to 5 and 8′to
9′. The proposed mechanism implicates a cyclobutylidene intermediate formed either via a regiospecific
[2+2] cycloaddition of two double bonds in the ruthenium vinylidene 4 or via a cyclization of 4 giving a
nonclassical ion intermediate followed by a 1,2-alkyl shift.
Introduction
Reactions of enynes catalyzed by electrophilic late-transition-metal complexes have attracted much attention because a variety of products can be obtained from fairly simple substrates under mild conditions.1 As a result of the compatibility and the
utilization of very diverse precursors, the metal-catalyzed cycloisomerization of enyne systems has recently expanded on synthetically versatile developments, including notably applica-tions in the total synthesis of natural products.2Metal-catalyzed
cycloisomerization of enynes often leads to various skeletal rearrangements because “nonclassical” cations may participate as reaction intermediates.3 For example,
transition-metal-catalyzed cycloisomerization of 1,6-enynes represents an ef-ficient strategy for a variety of five-membered and six-membered alkenes and dienes.4 The corresponding
metal-catalyzed cycloisomerizations of 1,5-enynes have been much less investigated; however, the known reactivities of 1,5-enynes also involve the same type of cycloisomerization.5On the other
hand, this very same diversity can be viewed as a disadvantage from the synthetic viewpoint that the outcome of a specific transformation may be difficult to predict. Therefore, further insight into the mechanism of these transformations is desirable.6
Herein, we describe the development of ruthenium-mediated isomerizations of enynes to furnish the other type of 1,5-enynes and cycloisomerizations to produce alkoxycyclohexenes. This process was supported by our recent discovery of the ruthenium-mediated skeletal rearrangement of
3,3-diphenyl-5-(1) (a) Chatani, N.; Kataoka, K.; Murai, S.; Furukawa, N.; Seki, Y. J. Am.
Chem. Soc. 1998, 120, 9104. (b) Trost, B. M.; Doherty, G. A. J. Am. Chem. Soc. 2000, 122, 3801. (c) Me´ndez, M.; Mun˜oz, M. P.; Nevado, C.; Ca´rdenas,
D. J.; Echavarren, A. M. J. Am. Chem. Soc. 2001, 123, 10511. (d) Martı´n-Matute, B.; Nevado, C.; Ca´rdenas, D. J.; Echavarren, A. M. J. Am. Chem.
Soc. 2003, 125, 5757. (e) Ma, S.; Yu, S.; Gu, Z. Angew. Chem., Int. Ed.
2006, 45, 200. (f) Zhang, L.; Sun, J.; Kozmin, S. A. AdV. Synth. Catal. 2006, 348, 2271.
(2) (a) Fu¨rstner, A.; Stelzer, F.; Szillat, H. J. Am. Chem. Soc. 2001, 123, 11863. (b) Fu¨rstner, A.; Szillat, H.; Gabor, B.; Mynott, R. J. Am. Chem. Soc. 1998,
120, 8305. (c) Charruault, L.; Michelet, V.; Taras, R.; Gladiali, S.; Geneˆt,
J. P. Chem. Commun. 2004, 850.
(3) (a) Aubert, C.; Buisine, O.; Malacria, M. Chem. ReV. 2002, 102, 813. (b) Trost, B. M.; Krische, M. J. Synlett 1998, 1. (c) Ojima, I.; Tzamarioudaki, M.; Li, Z.; Donovan, R. J. Chem. ReV. 1996, 96, 635. (d) Oi, S.; Tsukamoto, I.; Miyano, S.; Inoue, Y. Organometallics 2001, 20, 3704.
(4) (a) Driver, S. T.; Geissert, A. J. Chem. ReV. 2004, 104, 1317. (b) Nevado, C.; Ca´rdenas, D. J.; Echavarren, A. M. Chem.-Eur. J. 2003, 9, 2627. (5) (a) Mamane, V.; Gress, T.; Krause, H.; Fu¨rstner, A. J. Am. Chem. Soc.
2004, 126, 8654. (b) Harrak, Y.; Blaszykowski, C.; Bernard, M.; Cariou,
K.; Mainetti, E.; Mouries, V.; Dhimane, A. L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2004, 126, 8656. (c) Nieto-Oberhuber, C.; Mun˜oz, M. P.; Bun˜uel, E.; Nevado, C.; Ca´rdenas, D. J.; Echavarren, A. M. Angew.
Chem., Int. Ed. 2004, 43, 2402.
(6) (a) Me´ndez, M.; Mamane, V.; Fu¨rstner, A. Chemtracts: Org. Chem. 2003,
16, 397. (b) Lloyd-Jones, G. C. Org. Biomol. Chem. 2003, 1, 215. (c)
Nieto-Oberhuber, C.; Lo´pez, S.; Jime´nez-Nu´n˜ez, E.; Echavarren, A. M.
Chem.-Eur. J. 2006, 12, 5916. (d) Echavarren, A. M.; Nevadoa, C. Chem. Soc. ReV. 2004, 33, 431.
129, 14974
-Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
en-1-yne, which was proposed to proceed via an unusual mechanism involving a new metathesis process of the terminal vinyl group with the CdC of the vinylidene group.7Elucidation
of this novel reaction mechanism enabled us to extend the scope of the ruthenium fragment.
Results and Discussion
Cleavage of Two CdC Bonds. Metathesis of two CdC double bonds in a vinylidene complex of Cp(PPh3)2Ru (Cp )
η5-C
5H5) system containing a tethering vinyl group was recently
reported by us.7The same metathesis is also observed in similar
vinylidene complexes of Cp(P(OPh)3)(PPh3)Ru and
Cp(dppp)-Ru described below. The reaction of [Cp(dppp)-Ru]Cl ([Cp(dppp)-Ru] ) Cp-(P(OPh)3)(PPh3)Ru) with HCtCC(Ph)2OH in dichloromethane
at room temperature gives the ruby red allenylidene complex [[Ru]dCdCdC(Ph)2][PF6]8 (1) in high yield. Similarly, the
allenylidene complex [[Ru′]dCdCdC(Ph)2][PF6] (1′, [Ru′] )
Cp(dppp)Ru) is also obtained in high yield. Regioselective nucleophilic addition is commonly observed at Cγ of the
allenylidene ligand,9and by means of this reaction we synthesize
neutral σ-alkynyl complexes [M]-CtCC(Ph)2CH2CHdCH2
(2a, [M] ) [Ru] and 2a′, [M] ) [Ru′]) from the reactions of Grignard reagent CH2dCHCH2MgCl with 1 and 1′, respectively.
Treatment of theσ-alkynyl complex 2a′with HBF4in diethyl
ether at -20°C generates in high yield the vinylidene complex [[Ru′]dCdCHC(Ph)2CH2CHdCH2][BF4] (3a′), which in
dichlo-romethane transforms to the other vinylidene complex [[Ru′]d CdCHCH2C(Ph)2CHdCH2][BF4] (4′) at room temperature.
However, protonation of 2a yields the vinylidene complex 4 directly, and the anticipated vinylidene complex [[Ru]dCd CHC(Ph)2CH2CHdCH2][BF4] (3a) is too reactive to be isolated
(Scheme 1). The much faster rate of the formation of 4 as compared to that of the dppp analogue 4′ is ascribed to the
electronic as well as the steric effects of the ruthenium fragment. A series of 2D NMR studies, including COSY, HMQC, and HMBC experiments, unambiguously establish the structure of 4. Formation of 4′from 3a′, formally a migration of two phenyl substituents, could be interpreted by a metathesis process of two CdC bonds (Scheme 1) involving a double cleavage of two unsaturated carbon-carbon bonds.7Formation of 4 from
2a presumably proceeds via a similar pathway.
Reconstruction of Two CdC Bonds. Unexpectedly, treat-ment of 4 with MeOH affords the carbene complex 5 with an unsaturated six-membered ring ligand as a reddish orange solid almost quantitatively (Scheme 1). Complex 5 is characterized by1H,31P{1H},13C{1H}NMR, and 2D NMR spectroscopies
and analytical data. In the 13C{1H}NMR spectrum of 5, the
resonance corresponding to the RudCR carbon atom appears atδ 321.4 as a multiplet. In addition, two singlet resonances at δ 151.2 and 135.5 are assigned to the olefinic carbon atoms of the six-membered ring on the basis of the1H,13C-HMQC and 1H,13C-HMBC spectra. Complex 5 is also directly obtained via
protonation of 2a with HBF4 in MeOH at 0 °C. From a
mechanistic point of view, it should be noted that direct intramolecular cyclization of the vinylidene with the terminal vinyl group in 4 should have a preference to give the unsaturated cyclic carbene complex A (Scheme 1), which is not observed. Treatment of complex 4′with MeOH also affords analogous carbene complex with much lower yield. No attempt was made to isolate the pure product for 4′.
Slow diffusion of diethyl ether into a solution of 5 in dichloromethane allows collection of suitable single crystals for X-ray diffraction study. An ORTEP diagram is shown in Figure 1, and selected structure parameters are listed. The bond length of Ru(1)-C(1) is 1.957(4) Å showing a typical RudC double bond, and the C(2)-C(3) bond length is 1.334(5) Å showing a CdC double bond character. The bond length of C(1)-C(2) (1.441(5) Å) is shorter than 1.531(5) Å of C(1)-C(6), indicating delocalization of double bond character at Ru(1)-C(1)-C(2)-C(3).
The cyclization reaction of 4 in solvent other than MeOH gives an additional product. Treatment of complex 4 in aqueous acetone results in the formation of two products identified as 5 (7) Yen, Y. S.; Huang, S. L.; Liu, Y. H.; Sung, H. L.; Wang, Y.; Lin, Y. C.
J. Am. Chem. Soc. 2005, 127, 18037.
(8) Selegue, J. P. Organometallics 1982, 1, 217.
(9) (a) Esteruelas, M. A.; Go´mez, A.; Lo´pez, A. M.; Modrego, J.; On˜ate, E.
Organometallics 1997, 16, 5826. (b) Cadierno, V.; Gamasa, M. P.; Gimeno,
J.; Gonza´lez-Cueva, M.; Lastra, E.; Borge, J.; Garcı´a-Granda, S.; Pe´rez-Carreno˜, E. Organometallics 1996, 15, 2137.
atoms on the phosphorus ligands have been omitted for clarity. Selected bond distances (Å) and angles (deg): Ru(1)-C(1), 1.957(4); Ru(1)-P(1), 2.2349(10); Ru(1)-P(2), 2.3539(10); C(1)-C(2), 1.441(5); C(2)-C(3), 1.334(5); C(3)-C(4), 1.486(5); C(4)-C(5), 1.532(5); C(5)-C(6), 1.550-(5); C(1)-C(6), 1.5311.550-(5); C(2)-C(1)-C(6), 113.2(3); C(1)-C(2)-C(3), 124.3(3); C(2)-C(3)-C(4), 123.5(4); C(1)-C(6)-C(5), 116.9(3).
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
and the organic cyclic alkene 6a in ca. 1:5 ratio (Scheme 1).10
Structural assignment of 6a is based on 2D NMR studies, including COSY, HMQC, and HMBC experiments and mass spectroscopy. In the1H NMR spectrum, the relatively downfield
doublet resonance atδ 6.02 is assigned to the olefinic proton near the gem-diphenyl groups. The multiplet resonance corre-sponding to the proton of OCH appears atδ 3.78, demonstrating that OCH is directly bonded to two CH2units. The anticipated
product B shown in Scheme 1 is ruled out by the spectroscopic data. For the reaction in a mixture of CH2Cl2/MeOH (1:1), the
organic product 6b along with 5 was obtained. A similar outcome is obtained in the cyclization of 4 in CH2Cl2/EtOH
(1:1), affording a mixture of 5 and 6c in a 3:1 ratio. Subjection of 4 to CH2Cl2/iPrOH (1:1) also results in formation of 5 and
6d in a 3:1 ratio. For the reaction in CH2Cl2in the presence of
alcohol, lowering the concentration of alcohols (MeOH, EtOH, andiPrOH) results in the formation of 6 as the major product
(ca. 75%). Gold(I)-catalyzed 5-endo hydroxy- or alkoxycycliza-tion of 1,5-enynes provided access to funcalkoxycycliza-tionalized five-membered cyclopentenes where hydroxylation or alkoxylation took place at the external carbon of the ring skeletal.11 In
contrast, Kozmin described an intramolecular formation of either C-O or C-N bonds in the skeletal framework of the ring in gold(I)-catalyzed synthesis of heterobicyclic alkenes as a result of the 6-endo-dig carbocyclizations of 1,5-enynes.12The
forma-tion of 6 is similar to Kozmin’s result.
13C-Labeling Experiment. To obtain information regarding
the bonding relationship of the rearrangement of 3a to give 4 with subsequent transformation to 5 and 6, double labeled13
C-compounds are prepared. The13C double labeling at both CR
and Cβ for 2a-13C is readily achieved from alkylation of 1a-13C. The13C NMR spectrum of 2a-13C displays two intense
resonances atδ 92.8 and 112.9 with1J
C-C) 122.3 Hz assigned
to the two enriched CRand Cβcarbons, respectively, indicating
direct connectivity (Figure 2). Treatment of 2a-13C with HBF 4
exclusively affords 4-13C, in which the two labeled13C atoms
are at CR(a doublet of doublet resonance atδ 350.2 with2J C-P
) 21.0 Hz and2JC-P′) 14.0 Hz) and at the internal vinyl CH
unit (a singlet resonance atδ 143.6). Cleavage of two carbon atoms in the transformation of 2a to 4 is noticeably observed
by the disappearance of a C-C coupling between resonances of two enriched carbon atoms for 4-13C. As is also shown in
Figure 2, reconstruction of the two labeled carbon atoms is revealed in the13C NMR spectrum showing two resonances at
δ 321.1 and 77.2 with 1J
CC) 23.8 Hz of the product 5-13C
obtained from 4-13C. The formation of alkoxy cyclic alkene 6
presumably also involves the same skeletal rearrangement. The
13C NMR spectrum of 6a-13C obtained from 4-13C in aqueous
acetone shows two resonances atδ 134.4 and 125.2 with1J CC
) 69.4 Hz, indicating direct bonding of two 13C-enriched
carbons. The formation of product 6 is thus proposed to proceed via skeletal rearrangement of 4 rather than a simple direct C-C bond formation.
To further understand the mechanism of the formation of the unexpected finding of 5, we carry out the protonation of 2a using the deuterated tetrafluoroboric acid. The addition of excess amount of DBF4 to a methanol-d4solution of 2a produces a
mixture marked as 5-D, which 1H NMR displays deuterium
contents of 100% and 50% at the C6and C3carbons, respectively
(Scheme 2). The distribution of deuterium atoms in 5-D is supported by the1H NMR spectrum, which contains one 50%
intensity of proton signal atδ 6.34, and the results support the presence of skeletal rearrangement. Scheme 2 shows a plausible mechanism to account for the formation of 5-D. Deuteration of 2a generates 3a-d1, which could form either the cyclization
product 5-d2via pathway a or the metathesis product 4-d1via
(10) Nieto-Oberhuber, C.; Mun˜oz, M. P.; Lo´pez, S.; Jime´nez-Nu´n˜ez, E.; Nevado, C.; Herrero-Go´mez, E.; Raducan, M.; Echavarren, A. M. Chem.-Eur. J.
2006, 12, 1677.
(11) Buzas, A. K.; Istrate, F. M.; Gagosz, F. Angew. Chem., Int. Ed. 2007, 46, 1141.
(12) Zhang, L.; Kozmin, S. A. J. Am. Chem. Soc. 2005, 127, 6962.
Figure 2. Resonances of enriched13C in the13C spectra of 1-13C, 2a-13C, 4-13C, 5-13C, and 6a-13C. Scheme 2 A R T I C L E S
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
pathway b. Fast exchange of the vinylidene proton of 4-d1as
shown in pathway c followed by retro-metathesis and cyclization results in 5-d3as the final product. Thus, the deuteration of 2a
should yield 5-D: a mixture of 5-d2and 5-d3. Presumably, the
rate of proton exchange (pathway c) between the vinylidene proton and CD3OD should be much faster than the rate of the
metathesis reaction (pathway b) and the cyclization (pathway a), which are probably approximately comparable. This labeling experiment also indicates that the transformation of 4 to 5 proceeds via the reversed metathesis reaction pathway to 3a first. Therefore, subjection of 4 in CD3OD should give 5-d3
with complete D-labeling at C6and C3carbons (Scheme 2). As
shown in Scheme 3, for the formation of 4′, the double cleavage may proceed either via a [2+2] cycloaddition of the two double bond in 3a′leading to an unobserved species I followed by a retro-cycloaddition to give the observed product13 or via
formation of a bicyclic species III followed by 1,2-methylene shift.14Our studies establish that the presence of a ruthenium
vinylidene fragment is responsible for the novel skeletal reorganization to furnish another type of vinylidene complex possibly due to efficient stabilization of the cationic intermediate I. In the absence of the vinylidene fragment, the cyclization by gold-based catalyst afforded exclusively the [3.1.0] bicyclo-hexene.15
The formation of 5 shows an interesting ring closure that may be caused by two different pathways. The upper portion of Scheme 3 shows the skeletal rearrangement to give 5 with two labeled carbon atoms bound together. An alternative yet simpler
pathway is a C-C bond formation followed by 1,2-metal migration to give C with two separated labeled atoms as shown in the lower part of Scheme 3. According to the13C-labeling
results, we proposed that product 5 is derived from 4 via a skeletal rearrangement. Transformation of 4 to 5 could be interpreted by a reversed process of formation of 4 giving back 3a via the metathesis route followed by an intramolecular attack of the terminal olefin to CRgenerating the cation II (Scheme 3) containing a six-membered ring. Deprotonation at C6(see
Scheme 3) followed by protonation at C2leads to the product
5. Scheme 3 also shows an alternative pathway sequentially via V, IV, I, then the cation III, which also undergoes deprotonation and protonation to give the observed product 5.
Formation of the alkoxy-cyclic alkene products 6 can be rationalized using the mechanistic analysis presented also in Scheme 3. The transformation of complex 4 to the cationic species II is followed by a nucleophilic attack of an alkoxy group with subsequent cleavage of the ruthenium-cyclic alkene bond producing compound 6 and ruthenium species, which is not identified.
A Further Example. Two neutral σ-alkynyl complexes [M]-CtCC(Ph)2CH2C(Me)dCH2 (2b, [M] ) [Ru] and 2b′,
[M] ) [Ru′]) are synthesized from the reactions of the Grignard reagent CH2dC(Me)CH2MgCl with 1 and 1′, respectively.9With
only an additional methyl group on the internal carbon of the terminal vinyl functionality, complex 2b′, upon protonation, is converted to complex 9′containing a cyclic π-allenyl ligand (Scheme 4).7,16A mixture of diastereomers of 9 is also obtained
directly and instantaneously from protonation of 2b. In such a cyclization, no C-C bond cleavage was observed. We are surprised to observe the completely different reactivity between the allyl- and 2-methylallyl-substituted vinylidene complexes. It is therefore interesting to explore the chemical reactivity of similar metal vinylidene complex containing a methyl group, instead of a hydrogen, at Cβ, that is, [[Ru′
]dCdC(Me)-CH2C(Ph)2CHdCH2][BF4]. For this reason, the following
experiments shown in Scheme 4 are carried out. Deprotonation of 4′generates the acetylide complex 7′. Methylation of 7′using methyl triflate takes place at Cβ, giving the unstable vinylidene
complex 8′, which undergoes a cyclization to also generate 9′. (13) Sun, J.; Conley, M. P.; Zhang, L.; Kozmin, S. A. J. Am. Chem. Soc. 2006,
128, 9705.
(14) (a) Alvarez, P.; Lastra, E.; Gimeno, J.; Bassetti, M.; Falvello, L. R. J. Am.
Chem. Soc. 2003, 125, 2386. (b) Bran˜a, P.; Gimeno, J.; Sordo, J. A. J. Org. Chem. 2004, 69, 2544.
(15) Luzung, M. R.; Markham, J. P.; Toste, F. D. J. Am. Chem. Soc. 2004, 126, 10858.
(16) Cadierno, V.; Conejero, S.; Dı´ez, J.; Gamasa, M. P.; Gimeno, J.; Garcı´a-Granda, S. Chem. Commun. 2003, 840.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
The anticipated complex D with two separated labeled carbon atoms shown in Scheme 4 is not observed.
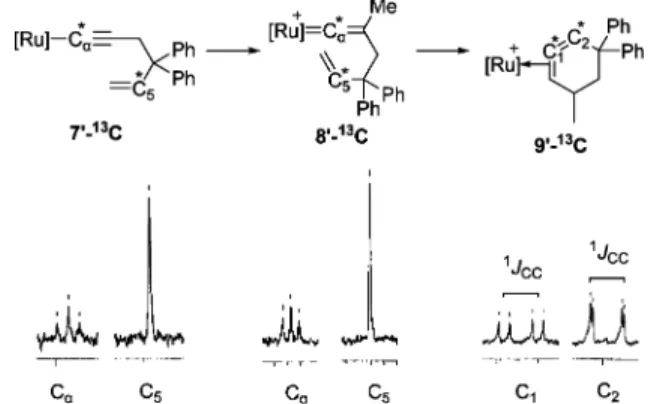
The13C-labeling experiment also elucidates the mechanism
of such a ring closure process. Using 25% doubly13C-enriched
sample, separation of two13C-labeled atoms in 4′-13C obtained
from 3a′-13C is confirmed by the disappearance of the1J C-C
(Figure 3). Deprotonation of 4′-13C gives 7′-13C, and then
methylation of 7′-13C at C
βyields the vinylidene complex 8′ -13C. A retro-metathesis process establishing the original C-C
bond of the two enriched13C atoms is followed by a cyclization
of the resulting vinylidene ligand giving complex 9′-13C. As
mentioned before, the cyclic allenyl complex 9′can also be readily prepared from the vinylidene complex 2b′.13C-labeling
studies clearly reveal the reestablishment of the CdC double bond again in the transformation of 8′ to 9′. The 13C-13C
coupling constant of 81.6 Hz of 9′-13C as shown in Figure 3
falls in the range of1J
C-Ccoupling.
Cyclization of the Alkynyl Vinylidene Moiety. The vi-nylidene complex 3c containing a terminal alkynyl group could also be obtained from alkylation of 1 using HCtCCH2MgBr
followed by protonation. Treatment of 3c with aqueous acetone gives the cyclic carbene complex 10a in high yield; see Scheme 5. The structure of 10a is established by its1H,13C, and 2D
NMR spectra. In the13C NMR spectrum, the broad resonance
atδ 300.5 is assigned to the carbene carbon. In 10a with two different phosphorus ligands, the restricted rotation of the Rud C bond caused by the presence of the gem-diphenyl group generates diastereoisomers making the CR carbon resonate as a broad peak. The same synthetic strategy is used to obtain other cyclic carbene complexes 10b and 10c containing methoxy and ethoxy functionalities from the reaction of 3c with MeOH and EtOH, respectively. In the presence of d4-methanol, complex
10b-D with a completely deuterated methylene group at Cβin
the ring is obtained. The presence of two deuterium atoms in the six-membered ring is consistent with facile protonation of the vinyl carbon in VI shown in Scheme 5. The anticipated cyclic organic compound E via a demetallation process was not observed. However, nucleophilic cyclization of enediynes catalyzed by [TpRu(PPh3)(CH3CN)2][PF6] resulting in
aroma-tized product similar to E has been reported by Liu.17
Conclusion
In summary, we report that the protonation of 2a in Et2O
affords 4 via an unusual metathesis process of the terminal vinyl group and the CdC of the vinylidene group of the proposed intermediate complex 3a. Two CdC bonds are reconstructed during the transformation of 4 in alcohols to 5 and 6, which involves a skeletal rearrangement followed by a C-C bond formation giving cyclic compounds, and the results are supported by13C- and2H-labeling experiments. However, protonation of
the acetylide complex 2b′with a 2-methylallyl group readily affords the cyclic allenyl complex 9′. Interestingly, reestablish-ment of two CdC double bonds could also be observed in the vinylidene complex with a methyl and the same butenyl group at Cβ, giving also 9′. Metathesis of two CdC bonds takes place
for the transformation of 3a′to 4′. Deprotonation of 4′yields the acetylide complex 7′, which undergoes methylation at Cβ
to give the vinylidene product 8′. The retro-metathesis process of 8′establishing two original CdC bonds in 3a′is followed by a cyclization of the resulting vinylidene ligand also producing 9′.13C-labeling studies clearly reveal reestablishment of the Cd
C double bond in the transformation of 8′to 9′. Importantly, the vinylidene moiety is uniquely responsible for the novel mechanism of the cycloisomerization, which is proposed to involve a regiospecific [2+2] cycloaddition of two double bonds in 4 or 4′, or a cyclization of ruthenium vinylidene 4 or 4′to give a nonclassical ion followed by a cascade of 1,2-alkyl shift to form the cyclobutylidene intermediate I.
Experimental Section
General Procedures. The manipulations were performed under an atmosphere of dry nitrogen using a vacuum-line and standard Schlenk techniques. Solvents were dried by standard methods and distilled under nitrogen before use. All reagents were obtained from commercial suppliers (TMS13Ct13CH from Isotec) and used without further purification. The complexes Cp(PPh3)2RuCl,18a Cp(P(OPh)3)(PPh3 )-RuCl,18b and Cp(dppp)RuCl18cwere prepared by literature methods. The C and H analyses were carried out with a Perkin-Elmer 2400 microanalyzer. Mass spectra (FAB) were recorded using a JEOL SX-102A spectrometer; 3-nitrobenzyl alcohol (NBA) was used as the matrix. NMR spectra were recorded on a Bruker AC-300 instrument at 300 MHz (1H), 121.5 MHz (31P), or 75.4 MHz (13C) using SiMe
4or 85% H3PO4as a standard or an Avance 500 FT-NMR spectrometer. Synthesis of Ruthenium Vinylidene Complexes 4 and 4′. [[Ru]d CdCHCH2C(Ph)2CHdCH2][BF4] (4). A solution of HBF4‚Et2O (0.200 mL, 1.0 M) in diethyl ether was added dropwise at -20°C to a stirred solution of 2a (0.100 g, 0.103 mol) in 20 mL of diethyl ether. Immediately, an insoluble solid formed, but the addition was continued until no solid was further formed. The solution was decanted, and the orange solid 4 was washed with diethyl ether (3× 5 mL) and dried in
(17) Odedra, A.; Wu, C. J.; Pratap, T. B.; Huang, C. W.; Ran, Y. F.; Liu, R. S.
J. Am. Chem. Soc. 2005, 127, 3406.
(18) (a) Bruce, M. I.; Wallis, R. C. Aust. J. Chem. 1979, 32, 1471. (b) Joslin, F. L.; Mague, J. T.; Roundhill, D. M. Organometallics 1991, 10, 521. (c) Oshima, N.; Suzuki, H.; Moro-Oka, Y. Chem. Lett. 1984, 1161. Figure 3. Signal of resonances of enriched13C in the13C spectra of 7′
-13C, 8′-13C, and 9′-13C. Scheme 5
A R T I C L E S
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
vacuum, and then the residue mixture was added to 30 mL of diethyl ether. The chestnut brown precipitate thus formed was filtered and washed with diethyl ether (2× 5 mL) and dried under vacuum to give 4′(0.09 g, yield 90%).1H NMR (δ, CDCl 3): 7.42-7.05 (m, 30H, Ph), 6.33 (dd,3J HH) 17.5 Hz,3JHH) 10.8 Hz, 2H, dCH2), 5.20 (d,3JHH ) 10.8 Hz, 1H, dCH2), 5.18 (s, 5H, Cp), 4.77 (d,3JHH) 17.5 Hz, 1H, dCH2), 4.21 (t,3JHH) 7.8 Hz, 1H, dCβH), 3.13 (d,3JHH) 7.8 Hz, 2H, CH2), 2.67-1.75 (m, 6H, dppp).13C{1H}NMR (δ, CD2Cl2): 343.2 (t,2J PC) 16.0 Hz, CR), 145.5-126.5 (m, Ph), 143.4 (s, -CHdCH2), 114.8 (s, dCH2), 108.0 (s, Cβ), 92.6 (s, Cp), 53.7 (s, Cγ), 29.7 (s, CH2), 26.7-20.3 (m, dppp).31P{1H}NMR (δ, CDCl 3): 36.6. Anal. Calcd for C50H47BF4P2Ru: C, 66.89; H, 5.28. Found: C, 66.74; H, 5.37.
Synthesis of Cyclic Carbene Complex 5. Method 1: A solution of HBF4‚Et2O in diethyl ether was added dropwise to a solution of 2a (0.100 g, 0.103 mol) in methanol (20 mL) at 0°C. The resulting solution was further stirred for 1 h at room temperature and then dried under vacuum. The resulting powder was washed with diethyl ether (3× 5 mL) and was identified as 5 (0.104 g, 92%). Method 2: A solution of complex 4 (0.100 g, 0.095 mmol) in MeOH was stirred for 1 h at room temperature under nitrogen. The resulting solution was dried under vacuum to give complex 5 quantitatively.1H NMR (δ, CDCl
3): 7.36-6.68 (m, 41H, Ph and dCβH), 6.31 (br, 1H, dCγH), 5.12 (s, 5H, Cp), 4.76 (d,2J HH) 18.1 Hz, 1H, CRCH2), 2.73 (d,2JHH) 20.0 Hz, 1H, CδH2), 2.68 (d,2JHH) 18.1 Hz, 1H, CRCH2), 1.75 (d,2JHH) 20.0 Hz, 4J HH) 5.5 Hz, 1H, CδH2).13C{1H}NMR (δ, CDCl3): 321.4 (br, CR), 151.2 (Cβ), 151.1-120.1 (Ph), 135.5 (Cγ), 94.7 (Cp), 70.6 (CRCH2), 47.5 (C), 39.2 (Cδ).31P{1H}NMR (δ, CDCl3): 135.2 (d,2JPP) 54.2 Hz, P(OPh)3), 51.2 (d,2JPP) 54.2 Hz, PPh3). MS (FAB+) m/z: 971.3 (M+), 739.3 (CpRuPPh3P(OPh)3). Anal. Calcd for C59H51BF4O3P2Ru: C, 66.99; H, 4.86. Found: C, 66.83; H, 4.85.
General Procedure for the Synthesis of 5-Alkoxy-3,3-diphenyl-cyclohex-1-ene (6). A solution of 4 (0.100 g, 0.095 mmol) in CH2Cl2 (5 mL) was treated with aqueous acetone (6a) or alcohol (0.2 mL, 6b, 6c, and 6d), and the resulting solution was stirred for 24 h at room temperature under nitrogen. After evaporation of the solvent, the residue was identified as the mixture of 5 and alkoxy cyclic alkene 6, which was extracted twice with 10 mL of hexanes and dried in vacuo to give the crude 6. The crude compound was purified by flash chromatography (silica gel, hexanes/EtOAc 20/1) to afford 6.
5,5-Diphenylcyclohex-3-enol (6a). Yield: 0.019 g (84%).1H NMR (δ, CDCl3): 7.31-6.87 (m, 10H, Ph), 6.02 (d,2JHH) 10.0 Hz, 1H, CH2CHdCH), 5.68 (m, 1H, CH2CHdCH), 3.80 (br s, 1H, OH), 3.78 (m, 1H, OCH), 2.55 (dd,2J HH) 12.3 Hz, 1H, CH2), 2.30-2.21 (m, 2H, CH2), 1.97 (m, 1H, CH2).13C{1H}NMR (δ, CDCl3): 134.4 (CH2 -CHdCH), 125.2 (CH2CHdCH), 149.5-120.4 (m, Ph), 64.6 (CHOH), 51.0 (C), 45.5 (CH2), 35.1 (CH2). EI-MS m/z (%, relative intensity): 250 (M+, 27%). Anal. Calcd for C18H18O: C, 86.36; H, 7.25. Found: C, 86.23; H, 7.15. 5-Methoxy-3,3-diphenylcyclohex-1-ene (6b). Yield: 0.016 g (64%). 1H NMR (δ, CDCl 3): 7.36-6.90 (m, 10H, Ph), 6.08 (d,2JHH) 9.2 Hz, 1H, CH2CHdCH), 5.71 (m, 1H, CH2CHdCH), 3.50 (m, 1H, OCH), 3.10 (s, 3H, OCH3), 2.91 (d,2JHH) 12.5 Hz, 1H, CH2), 2.40 (dt,2JHH
relative intensity): 278 (M+, 14%). Anal. Calcd for C20H22O: C, 86.29; H, 7.97. Found: C, 86.34; H, 7.86. 5-Isopropoxy-3,3-diphenylcyclohex-1-ene (6d). Yield: 0.020 g (75%).1H NMR (δ, CDCl 3): 7.32-6.98 (m, 10H, Ph), 6.11 (d,2JHH ) 9.4 Hz, 1H, CH2CHdCH), 5.88 (m, 1H, CH2CHdCH), 3.65 (m, 1H, OCH(Me)2), 3.52 (m, 1H, OCH), 2.69 (d,2JHH) 12.3 Hz, 1H, CH2), 2.42 (dt,2JHH) 17.3 Hz,3JHH) 5.2 Hz, 1H, CH), 2.16 (m, 1H, CH2), 2.05 (dd,2JHH) 17.3 Hz,2JHH) 9.5 Hz, 1H, CH2), 1.05 (m, 6H, 2CH3).13C{1H}NMR (δ, CDCl3): 134.2 (CH2CHdCH), 125.4 (CH2CHdCH), 149.5-120.2 (Ph), 69.1 (OCH), 68.1 (OCH(CH3)2), 50.5 (C), 42.8 (CH2), 32.2 (CH2), 22.4 (CH3), 22.2 (CH3). EI-MS m/z (%, relative intensity): 292 (M+, 5%). Anal. Calcd for C21H24O: C, 86.26; H, 8.27. Found: C, 86.33; H, 8.19.
Preparation of [Ru′]-CtCCH2C(Ph)2CHdCH2(7′). Complex 4′
(0.300 g, 0.334 mmol) was treated with excess sodium methoxide (0.020 g, 0.370 mmol) in methanol (10 mL), and the light-yellow precipitate formed immediately. Stirring was continued until no further solid was formed. The precipitate was filtered, washed with methanol (2× 5 mL), and dried under vacuum to give complex 7′(0.242 g, yield 89%). 1H NMR (δ, CDCl 3): 7.88-7.00 (m, 30H, Ph), 5.22 (d,2JHH) 17.5 Hz, 1H, dCH2), 5.13 (d,2JHH) 10.7 Hz, 1H, dCH2), 4.67 (s, 5H, Cp), 3.81 (s, 2H, CH2), 1.99-2.70 (m, 6H, dppp).13C{1H}NMR (δ, CD2Cl2): 147.3-125.6 (m, Ph), 147.1 (s, dCH), 114.8 (s, dCH2), 104.3 (s, Cβ), 97.9 (t,2JPC) 24.5 Hz, CR), 83.7 (s, Cp), 55.7 (s, CPh2), 35.5 (s, CH2), 26.4-21.5 (m, dppp).31P{1H}NMR (δ, CDCl3): 46.9. Anal. Calcd for C50H46P2Ru: C, 74.15; H, 5.72. Found: C, 74.32; H, 5.88. Preparation of [[Ru′]dCdC(Me)CH2C(Ph)2CHdCH2][BF4] (8′).
A flask was charged with 7′(0.810 g, 1.00 mmol) in dichloromethane (50 mL) under nitrogen. MeOTf (0.15 mL, 1.1 mmol) was added dropwise at room temperature, and the color of the solution changed from yellow to orange immediately. After the mixture was stirred about 10 min, the volume of the solvent was reduced to 5 mL under vacuum, and then the residue mixture was added to 30 mL of diethyl ether. The precipitate thus formed was filtered, washed with diethyl ether (2× 5 mL), and dried under vacuum to give the final product 8′as a chestnut brown powder (0.870 g, yield 89%).1H NMR (δ, CDCl
3): 7.39-6.70 (m, 30H, Ph), 6.54 (dd,3J HH) 17.5 Hz,3JHH) 10.7 Hz, 1H, dCH2), 5.28 (s, 5H, Cp), 5.25 (d,3J HH) 10.7 Hz, 1H, dCH2), 4.52 (d,3JHH) 17.5 Hz, 1H, dCH2), 2.80 (s, 2H, CH2), 2.87-1.93 (m, 6H, dppp), 1.07 (s, 3H, CH3).13C{1H}NMR (δ, CD2Cl2): 348.2 (t,2JCP) 15.5 Hz, CR), 147.3-117.8 (Ph), 146.2 (s, dCH), 117.6 (s, dCH2), 115.7 (s, Cβ), 93.1 (s, Cp), 54.0 (s, Cγ). 31P{1H} NMR (δ, CDCl3): 35.5. Anal. Calcd for C50H47BF4P2Ru: C, 64.12; H, 5.07. Found: C, 64.23; H, 5.21.
Synthesis ofπ-Cyclic Allene Complex 9. A solution of HBF4‚Et2O in diethyl ether was added dropwise at -20°C to a stirred solution of 2b (0.100 g, 0.102 mmol) in 20 mL of diethyl ether. Immediately, an insoluble solid formed, but the addition was continued until no further solid was formed. The solution was then decanted, and the brown solid was washed with diethyl ether (3× 5 mL) and dried in vacuo to give 9. Yield: 0.100 g (88%). Spectroscopic data for 9, 1H NMR (δ, CDCl3): 7.80-6.73 (m, 40H, Ph), 6.50 (s, 1H, CH), 5.30 (s, 5H, Cp),
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
2.75 (d,2J HH) 12.9 Hz, 1H, CH2), 2.43 (m, 1H, dCH), 2.18 (br, 1H, CHCH3), 1.74 (m, 1H, CH2), 1.38 (d,3JHH) 6.1 Hz, 3H, CH3).13 C-{1H}NMR (δ, CDCl 3): 151.5-120.1 (Ph), 90.4 (s, Cp), 58.4 (s, Cγ), 51.3 (s, CH2), 42.5 (s, CH), 35.6 (s, CH), 20.3 (s, CH3).31P{1H}NMR (δ, CDCl3): 121.7 (d,2JPP) 60.5 Hz, P(OPh)3), 45.0 (d,2JPP) 60.5 Hz, PPh3). Spectroscopic data of the other diastereomer,1H NMR (δ, CDCl3): 7.80-6.73 (m, 40H, Ph), 5.27 (s, 5H, Cp), 4.96 (s, 1H, CH), 2.85 (m, 1H, dCH), 2.71 (d,2J HH) 13.0 Hz, 1H, CH2), 2.06 (m, 1H, CH2), 1.68 (br, 1H, CHCH3), 1.37 (d,3JHH) 6.0 Hz, 3H, CH3).13 C-{1H}NMR (δ, CDCl 3): 151.5-120.1 (Ph), 91.2 (s, Cp), 58.2 (s, Cγ), 50.9 (s, CH2), 42.6 (s, CH), 35.5 (s, CH), 19.8 (s, CH3).31P{1H}NMR (δ, CDCl3): 124.7 (d,2JPP) 58.2 Hz, P(OPh)3), 45.4 (d,2JPP) 58.2 Hz, PPh3). MS (FAB+) m/z: 985.3 (M+), 739.3 (CpRuPPh3P(OPh)3). Anal. Calcd for C60H53BF4O3P2Ru: C, 67.23; H, 4.98. Found: C, 67.29; H, 4.87.
General Procedure for the Synthesis of Alkoxy Cyclic Carbene Complexes 10. A solution of complex 3c (0.100 g, 0.095 mmol) in alcohol or aqueous acetone (20 mL)was stirred for 1 h at room temperature under a nitrogen atmosphere. The resulting solution was reduced to about 3 mL, which was slowly added to 50 mL of vigorously stirred diethyl ether to afford the reddish orange precipitate. The powder was filtered off and washed with diethyl ether and hexanes, to give the corresponding alkoxy cyclic carbene complex 10.
Complex 10a. Yield: 0.093 g (91%).1H NMR (δ, CDCl
3): 7.51-6.67 (m, 41H, Ph and CβH), 5.24 (s, 5H, Cp), 4.82 (d,2JHH) 18.2 Hz, 1H, CRCH2), 3.41 (d,2JHH) 18.2 Hz, 1H, CRCH2), 2.95 (d,2JHH) 17.7 Hz, 1H, CδH2), 2.20 (d,2JHH) 17.7 Hz, 1H, CδH2).13C{1H}NMR (δ, CDCl3): 300.5 (br, CR), 172.2 (Cγ), 146.6 (Cβ), 151.2-120.8 (Ph), 92.2 (Cp), 66.0 (CRCH2), 48.4 (C), 42.7 (Cδ). 31P{1H} NMR (δ, CDCl3): 136.9 (d,2JPP) 59.5 Hz, P(OPh)3), 55.7 (d,2JPP) 59.5 Hz, PPh3). MS (FAB+) m/z: 987.3 (M+), 739.3 (CpRuPPh3P(OPh). Anal. Calcd for C59H51BF4O4P2Ru: C, 65.99; H, 4.79. Found: C, 65.88; H, 4.75. Complex 10b. Yield: 0.096 g (93%).1H NMR (δ, CDCl 3): 7.78-6.52 (m, 41H, Ph and CβH), 5.05 (s, 5H, Cp), 4.72 (d,2JHH) 18.3 Hz, 1H, CRCH2), 3.38 (s, 3H, CH3), 3.36 (d,2JHH) 18.3 Hz, 1H, CRCH2), 2.78 (d,2J HH) 18.0 Hz, 1H, CδH2), 2.12 (d,2JHH) 18.0 Hz, 1H, CδH2).13C{1H}NMR (δ, CDCl3): 300.5 (br, CR), 168.4 (Cγ), 145.2 (Cβ), 151.5-120.1 (m, Ph), 92.2 (Cp), 67.1 (CRCH2), 57.6 (OCH3), 47.5 (C), 42.0 (Cδ).31P{1H}NMR (δ, CDCl3): 136.8 (d,2JPP) 58.5 Hz, P(OPh)3), 55.3 (d,2JPP) 58.5 Hz, PPh3). MS (FAB+) m/z: 1001.3 (M+), 739.3 (CpRuPPh3P(OPh)3). Anal. Calcd for C60H53BF4O4P2Ru: C, 66.24; H, 4.91. Found: C, 66.27; H, 4.82. Complex 10c. Yield: 0.093 g (89%).1H NMR (δ, CDCl 3): 7.68-6.52 (m, 41H, Ph and CβH), 4.94 (s, 5H, Cp), 4.64 (d,2JHH) 18.0 Hz, 1H, CRCH2), 3.73 (q, 2H,2JHH) 6.5 Hz, OCH2CH3), 3.48 (d,2JHH) 18.0 Hz, 1H, CRCH2), 2.77 (d,2JHH) 18.1 Hz, 1H, CδH2), 2.25 (d, 2J HH) 18.1 Hz, 1H, CδH2), 1.32 (t,2JHH) 6.5 Hz, 3H, CH3).13C{1H} NMR (δ, CDCl3): 300.7 (br, CR), 168.0 (Cγ), 145.3 (Cβ), 151.5-121.7 (m, Ph), 91.8 (Cp), 67.0 (CRCH2), 66.6 (OCH2CH3), 47.6 (C), 42.2 (Cδ), 14.1 (CH3).31P{1H}NMR (δ, CDCl3): 136.8 (d,2JPP) 58.8 Hz, P(OPh)3), 54.8 (d,2JPP) 58.8 Hz, PPh3). MS (FAB+) m/z: 1015.3 (M+), 739.3 (CpRuPPh3P(OPh)3). Anal. Calcd for C61H55BF4O4P2Ru: C, 66.49; H, 5.03. Found: C, 66.55; H, 5.12.
Single-Crystal X-ray Diffraction Analysis of 5. Single crystals of 5 suitable for an X-ray diffraction study were grown as mentioned above. A single crystal of dimensions 0.20× 0.15 × 0.10 mm3was glued to a glass fiber and mounted on an SMART CCD diffractometer. The diffraction data were collected using 3 kW sealed-tube Mo KR radiation (T ) 295 K). Exposure time was 5 s per frame. SADABS19 (Siemens area detector absorption) absorption correction was applied, and decay was negligible. Data were processed, and the structure was solved and refined by the SHELXTL20 program. The structure was solved using direct methods and confirmed by Patterson methods refining on intensities of all data to give R1 ) 0.1050 and wR2 ) 0.1291 for 11 596 unique observed reflections (I > 2σ(I)). Hydrogen
atoms were placed geometrically using the riding model with thermal parameters set to 1.2 times that for the atoms to which the hydrogen is attached and 1.5 times that for the methyl hydrogens.
Acknowledgment. This research is supported by the National Science Council and National Center of High-Performance Computing of Taiwan, the Republic of China.
Supporting Information Available: Complete crystallo-graphic data for 5 (CIF) and preparations and spectroscopic data of complexes 1, 1′, 2a, 2a′, 2b, 2b′, 2c, 3a′, 3c, and 9′. This material is available free of charge via the Internet at http://pubs.acs.org.
JA074951P
(19) The SADABS program is based on the method of Blessing, see: Blessing, R. H. Acta Crystallogr., Sect. A 1995, 51, 33.
(20) SHELXTL: Structure Analysis Program, version 5.04; Siemens Industrial Automation Inc.: Madison, WI, 1995.
A R T I C L E S
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009