Abstract. The present data showed that a novel synthesized compound, 6-acetyl-9-(3,4,5-trimethoxybenzyl)-9H-pyrido [2,3-b]indole (HAC-Y6), exhibited potent antitumor activity against human hepatocellular carcinoma (HCC) cells in vitro. Western blot and immunofluorescence experiments showed that HAC-Y6 depolymerized microtubules similarly to the effects of colchicine. HAC-Y6-treatment in Hep3B cells resulted in the accumulation of the G2/M phase and induced apoptosis. In addition, HAC-Y6-treatment influenced the expression of cell cycle and apoptosis related proteins in Hep3B cells. HAC-Y6 exposure increased caspases-3, -8, -9 and Bax protein levels, while reducing levels of Bcl-2 family proteins. Moreover, Bid, a substrate of caspase-8, was also activated by HAC-Y6. Treatment of cells caused the up-regulation of the death receptor 4 (DR4) and phosphorylation of p38. Taken together, we show that HAC-Y6 exhibited its antitumor activity by disrupting microtubule assembly,
causing cell cycle arrest and apoptosis through both extrinsic and intrinsic pathways in Hep3B cells. Therefore, the novel compound HAC-Y6 is a promising microtubule inhibitor that has great potential for treatment of HCC.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common type of cancer and the third leading cause of cancer mortality in the world (1). HCC prevails in developing countries of Asia and Africa, with high occurrence in Japan, Western Europe and the United States (2). Current treatments, such as surgical excision and liver transplantation, have generally not been effective (3). Significant advances have been made in chemo-therapy of HCC but adverse side-effects have limited its overall usefulness. There is a critical need to develop alterna-tive strategies for treating HCC.
Microtubules, major dynamic components of the eukaryotic cytoskeleton, are composed of a backbone of tubulin hetero-dimers and microtubule-associated proteins (4). Microtubules and their dynamics are directly involved in a variety of bio-logical processes, including the maintenance of cell shape, mitosis, signaling, intracellular transport and cell movement (5). Since microtubules are extremely important in the process of mitosis, disrupting the assembly of microtubules can induce cell cycle arrest in M phase and trigger signals for programmed cell death. Their importance in mitosis and cell division has made microtubules a target of numerous anti-tumor drugs (6). Anti-microtubule drugs generally fall into two main groups: promoters and inhibitors of microtubule assembly. The microtubule inhibitors, such as the vinca alkaloids, cryptophycins and colchicines, inhibit the poly-merization of microtubules (7). In contrast, the microtubule promoters, such as taxanes (taxol and taxotere), epothilones, eleutherobins, laulimalide and discodermolide, promote the polymerization of microtubules (8).
Apoptosis plays an important role in embryo develop-ment. A balance between survival and apoptosis is critical for the maintenance of physiological functions. Imbalance towards cell survival can result in cancer development and
Induction of apoptosis by HAC-Y6, a novel microtubule inhibitor,
through activation of the death receptor 4 signaling pathway
in human hepatocellular carcinoma cells
JUI-YING TSAI1, CHAO-MING HUNG4, SHIH-TING BAI3, CHI-HUNG HUANG5, WEI-CHIH CHEN2, JINH-GUNG CHUNG3, SHENG-CHU KUO1, TZONG-DER WAY3 and LI-JIAU HUANG1
1Graduate Institute of Pharmaceutical Chemistry and 2The PhD Program for Cancer Biology and Drug Discovery, College of Pharmacy, 3Department of Biological Science and Technology, College of Life Sciences, China Medical University, Taichung; 4Department of General Surgery, E-Da Hospital, I-Shou University,
Kaohsiung; 5Taiwan Advance Biopharm, Inc., Xizhi City, Taipei, Taiwan, R.O.C. Received April 14, 2010; Accepted June 2, 2010
DOI: 10.3892/or_00000969
_________________________________________
Correspondence to: Dr Tzong-Der Way, Department of Bio-logical Science and Technology, College of Life Sciences; or Dr Li-Jiau Huang, Graduate Institute of Pharmaceutical Chemistry, College of Pharmacy, China Medical University, no. 91 Hsueh-Shih Road, Taichung, Taiwan 40402, R.O.C.
E-mail: [email protected] E-mail: [email protected]
Abbreviations: DAPI, 4,6-diamidino-2-phenylindole; DMEM, Dulbecco's modified Eagle's medium; DR, death receptor; EDTA, ethylenediaminetetraacetic acid; ECL, enhanced chemiluminescence; FBS, fetal bovine serum; HCC, hepatocellular carcinoma; PBS, phosphate buffer saline; PI, propidium-iodide; rpm, revolutions per minute; Tris, Tris(hydroxymethyl)aminomethane; TCA, trichloroacetic acid; TNFR1, tumor necrosis factor receptor 1; μM, micromolar
Key words:HAC-Y6, hepatocellular carcinoma cells, microtubule inhibitor, death receptor 4, p38
resistance to anticancer therapies such as radiotherapy and chemotherapy (9). Apoptosis can be triggered in a cell through either the extrinsic pathway or the intrinsic pathway. The extrinsic pathway is initiated by death receptors located on the cell membrane [e.g., Fas/CD95, death receptor 4/5 (DR4/5) and tumor necrosis factor receptor 1 (TNFR1)] with sequential activation of the initiator caspase-8 and the effector caspase-3. The intrinsic pathway is mitochondria-dependent for ampli-fication of the caspase cascade and cell death (10). Both pathways are activated by specific stimuli independently and through cross-talk between the two pathways. The growing knowledge on the relation between apoptosis and carcino-genesis has made it possible to develop novel approaches to treat cancer.
Carbazole alkaloids are well-known in nature (11) and some of these compounds possess anticancer activity (12-16). ·-Carboline can be regarded as a bioisostere of carbazole with the benzene ring of carbazole replaced by a pyridine ring. This study investigated effects of a newly synthesized · -carboline analog 6-acetyl-9-(3,4,5-trimethoxybenzyl)-9H-pyrido[2,3-b]indole (HAC-Y6), on microtubule assembly and HCC cell growth. We demonstrated that HAC-Y6 is a potent anti-microtubule agent that arrests mitotic cells in the G2/M phase, by depolymerizing microtubules in Hep3B cells. Moreover, HAC-Y6 induced apoptosis in HCC via the extrinsic DR4-dependent pathway. HAC-Y6 induced apoptosis via the mitochondrial pathway involving trans-location of Bax, and the cleavage of Bid and caspase activation. We also show that Bid is crucial for the extrinsic ‘component’ of the process, which suggests that the protein may act as a point of convergence of the intrinsic and extrinsic pathways. Materials and methods
Chemicals. Cell culture materials were obtained from Invitrogen (Burlington, Ontario, Canada). Antibodies and reagents were purchased from commercial sources: anti-bodies against cyclin B1, securin, poly(ADP)ribose poly-merase (PARP), Bcl-2, and cleaved caspase-3, caspase-8, caspase-9, Fas/CD95, TNFR1, DR4, DR5, ERK1/2, JNK and p-JNK were purchased from Cell Signaling Technology (Beverly, MA); antibodies against BuBR1 and Bid were from BD Biosciences (Los Angeles, CA); antibody against ·-tubulin was from Abcam plc (Cambridge, UK); antibody against Bax was from Upstate Biotechnology (Charlottesville, VA); antibodies against Mcl-1, Bcl-xL, p-ERK1/2, p38, p-p38, anti-mouse and anti-rabbit antibodies conjugated to horse-radish peroxidase were obtained from Santa Cruz Biotechno-logy (Santa Cruz, CA); ß-actin antibody, 4,6-diamidino-2-phenylindole (DAPI), propidium-iodide (PI), hydroxyurea, SB203580, paclitaxel and colchicine were from Sigma Chemical Co. (St. Louis, MO). The caspase inhibitor z-VAD-fmk, z-DEVD-z-VAD-fmk, z-IETD-fmk and z-LEHD-fmk were from Promega Corp. (Madison, WI).
Cell culture. All cell lines were purchased from the American Type Culture Collection (Manassas, VA). Hep3B, MCF-7, SKOV3 and HeLa cell lines were cultured in DMEM. Normal human diploid skin fibroblast cell lines Detroit 551 cells were cultured in DME. PC-3 cells were cultured in RPMI-1640.
H23 cells were cultured in RPMI-1640 medium with 2 mM L-glutamine and supplemented with 1 mM sodium pyruvate, 4.5 g/l glucose. All media were supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Invitrogen Carlsbad, CA) at 37˚C. Cells were incubated in 75-cm2flasks at 37˚C with 5% CO
2and were passaged by
0.25% Trypsin-EDTA when they reached 80% confluence. Cell viability. Cells were treated with different concentrations of HAC-Y6 for 48 h before examining cell proliferation by MTT assay according to the manufacturer's instructions (Promega). Absorbance was measured at 590 nm.
Flow cytometry. Cell cycle analysis was determined by flow cytometry. Cells were cultured in 60-mm Petri dishes and incubated with HAC-Y6 for various times. After being trypsinized and washed twice with ice-cold PBS, the cells were fixed with 70% ice-cold ethanol overnight at -20˚C. After centrifugation, the cell pellets were treated by RNAase A and exposed to propidium iodide (PI), and then analysed by flow cytometry (FACScan, BD Biosciences, Mountain View, CA). The percentage was analyzed by Cell Quest software (BD Biosciences, Mountain View, CA).
In vivo microtubule assembly assay. The cell lysate in which the cytosolic and cytoskeletal fractions containing soluble (s) and polymerized (p) tubulin, respectively, was separated by centrifugation, resolved by electrophoresis through SDS-PAGE and immunoblotted with an antibody against ·-tubulin. Confocal microscopy. After treatment, cells were fixed with methanol, blocked with 3% bovine serum albumin, first stained with anti-·-tubulin monoclonal antibody and then FITC-conjugated anti-mouse IgG antibody. Nuclear staining was done with DAPI. Cells were imaged with a Leica TCS SP2 Spectral Confocal System.
Individual protein levels determined by Western blotting. Cells were treated with various agents. After treatment, Western blotting was done as described previously (17). Expression levels of tubulin, cyclin B1, securin, BubR1, PARP, caspase 3, caspase 8, caspase 9, Bax, Bcl-XL, Bcl-2, Mcl-1, Bid, JNK, p-JNK, p38, p-p38, ERK1/2, p-ERK1/2, DR4, DR5, TNFR1, Fas/CD95 and ß-actin were detected by using specific anti-bodies in combination with enhanced chemiluminescence (ECL, Amersham, Arlington Heights, IL).
In vivo studies. Male BALB/c nude mice (18-20 g; 6-8 weeks of age) were purchased from the National Animal Center and maintained in pressurized ventilated cage according to insti-tutional regulations. Hep3B cells (5x106) were inoculated
s.c. into the right flank of mice. When tumors reached an approximate volume of 100 mm3, mice were selected and
used for drug studies. HAC-Y6 was dissolved completely in DMSO. Mice were weighed and tumors were measured using calipers every 3 days. Tumor volumes were determined by measuring the length (l) and the width (w) and the volumes were calculated as V = lw2/2. On the final day of
treatment, mice were sacrificed, tumors were excised, weighted and sectioned and the tumor sections were
embedded in OCT (optimum cutting temperature) compound and frozen at -70˚C.
Statistics. Data are expressed as mean ± SD of at least three separate experiments in each group. Student's t-test was used for statistical comparisons. Asterisks indicate that the values are significantly different from the control (*P<0.05; **P<0.01).
Results
HAC-Y6 has antiproliferative activity in human cancer cells. The MTT assay was used to evaluate the antiproliferative effects of HAC-Y6 (Fig. 1) in various human cancer cells including hepatocellular carcinoma (Hep3B), ovarian carci-noma (SKOV3), cervical carcicarci-noma (HeLa), breast carcicarci-noma (MCF-7), prostate carcinoma (PC3), lung carcinoma (H23) and normal human diploid skin fibroblast cells Detroit 551. Table I shows the IC50values of HAC-Y6 tested in six human
cancer cell lines. All the tested cancer cell lines showed susceptibility to HAC-Y6 with IC50values ranging from 0.08
to 9.93 μM. HAC-Y6 had less effect on human diploid skin fibroblast cells Detroit 551 with an IC50of 21.39 μM compared
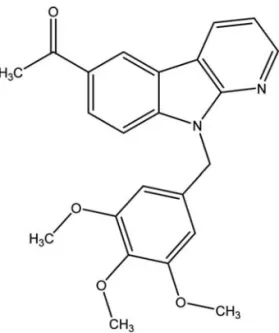
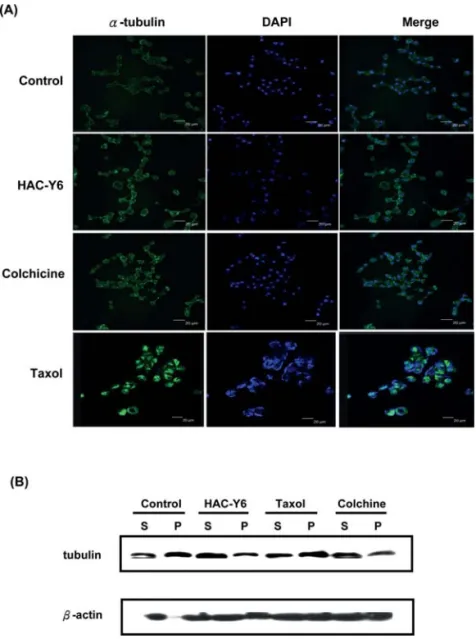
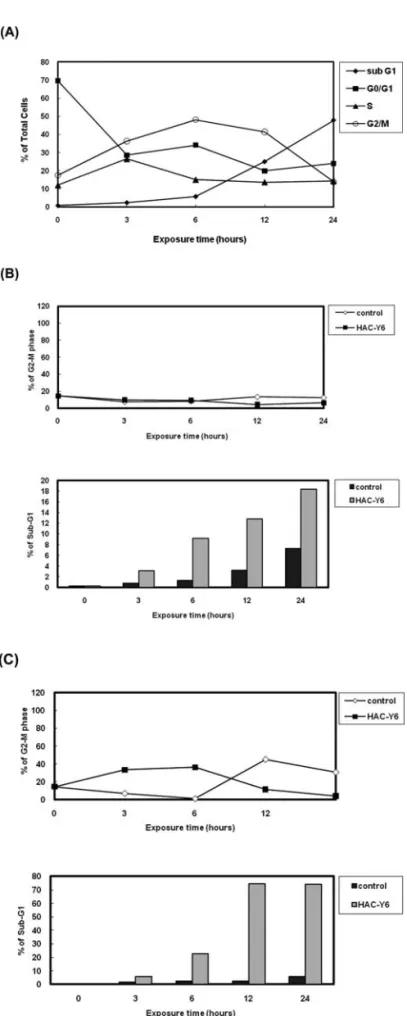
with the other cell lines. Our result show that HAC-Y6 exhibited potent antiproliferative activity against HCC cells. HAC-Y6 caused disruption of tubulin polymerization. To investigate whether HAC-Y6 affected microtubule function, we treated Hep3B cells with 0.08 μM HAC-Y6 for 24 h. Cells were imaged using confocal microscopy. As shown in Fig. 2A, the microtubule network exhibited normal arrange-ment and organization in Hep3B cells in the absence of drug treatment. Treatment with HAC-Y6 for 24 h resulted in findings similar to those of colchicine-induced microtubule changes. Colchicine caused cellular microtubule depolymeri-zation with short microtubule fragments scattered throughout the cytoplasm. In contrast, taxol induced dramatic promotion of tubulin polymerization. The effect of HAC-Y6 on micro-tubule assembly was compared with that of colchicine and taxol. After cells were treated with various antimitotic agents for 24 h, HAC-Y6, and colchicine caused inhibition of micro-tubule assembly (Fig. 2B). In contrast, taxol induced dramatic promotion of tubulin polymerization. Therefore, our data demonstrated that HAC-Y6 induced tubulin depolymerization. HAC-Y6 induced G2/M arrest and apoptosis. We examined effects of HAC-Y6 on the cell cycle to clarify the mechanism of HAC-Y6-induced inhibition of proliferation. HAC-Y6 caused the accumulation of the G2-M phase with concomitant losses in the G0-G1 phase with a maximum effect observed at 6 h (Fig. 3A). There was an accumulation of G2-M DNA content which was followed by an increase in hypodiploid cells at 24 h as indicated by apoptotic cells. We further determined whether HAC-Y6-induced apoptosis was preceded by G2/M phase arrest. The time course of G2/M phase arrest and apoptosis was examined in synchronized cells. When cells were continuously incubated with hydroxyurea, most cells stayed in the G1 phase regardless of HAC-Y6 treatment; in this situation, hydroxyurea per se produced apoptosis but HAC-Y6 failed to augment this apoptosis (Fig. 3B). On the other hand, when cells were treated with HAC-Y6 immediately after removing the hydroxyurea, the majority of cells stayed Figure 1. Chemical structure of HAC-Y6.
Table I. Effects of HAC-Y6 on cell viability in various cell lines.
–––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––
Origin Cell line Viability (μM) IC50(48 h)
–––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––
Hepatocellular carcinoma Hep3B 0.08
Ovarian carcinoma SKOV3 0.2
Cervical carcinoma HeLa 2.37
Breast carcinoma MCF-7 9.14
Prostate carcinoma PC3 9.93
Lung carcinoma H23 0.32
Human diploid skin fibroblast Detroit 551 21.39
––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––– Cell viability of intact cells were determined after 48 h. The concentration for IC50is shown. Each value is based on at least three different
experiments.
in the G2/M phase for 6 h without showing distinct apoptosis. After 24 h, dead cells gradually increased in the HAC-Y6 group as compared with the control group (Fig. 3C). These results suggest that delayed-onset apoptosis induced by HAC-Y6 is limited to cells arrested in the G2/M phase. Up-regulation of the mitotic spindle checkpoint protein BubR1 was associated with a sustained G2/M cell cycle arrest. To better understand the mechanisms of HAC-Y6-induced G2/M arrest, abundance of cell cycle-related proteins was analyzed. Cyclin B1 and securin serve as markers for mitotic arrest induction. Treatment of Hep3B cells with 0.08 μM HAC-Y6 produced time-dependent increases in protein levels of cyclin B1 and securin (Fig. 4A). BuBR1 are conserved multidomain protein kinases that play a key role in the mitotic checkpoint, HAC-Y6 caused the accumulation of BuBR1 (Fig. 4B). Our result suggested that BubR1 contributed to the mitotic checkpoint induced by HAC-Y6.
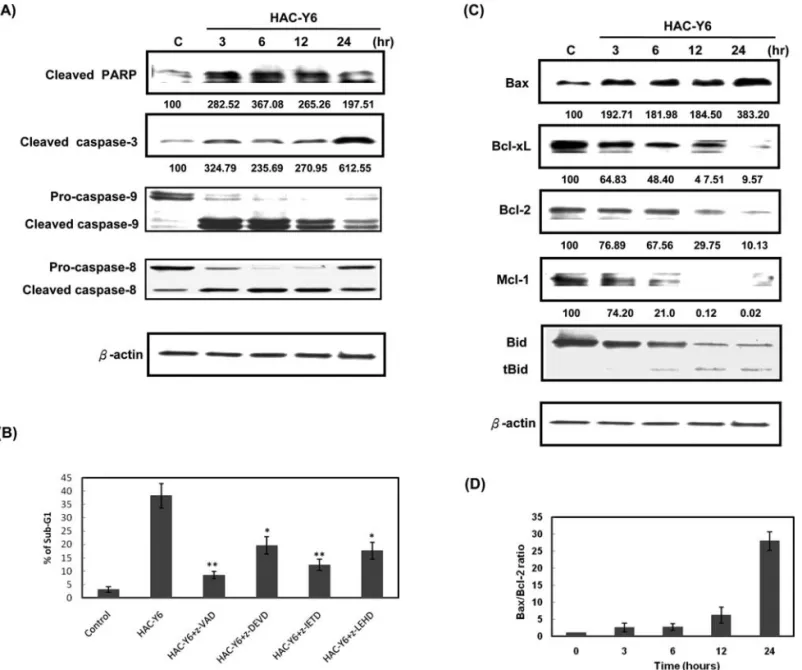
Effects of HAC-Y6 on caspase activation and levels of Bcl-2 family proteins. Caspase activation plays a crucial role in the initiation and execution of apoptosis. We showed that HAC-Y6 can cause apoptosis after cell cycle arrest in HCC cells, we next determined levels of certain proteins associated with apoptosis. Rapid activation of caspase-3, caspase-8 and caspase-9 was detected as early as 3 h after HAC-Y6 treatment. Clevage of PARP, a well-known substrate for caspase-3 was also observed (Fig. 5A). To verify which caspase plays a key role in HAC-Y6-induced apoptosis, we inhibited caspase activity with specific caspase inhibitors such as z-VAD-fmk (a pancaspase inhibitor), z-DEVD-fmk (a caspase-3 inhibitor), z-IETD-fmk (a caspase-8 inhibitor) and z-LEHD-fmk (a caspase-9 inhibitor) for 1 h, followed by treatment with HAC-Y6 (0.08 μM) for an additional 24 h. As shown in Fig. 5B, pancaspase inhibitor z-VAD-fmk blocked HAC-Y6-induced apoptosis. Caspase-8 inhibitor z-IETD-fmk reduced HAC-Y6 induced apoptosis by up to Figure 2. HAC-Y6 inhibits microtubule polymerization. (A) Effect of HAC-Y6 on the organizations of cellular microtubule network. Confocal staining was used in Hep3B cells treated with vehicle (DMSO) or 0.08 μM HAC-Y6, 1 μM taxol or 3 μM colchicine for 24 h. Cells were fixed, permeabilized and stained with anti-·-tubulin monoclonal antibody. Cells were analyzed by confocal microscopy. Middle, DAPI; left, microtubule network; right, merged microtubule network and DAPI. (B) Effect of HAC-Y6 on in vivo microtubule polymerization. Cytosolic (S, soluble) and cytoskeletal (P, polymerized tubulin) tubulin fractions were extracted from Hep3B cells treated with 0.08 μM of HAC-Y6 for 24 h. Colchicine 3 μM and 1 μM taxol were included as controls. Cells were lysed to separate cytosolic and cytoskeletal fractions as described in Materials and methods. Both polymerized tubulin and unpolymerized ones were loaded on SDS-PAGE. After electrophoresis and transfer to nitrocellulose membrane, ·-tubulin was visualized by Western blot analysis. Western blot data presented are representative of those obtained in at least three separate experiments.
one-third. Caspase-3 inhibitor z-DEVD-fmk and caspase-9 inhibitor z-LEHD-fmk also reduced HAC-Y6 induced apoptotic cells. Data in Fig. 5C showed that HAC-Y6 increased Bax protein levels in a time-dependent manner. However, the treatment with HAC-Y6 decreased total Bcl-2, Mcl-1, and Bcl-xL protein levels. Optical densitometric analysis revealed a 6- and 28-fold increase in the ratio of Bax to Bcl-2 protein expression treatment with HAC-Y6 (0.08 μM) for 12 and 24 h, respectively (Fig. 5D). Bid, a substrate of caspase-8, also was activated by HAC-Y6 (Fig. 5C).
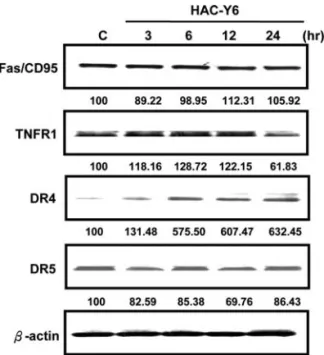
HAC-Y6 induced apoptosis via activation of the DR4 signaling pathway in Hep3B cells. To further characterize the apoptotic pathway activated by HAC-Y6, we examined levels of certain death receptors including Fas/CD95, death receptor 4 (DR4 or TRAILR1), death receptor 5 (DR5 or TRAILR2) and TNFR1 in HAC-Y6-treated Hep3B cells. HAC-Y6 treatment induced an increase in DR4 but did not alter levels of Fas, DR5 and TNFR1 (Fig. 6). Together, these results suggest that DR4 up-regulation plays an important role in HAC-Y6-mediated apoptosis in Hep3B cells.
HAC-Y6 activated the p38 pathway. Mitogen-activated protein kinases (MAPK) respond to extracellular stimuli and regulate cellular activities, such as gene expression, mitosis, differen-tiation, and cell survival/apoptosis (18). We proposed that extracellular signal-regulated kinases (ERK1/2), JNK and/or p38 signaling pathways may be involved in HAC-Y6 induced apoptosis. Fig. 7A shows that HAC-Y6 treatment (0.08 μM) Figure 3. HAC-Y6 induced apoptosis is preceded by G2/M phase arrest. (A)
Hep3B cells were treated with 0.08 μM HAC-Y6 for indicated durations and analyzed for PI-stained DNA content by flow cytometry. (B) Hep3B cells were arrested at G1 phase by treatment with 1 mM hydroxyurea for 12 h and then treated with 0.08 μM HAC-Y6 for indicated durations and analyzed for PI-stained DNA content by flow cytometry. (C) Hep3B cells were arrested at G1 by treating with 1 mM hydroxyurea for 12 h and allowed to re-enter the cell cycle by washing out the hydroxyurea. The cells were then incubated in the presence of 0.08 μM HAC-Y6 for indicated durations, stained with PI, and subjected to cell cycle analysis. Apoptotic cell death is represented by the sub-G1 population.
Figure 4. HAC-Y6 increases G2/M phase checkpoint protein expression. (A) Hep3B cells were treated with 0.08 μM HAC-Y6 for indicated durations. Cells were then harvested and lysed for detection of cyclin B1 and securin. (B) Cells were then harvested and lysed for detection of BubR1. Western blot data are representative of those obtained in at least three separate experiments. The values below the figures represent change in protein expression normalized to ß-actin.
resulted in phospho-JNK levels similar to control levels but that phospho-ERK1/2 levels were moderately repressed. In contrast, HAC-Y6-induced phosphorylation of p38 after 30 min and more distinct response being observed at 1 and 2 h. The total amount of p38 remained unchanged during the treatment period (Fig. 7A). These observations suggest that activation of p38, but not ERK1/2 or JNK, is associated with HAC-Y6-induced cellular apoptosis.
SB203580 reduced HAC-Y6-induced apoptosis in Hep3B cells. SB203580, a pyridinyl imidazole derivative, specifi-cally inhibits p38a and p38b in the p38 signaling transduction pathway (19). As shown in Fig. 7B, flow cytometric analysis revealed that 34.86% of Hep3B cells were at sub-G1 phase (apoptotic cells) after a 24 h treatment with 0.08 μM HAC-Y6.
However, when Hep3B cells were pre-incubated with 10 μM SB203580 for 1 h, <6.5% of the cells were at sub-G1 phase in the HAC-Y6 treatment group (Fig. 7B). These results strongly support the notion that HAC-Y6-mediated cellular apoptosis is closely involved with activation of the p38 signaling trans-duction pathway.
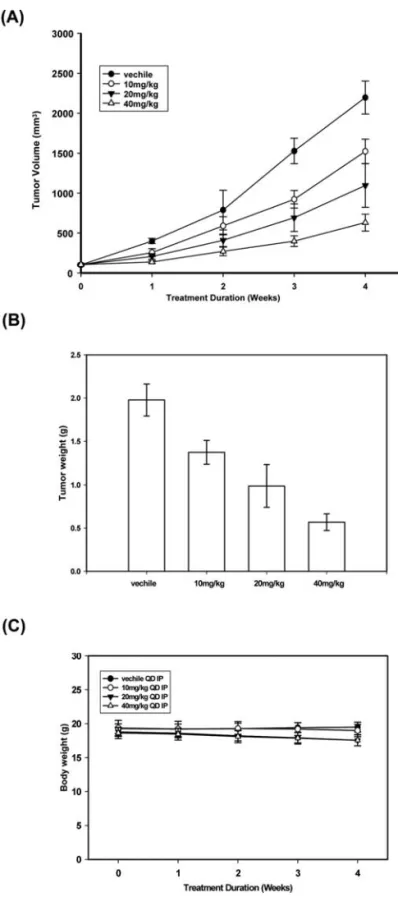
HAC-Y6 inhibited the growth of Hep3B xenografts in nude mice. Our in vitro findings demonstrated that HAC-Y6 induced apoptosis and cell death. Based on these findings, we proposed that HAC-Y6 would inhibit tumor growth in vivo. Hep3B cell xenografts were produced in athymic nude mice. Male BALB/c nude mice (18-20 g; 6-8 weeks of age) were inoculated with 5x106Hep3B cells subcutaneously in the
right flank region. When the average tumor volume reached Figure 5. Bcl-2 family proteins are involved in HAC-Y6-induced apoptosis. (A) Hep3B cells were treated with 0.08 μM HAC-Y6 for indicated durations. Cells were then harvested and lysed for detection of cleaved PARP, cleaved caspase-3, -9 and -8. (B) Caspase inhibitors reduce apoptosis in Hep3B cells. Hep3B cells were pretreated with the caspase inhibitors including pancaspase inhibitor (z-VAD-fmk, 50 μmol/l), caspase-3 inhibitor (z-DEVD-fmk, 100 μmol/l), caspase-8 inhibitor (z-IETD-fmk, 100 μmol/l), and caspase-9 inhibitor (z-LEHD-fmk, 100 μmol/l) for 1 h followed by treatment with HAC-Y6 (0.08 μM) for an additional 12 h. Sub-G1 was determined in triplicate from three independent experiments by flow cytometry analysis. Data are presented as means ± SD.
*P<0.05; **P<0.01 vs. HAC-Y6-treated cells. (C) Hep3B cells were treated with 0.08 μM HAC-Y6 for indicated durations. Cells were then harvested and lysed
for detection of Bax, Bcl-xL, Bcl-2, Mcl-1 and Bid. (D) Bottom graph shows the ratio of relative expression of Bax to Bcl-2. Data are presented as means ± SD. Western blot data presented are representative of those obtained in at least three separate experiments. The values below the figures represent change in protein expression normalized to ß-actin.
~100 mm3, the animals were randomly allocated into four
groups, which were control and HAC-Y6 (10, 20, 40 mg/kg, respectively). Each group contained eleven mice. The animals were given HAC-Y6 (i.p.) five days a week for 4 weeks. Animals, monitored for vital signs and weight changes for the duration of the experiment, did not lose weight (Fig. 8C). After 4 weeks, the animals were sacrificed and no pathologic signs were seen. Administration of HAC-Y6 induced a dose-dependent inhibition of Hep3B tumor growth (Fig. 8A) (40 mg/kg HAC-Y6, 825.93 mm3, P<0.01 vs. vehicle;
vehicle-treated group = 2113.28 mm3on day 28) and tumor weight
(Fig. 8B). Discussion
Carbazole analogs have several protective effects including antiplatelet (20) and anti-inflammatory activities (21,22). In the present study, the potent antiproliferative activity of a newly synthesized ·-carboline analog, HAC-Y6, was demon-strated in different human cancer cell lines. HAC-Y6 exhibits potent anti-hepatocellular carcinoma cell activity compared with other cancer cell lines. Hepatocellular carcinoma is a phenotypically and genetically heterogeneous polyclonal disease and resistant to most chemotherapeutic agents (23). Specifically, HAC-Y6 shows less effect on human diploid skin fibroblast cells indicating that HAC-Y6 is a novel outstanding agent in treating HCC cells in vitro.
HAC-Y6 had multiple effects. For example, HAC-Y6 strongly inhibited tubulin polymerization and induced the depolymerization of microtubules and degradation of the microtubule network in Hep3B cells. Thus, HAC-Y6 can be classified as a microtubule-depolymerizing agent. Our data also indicate that like the other microtubule inhibitors,
HAC-Y6 initially inhibits tubulin polymerization, then induces cell cycle arrest and finally triggers apoptosis in the Hep3B cells.
It has been widely reported that cyclin B1/CDK1 com-plexes are involved in the regulation of the G2/M phase and the M-phase transition. Many reports have demonstrated that microtubule inhibitor induced M-phase arrest and inappro-priate accumulation of B-type cyclins were associated with the initiation of apoptotic pathways (24,25). In the present study, HAC-Y6 arrested the growth of cancer cells at the Figure 6. HAC-Y6 increases DR4 expression. Hep3B cells were treated with
0.08 μM HAC-Y6 for indicated durations. Cells were then harvested and lysed for detection of Fas/CD95, TNFR1, DR4 and DR5. Western blot data presented are representative of those obtained in at least three separate experi-ments. The values below the figures represent change in protein expression normalized to ß-actin.
Figure 7. Expression of MAPKs in the HAC-Y6-treated Hep3B cells. (A) Hep3B cells were treated with 0.08 μM HAC-Y6 for indicated durations. Cells were then harvested and lysed for detection of JNK, p-JNK, p38, p-p38, ERK1/2 and p-ERK1/2. Western blot data are representative of those obtained in at least three separate experiments. The values below the figures represent change in protein expression normalized to ß-actin. (B) Flow cytometric analysis of sub-G1 population of HAC-Y6-treated Hep3B cells in the presence and absence of SB203580. Flow cytometric detection revealed that pre-treatment with 10 μM SB203580 markedly reduced apoptosis among HAC-Y6-treated Hep3B cells. Sub-G1 was determined in triplicate from three independent experiments by flow cytometry analysis. Data are presented as means ± SD. **P<0.01 vs. HAC-Y6-treated cells.
G2-M phase and then induced apoptotic cell death via the accumulation of cyclin B1.
The endogenous level of the mitotic spindle checkpoint protein BubR1 directly correlated with the cellular response to microtubule disruption. A recent study demonstrated that
knockdown of BubR1 accelerates the normal progression of mitosis (26). The mitotic spindle checkpoint is a complex pathway conserved across species and monitors the metaphase to anaphase transition. The basic model for the spindle check-point defines that tension defects or unattached chromosomes activate the checkpoint, delaying the onset of anaphase until such aberrations are corrected. We found that HAC-Y6 induced G2/M block was maintained until endogenous levels of BubR1 protein became undetectable after which a marked increase in apoptosis occurred. BubR1 cellular levels inversely correlated with the onset of apoptosis. Together, these results suggest that endogenous levels of BubR1 may predict the apoptotic efficacy and potential chemotherapeutic benefit of microtubule-targeted drugs in human cancers.
Induction of apoptosis is an important target for cancer therapy (27). Agents that induce apoptosis can be divided into two broad categories according to their site of action and classified as the extrinsic and the intrinsic pathways (28,29). The extrinsic pathway is initiated by death receptors (e.g., Fas/CD95, DR4/5 and TNFR1) on the cell surface. Interaction of a death receptor with its ligand triggers the formation of a death-inducing signaling complex, which in turn recruits pro-caspase-8. Pro-caspase-8 undergoes autoproteolytic cleavage, forming active caspase-8, which then activates other pro-caspases such as pro-caspase-3 (28). The intrinsic pathway is initiated with loss of membrane potential in mitochondria and then the release of cytochrome c from the mitochondria into the cytosol and binding to the adaptor protein Apaf-1. Activation of caspase-9, an initiator caspase of the intrinsic pathway leads to the activation of caspase-3, the effector caspase (30).
In the present report, we investigated HAC-Y6 induction of the the intrinsic pathway and its interaction with the extrinsic pathway in the human hepatocellular carcinoma cell line Hep3B. Proteolytic degradation of PARP, a substrate of caspase-3, indicated that activation of caspases was involved in HAC-Y6-induced apoptosis in Hep3B cells. Correspond-ingly, we found that caspase-3, -8 and -9 were activated in HAC-Y6-treated Hep3B cells. We also found an increased level of the intrinsic pathway activator Bax as well as a reduction in the expression of the apoptosis inhibitor Bcl-2 in HAC-Y6-treated Hep3B cells. Mitochondria-dependent apoptosis is regulated positively and negatively by Bcl-2 family members, which include three subfamilies based on their structural and functional characteristics. The anti-apoptotic subfamily comprises proteins such as Bcl-2, Bcl-xL and Mcl-1 that have four Bcl-2 homology (BH) domains. The pro-apoptotic subfamily includes Bax, Bak and Bok proteins which contain BHl-3 domains and they are capable of forming pores in the mitochondrial outer membrane.
Caspase-8 is activated by the death receptor. Activated caspase-8 can cleave and activate effector caspase-3. On the other hand, caspase-8 can induce Bid cleavage. The cleaved Bid causes the cytochrome c efflux from mitochondria, then activation of caspase-9 and -3 (31). We showed that HAC-Y6 induced the cleavage of full-length Bid producing the truncated Bid (tBid, 15 kDa) that translocated to the mito-chondria. These results indicate that activation of caspase-3 may be due to direct caspase-8 cleavage and caspase-8-induced caspase-9 activation. Both the extrinsic signaling Figure 8. Effect of HAC-Y6 on Hep3B tumor growth in a mouse xenograft
model. Male nude mice s.c. received Hep3B transfectants to induce tumor xenografts. Seven days later, mice were divided into four groups (11 mice/ group). The first group was a control group treated with vehicle. The other groups were given i.p. with HAC-Y6 (10, 20 and 40 mg/kg, respectively) every day for a total of 28 days. Tumor volume was measured and calculated as described in Materials and methods. Points, mean tumor volume; bars, SD. (A) tumor volume (mm3), (B) tumor weight (g), (C) body weight (g).
pathway and the intrinsic signaling pathway would appear to contribute to HAC-Y6-induced apoptosis.
Activation of caspase-8 is required for death receptor-induced apoptosis (32). Enhancement of TRAIL and induction of DR4 and/or DR5 induced apoptosis have been shown in certain types of cancer cells, including colon cancer, prostate cancer, bladder cancer and chronic lymphocytic leukemia cells (33,34). In the present study, we found that HAC-Y6 treatment increased DR4 protein levels but did not alter protein levels of Fas, TNFR1 and DR5. In addition, DR4 monoclonal antibody strongly blocked HAC-Y6-induced apoptosis in Hep3B cells. These results indicate that DR4 signaling system plays a central role in apoptosis induced by HAC-Y6 in Hep3B cells.
Mitogen-activated protein kinases (MAPKs) are critical mediators of the cell membrane to nucleus signal transduction in response to various extracellular stimuli (28). They include the extracellular-signal-related kinases (ERKs), c-Jun N-terminal protein kinase (JNK) and p38. These kinases play an important role in the control of cell growth, differentiation and programmed cell death (35). Several chemotherapeutic agents, such as cisplatin, doxorubicin and taxol, were shown to induce p38 activation in human cervical, ovarian and skin epidermoid carcinoma cells (36-38). We observed in the present study that HAC-Y6 after a 30 min incubation increased levels of phosphorylated p38 in Hep3B cells. This increase in p38 was followed by activation of caspases suggesting that p38 acts upstream of caspase activation and that p38 is associ-ated with pro-apoptotic activity of HAC-Y6. Further support for the hypothesis that HAC-Y6 stimulates p38 is that pre-incubation with the p38 inhibitor SB203580 prevented HAC-Y6-induced apoptotic cell death as seen by a reduction of the subG1 peak. Recent studies have shown that JNKs and p38 pathways are associated with increased apoptosis, whereas the ERK1/2 pathway is shown to suppress apoptosis (39-42).
In conclusion, we have demonstrated that HAC-Y6, a novel synthesized compound, possesses potent antitumor activity against human tumor cells via disruption of micro-tubules. HAC-Y6 exhibits better antiproliferative activity against human hepatocellular carcinoma cells. We propose that death stimuli generated by HAC-Y6 are transduced from the cell membrane to the nucleus through the p38 signaling pathway. Both apoptotic pathways are activated following p38 phosphorylation, and ultimately, the effector caspase 3 is activated causing irreversible apoptotic cell death. HAC-Y6 is a promising compound against hepatocellular carcinoma. Acknowledgements
This study was supported by research grants from the National Science Council (NSC) of the R.O.C. (NSC97-2320-B-039-008-MY3 and NSC98-2628-B-039-018-MY3) awarded to T.D.W. and L.J.H.
References
1. Parkin DM, Bray F, Ferlay J and Pisani P: Global cancer statistics, 2002. CA Cancer J Clin 55: 74-108, 2005.
2. Jemal A, Siegel R, Ward E, Hao Y, Xu J and Thun MJ: Cancer statistics, 2009. CA Cancer J Clin 59: 225-249, 2009.
3. Llovet JM, Burroughs A and Bruix J: Hepatocellular carcinoma. Lancet 362: 1907-1917, 2003.
4. Kirschner MW: Microtubule assembly and nucleation. Int Rev Cytol 54: 1-71, 1978.
5. Wittmann T, Hyman A and Desai A: The spindle: a dynamic assembly of microtubules and motors. Nat Cell Biol 3: E28-E34, 2001.
6. Sudakin V and Yen TJ: Targeting mitosis for anti-cancer therapy. Bio Drugs 21: 225-233, 2007.
7. Pellegrini F and Budman DR: Review: tubulin function, action of antitubulin drugs and new drug development. Cancer Invest 23: 264-273, 2005.
8. Schiff PB and Horwitz SB: Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci USA 77: 1561-1565, 1980.
9. Cotter TG: Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer 9: 501-507, 2009.
10. Fulda S and Debatin KM: Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25: 4798-4811, 2006.
11. Chakraborty DP, Barman BK and Bose PK: On the structure of girinimbine, a pyranocarbazole derivative isolated from Murraya
koenigii Spreng. Sci Cult 30: 445, 1964.
12. Ramsewak RS, Nair MG, Strasburg GM, DeWitt DL and Nitiss JL: Biologically active carbazole alkaloids from Murraya
koenigii. J Agric Food Chem 47: 444-447, 1999.
13. Ito C, Itoigawa M, Nakao K, Murata T, Tsuboi M, Kaneda N and Furukawa H: Induction of apoptosis by carbazole alkaloids isolated from Murraya koenigii. Phytomedicine 13: 359-365, 2006.
14. Ito C, Itoigawa M, Sato A, Hasan CM, Rashid MA, Tokuda H, Mukainaka T, Nishino H and Furukawa H: Chemical consti-tuents of Glycosmis arboera: three new carbazole alkaloids and their biological activity. J Nat Prod 67: 1488-1491, 2004. 15. Itoigawa M, Kashiwada Y, Ito C, Furukawa H, Tachibana Y,
Bastow KF and Lee KH: Antitumor agents. 203. Carbazole alkaloid murrayaquinone A and related synthetic carbazole-quinones as cytotoxic agents. J Nat Prod 63: 893-897, 2000. 16. Ito C, Katsuno S, Itoigawa M, Ruangrungsi N, Mukainaka T,
Okuda M, Kitagawa Y, Tokuda H, Nishino H and Furukawa H: New carbazole alkloids from Clausena anisata with antitumor promoting activity. J Nat Prod 63: 125-128, 2003.
17. Lee JC, Tsai CY, Kao JY, Kao MC, Tsai SC, Chang CS, Huang LJ, Kuo SC, Lin JK and Way TD: Geraniin-mediated apoptosis by cleavage of focal adhesion kinase through up-regulation of Fas ligand expression in human melanoma cells. Mol Nutr Food Res 52: 655-663, 2008.
18. Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K and Cobb MH: Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22: 153-183, 2001.
19. Saklatvala J, Rawlinson L, Waller RJ, Sarsfield S, Lee JC, Morton LF, Barnes MJ and Farndale RW: Role for p38 mitogen-activated protein kinase in platelet aggregation caused by collagen or a thromboxane analogue. J Biol Chem 22: 6586-6589, 1996. 20. Lee CY, Kuo SC, Teng CM and Huang LJ: Synthesis and
anti-platelet activity of 9-benzyl-3-hydroxymethylcarbazoles. Chin Pharm J 54: 25-34, 2002.
21. Lee CY, Huang LJ, Wang JP and Kuo SC: Anti-inflammatory activity of 9-substituted benzyl-3-substituted carbazole deriva-tives. Chin Pharm J 54: 35-40, 2002.
22. Ho FM, Kang HC, Lee ST, Chao Y, Chen YC, Huang LJ and Lin WW: The anti-inflammatory actions of LCY-2-CHO, a carbazole analogue, in vascular smooth muscle cells. Biochem Pharmacol 74: 298-308, 2007.
23. Hui KM: Human hepatocellular carcinoma: expression profiles based molecular interpretations and clinical applications. Cancer Lett 10: 1-7, 2008.
24. Shi YQ, Zhu CJ, Yuan HQ, Li BQ, Gao J, Qu XJ, Sun B, Cheng YN, Li S, Li X and Lou HX: Marchantin C, a novel microtubule inhibitor from liverwort with anti-tumor activity both in vivo and in vitro. Cancer Lett 276: 160-170, 2009. 25. Wang SW, Pan SL, Huang YC, Guh JH, Chiang PC, Huang DY,
Kuo SC, Lee KH and Teng CM: CHM-1, a novel synthetic quinolone with potent and selective antimitotic antitumor activity against human hepatocellular carcinoma in vitro and in vivo. Mol Cancer Ther 7: 350-360, 2008.
26. Meraldi P, Draviam VM and Sorger PK: Timing and check-points in the regulation of mitotic progression. Dev Cell 7: 45-60, 2004.
27. Fesik SW: Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer 5: 876-885, 2005.
28. Ghobrial IM, Witzig TE and Adjei AA: Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin 55: 178-194, 2005.
29. Putcha GV, Harris CA, Moulder KL, Easton RM, Thompson CB and Johnson EM Jr: Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J Cell Biol 157: 441-453, 2002.
30. Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G and Vandenabeele P: Toxic proteins released from mitochondria in cell death. Oncogene 23: 2861-2874, 2004.
31. Brenner C and Kroemer G: Apoptosis: mitochondria - the death signal integrators. Science 289: 1150-1151, 2000.
32. Lavrik I, Golks A and Krammer PH: Death receptor signaling. J Cell Sci 118: 265-267, 2005.
33. Kabore AF, Sun J, Hu X, McCrea K, Johnston JB and Gibson SB: The TRAIL apoptotic pathway mediates proteasome inhibitor induced apoptosis in primary chronic lymphocytic leukemia cells. Apoptosis 11: 1175-1193, 2006.
34. Johnson TR, Stone K, Nikrad M, Yeh T, Zong WX, Thompson CB, Nesterov A and Kraft AS: The proteasome inhibitor PS-341 overcomes TRAIL resistance in Bax and caspase 9-negative or Bcl-xL overexpressing cells. Oncogene 22: 4953-4963, 2003.
35. Wagner EF and Nebreda AR: Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9: 537-549, 2009.
36. Deacon K, Mistry P, Chernoff J, Blank JL and Patel R: p38 mitogen-activated protein kinase mediates cell death and p21-activated kinase mediates cell survival during chemotherapeutic druginduced mitotic arrest. Mol Biol Cell 14: 2071-2087, 2003.
37. Holmes WF, Soprano DR and Soprano KJ: Early events in the induction of apoptosis in ovarian carcinoma cells by CD437: activation of the p38 MAP kinase signal pathway. Oncogene 22: 6377-6386, 2003.
38. Losa JH, Parada Cobo C, Viniegra JG, Sánchez-Arevalo Lobo VJ, Ramón y Cajal S and Sánchez-Prieto R: Role of the p38 MAPK pathway in cisplatin-based therapy. Oncogene 22: 3998-4006, 2003.
39. Frey RS and Singletary KW: Genistein activates p38 mitogen-activated protein kinase, inactivates ERK1/ERK2 and decreases Cdc25C expression in immortalized human mammary epithelial cells. J Nutr 133: 226-231, 2003.
40. Lavelle D, DeSimone J, Hankewych M, Kousnetzova T and Chen YH: Decitabine induces cell cycle arrest at the G1 phase via p21(WAF1) and the G2/M phase via the p38 MAP kinase pathway. Leuk Res 27: 999-1007, 2003.
41. Wang X, McGowan CH, Zhao M, He L, Downey JS, Fearns C, Wang Y, Huang S and Han J: Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest. Mol Cell Biol 20: 4543-4552, 2000.
42. Zhang Z, Leonard SS, Huang C, Vallyathan V, Castranova V and Shi X: Role of reactive oxygen species and MAPKs in vanadate-induced G(2)/M phase arrest. Free Rad Biol Med 34: 1333-1342, 2003.