行政院國家科學委員會專題研究計畫 成果報告
血管收縮素誘發心臟纖維化之分子機
制:Thiazolidinedione 之抑制作用
計畫類別: 個別型計畫 計畫編號: NSC92-2314-B-002-309- 執行期間: 92 年 08 月 01 日至 93 年 07 月 31 日 執行單位: 國立臺灣大學醫學院內科 計畫主持人: 陳錦澤 共同主持人: 鄭志鴻 報告類型: 精簡報告 處理方式: 本計畫可公開查詢中 華 民 國 93 年 11 月 2 日
中文摘要 血管收縮素誘發心臟纖維化之分子機制: Thiazolidinedione 之抑制作用 心臟纖維化為由高血壓及心肌梗塞等疾 患所引發的基本反應,導致心臟舒張功能失 常及衰竭,臨床上由心臟舒張功能不良所引 起之心臟衰竭約佔 30 到 50%,心臟纖維化的 過程為心臟纖維細胞的增生及胞外基質的堆 積。血管收縮素會造成心臟纖維細胞的增 生,然而其分子機制目前尚不清楚;本計劃 的部份研究結果發現:血管收縮素會增加內 皮素基因的表現,並於纖維細胞增生過程中 扮 演 重 要 的 角 色 。 近 期 , 糖 尿 病 用 藥 rosiglitazone 經由活化細胞內過氧小體增生 活 化 受 體 (peroxisome
proliferator-activated receptor, PPAR) 能產生許多有益於心血管系統的作用,引起 醫界的高度重視。最新的文獻報告在鼠的實 驗觀察發現:rosiglitazone 有改善心臟肥大及 心 肌 梗 塞 所 造 成 的 損 傷 等 作 用 , 然 rosiglitazone 是否會影響血管收縮素誘發心 臟纖維細胞增生以及內皮素基因表現,進而 改善心臟纖維化的現象,目前仍然未知。所 以本研究同時深入探討內皮素在血管收縮素 誘發心臟纖維細胞增生所扮演的角色及血管 收縮素誘發內皮素基因表現的調控機制,探 究 rosiglitazone 對於血管收縮素誘發心臟纖 維細胞增生以及內皮素基因表現的影響。於 培養的初生鼠心臟纖維細胞給予血管收縮素 的處理後,分析去氧核醣核酸合成的增加以 及內皮素的基因表現,同時觀察rosiglitazone 對血管收縮素誘發心臟纖維細胞增生及內皮 素基因表現的作用。實驗結果顯示:血管收縮 素 增 加 細 胞 增 生 的 作 用 可 為 其 拮 抗 劑 losartan 及內皮素 A 型受體拮抗劑 BQ485 所 阻斷。以北方式點墨法搭配啟動子活性分析 法確定血管收縮素會誘發內皮素基因表現, 胞外訊號調節激酶(ERK)抑制劑 PD98059 可完全抑制血管收縮素誘發內皮素基因的表 現,進一步共轉染 Ras、 Raf 以及 MEK1 的負 向突變可減弱血管收縮素之增加內皮素基因 啟動子活性,顯示血管收縮素的誘發內皮素 基因須要經由 Ras-Raf-ERK 路徑,去除部份 啟動子區域搭配突變分析法進一步發現在血 管收縮素誘發內皮素基因表現時,內皮素基 因啟動子區域內的活化蛋白 1(activator protein-1;AP-1)的結合部位扮演重要的角 色;rosiglitazone 則有抑制血管收縮素增加細 胞增生以及內皮素基因表現的作用。 關鍵詞:血管收縮素; 內皮素; rosiglitazone; 細胞增生; 訊息傳遞; 心臟纖維細胞;過氧 小體增生活化受體 英文摘要
Molecular Mechanism of Angiotensin II-induced Cardiac Fibrosis: the Inhibitory Effect of Thiazolidinedione
Fibroblasts play an important role in maintaining cardiac function by providing structural support for the cardiomyocytes and serving as a source for autocrine/paracrine
II
growth factors. After myocardial infarction, reactive fibrosis results in excessive scar formation as proliferating fibroblasts invade the necrotic area. This remodeling leads to an increase of the ventricular stiffness and ultimately compromises the function of the heart. Recent studies in humans and animal models have shown that the expression of myocardial endothelin-1 (ET-1) is increased during cardiacfibrosis. It is suggested that ET-1 might contribute to cardiac fibroblast proliferation resulting in cardiac fibrosis. ET-1 works as a paracrine as well as an autocrine. In the first part of this project, our data revealed that angiotensin II (Ang II)-stimulated fibroblast proliferation is mediated by release of ET-1 from fibroblasts rather than by direct action.
Rosiglitazone, approved by the FDA during the spring of 1999, was the second thiazolidinedione to be marketed in the United States. Rosiglitazone improves insulin sensitivity in patients with type 2 diabetes by activating peroxisome proliferator-activated receptor-γ (PPARγ) receptors in adipose tissues, skeletal muscles, and the liver. Using Northern
blot analysis and reverse transcriptase-polymerase chain reaction in samples of rat heart, Wayman et al. (2002) documented the expression of the mRNA for PPARγ (isoform 1 but not isoform 2) in freshly isolated cardiac myocytes and cardiac fibroblasts and in the left and right ventricles of the heart. Using a rat model of regional myocardial ischemia and reperfusion (in vivo), Wayman et al. (2002) discovered that rosiglitazone causes a substantial reduction of myocardial infarct size in the rat. Treatment of normal and diabetic rat hearts with rosiglitazone, also improves postischemic
functional recovery. Using Western immunoblotting, it was demonstrated that the acute cardioprotective effect of rosiglitazone is associated with an inhibition of Jun NH(2)-terminal kinase phosphorylation in both normal and diabetic rat hearts. Furthermore, rosiglitazone also inhibited activating protein-1(AP-1) DNA-binding activity.
Our data indicate that rosiglitazone inhibits Ang II–induced proliferation in cardiac fibroblasts. Ang II increased DNA synthesis which was inhibited with rosiglitazone. Rosiglitazone inhibited the Ang II-induced ET-1 gene expression as revealed by Nothern blotting and promoter activity assay. Rosiglitazone also inhibited Ang II-increased intracellular ROS generation as measured by a redox sensitive fluorescent dye, 2' 7'-dichlorofluorescin diacetate, and ERK phosphorylation. Furthermore, rosiglitazone and antioxidants such as N-acetyl-cysteine and diphenylene iodonium decreased Ang II-induced cell proliferation, ET-1 promoter activity, ET-1 mRNA, ERK phosphorylation, and activator protein-1-mediated reporter activity. In summary, our results suggest that rosiglitazone inhibits Ang II-induced cell proliferation and ET-1 gene expression, partially by interfering with the ERK pathway via attenuation of ROS generation.
Key Words: angiotensin II; endothelin-1; rosiglitazone; proliferation; signal transduction; cardiac fibroblasts;peroxisome
proliferator-activated receptor
報告內容:
前言
The thiazolidinedione class of medications was first introduced to the United States when troglitazone was marketed during early 1997.
III
Rosiglitazone, approved by the FDA during the spring of 1999, was the second thiazolidinedione to be marketed in the United States. Similar to troglitazone, rosiglitazone improves insulin sensitivity in patients with type 2 diabetes by activating peroxisome proliferator-activated receptor γ (PPARγ) receptors in adipose tissues, skeletal muscles, and the liver. The efficacy and safety of rosiglitazone therapy in patients with type 2 diabetes have been demonstrated in a number of clinical studies (Cheng-Lai and Levine, 2000). Peroxisome proliferator-activated receptors (PPARs) are a family of 3 nuclear hormone receptors, PPARα, PPARδ, and PPARγ (Schoonjans et al., 1997). PPARs act as transcription factors on ligand-induced heterodimerization with the common nuclear receptor binding partner, the retinoid X receptor (RXR). When combined as a PPAR:RXR heterodimer, PPAR ligands and 9-cis retinoic acid can act synergistically on PPAR responses. Different dimers of RXR induce specific responses by binding highly specific sequences in the promoter regions of the various genes. PPAR activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 (AP-1) signaling pathway (Delerive et al., 1999). PPARγ is activated by the natural ligand 15-deoxy- ∆12,14-prostaglandin J2 (15d-PGJ2) (Forman et al., 1995) as well as the synthetic ligand thiazolidinedione (rosiglitazone) (Asakawa et al., 2002). The thiazolidinediones decrease blood pressure in a hypertensive rat model (Dubey et al., 1993). Recently, Takano et al (2000) demonstrated that PPAR activators inhibit tumor necrosis factor-α expression at the transcriptional level in part by preventing
nuclear factor (NF)-kB activity in cardiac myocytes. Barger et al (Barger et al., 2000) reported that PPARa is deactivated during cardiac hypertrophic that PPARα is deactivated during cardiac hypertrophic and energy homeostasis. However, it remains unclear whether PPARs participates in cardiac fibroblast proliferation and the process of cardiac fibrosis.
研究目的
In this project, we set out to study whether rosiglitazone affects the cardiac fibroblast proliferation and the Ang II–induced augmentation of ET-1 gene expression in cultured cardiac fibroblasts of neonatal rats. The intracellular signal regulation pathway such as MAPK pathway is modulated by rosiglitazone or not will be also investigated. 文獻探討
Cardiac remodeling is manifested clinically as changes in the size, shape, and function of the heart (Cohn et al., 2000). Histopathologically, it is characterized by a structural rearrangement of components of the normal chamber wall that involves cardiomyocyte hypertrophy, cardiac fibroblast proliferation, fibrosis, and cell death (Swynghedauw, 1999). Fibroblasts play an important role in maintaining cardiac function by providing structural support for the cardiomyocytes(Marsen et al., 2000) and serving as a source for autocrine/paracrine growth factors(Ancey et al., 2002). Cardiac fibroblasts, along with cardiomyocytes, play an essential role in the progression of cardiac remodeling. After myocardial infarction, reactive fibrosis results in excessive scar formation as proliferating fibroblasts invade the necrotic area. Fibrosis, which is a disproportionate accumulation of fibrillar
IV
collagen, is an integral feature of the remodeling characteristic of the failing heart. Accumulation of type I collagen, the main fibrillar collagen found in cardiac fibrosis, stiffens the ventricles and impedes both contraction and relaxation (Ichiro et al., 2002). This remodeling leads to an increase of the ventricular stiffness and ultimately compromises the function of the heart(Borer et al., 2002). Fibrosis can also impair the electrical coupling of cardiomyocytes by separating myocytes with extracellular matrix (ECM) proteins (Swynghedauw, 1999). Furthermore, fibrosis results in reduced capillary density and an increased oxygen diffusion distance that can lead to hypoxia of myocytes (Sabbah et al., 1995). Thus, fibrosis profoundly affects myocyte metabolism and performance and ultimately ventricular function (Schnee et al., 2000). Excessive myocardial fibrosis has been found in the progression of cardiac dysfunction, especially in the case of diastolic dysfunction, in hypertensive hearts(Mann, 1999).
Humoral factors that affect the phenotype and function of cardiac fibroblasts include angiotensin II (Ang II), basic fibroblast growth factor (bFGF/FGF-2), transforming growth factor-β (TGF-β), catecholamines, and insulin-like growth factor-1 (IGF-1) (Booz et al., 1995; Bouzegrhane and Thibault, 2002). Among these factors, Ang II appears to be one of the most important regulating cardiac fibrosis and remodeling, although other factors are also important, as clearly demonstrated in recent studies in which pressure overload caused cardiac hypertrophy and fibrosis in type 1a Ang II (AT1a) receptor knockout mice (Harada et al., 1998). In vitro studies of cultured cardiac fibroblasts have shown that
Ang II directly stimulates fibroblast proliferation, collagen synthesis, and the expression of ECM proteins (collagen, fibronectin, and laminin) via AT1 receptors (Bouzegrhane and Thibault, 2002). Ang II also regulates cardiac fibroblast function indirectly through induction of endothelin-1 (ET-1), TGF-β, interleukin-6 (IL-6), and osteopontin, which function in autocrine and paracrine manners (Bouzegrhane and Thibault, 2002; Kawano et al., 2000;101:1130–1137. Fujisaki et al., 1995; Sano et al., 2000; Ashizawa et al., 1996) Treatment of normal and diabetic rat hearts with rosiglitazone, improves postischemic functional recovery (Khandoudi et al., 2002). The effects of acute rosiglitazone administration were investigated using working hearts isolated from normal rat or rats diabetic for 4 weeks after streptozotocin (STZ) injection. Rosiglitazone (1 micromol/l) administered before ischemia had no effect on cardiac function during baseline perfusion, but it significantly improved aortic flow during reperfusion in both normal and diabetic hearts. In a chronic protocol in which rosiglitazone was given by daily gavages (10 micromol/kg body wt) immediately after STZ injection, rosiglitazone also prevented postischemic injury and significantly improved functional recovery. Using Western immunoblotting, it was demonstrated that the acute cardioprotective effect of rosiglitazone is associated with an inhibition of Jun NH(2)-terminal kinase phosphorylation in both normal and diabetic rat hearts. Furthermore, rosiglitazone also inhibited activating protein-1 (AP-1) DNA-binding activity (Khandoudi et al., 2002). These data, demonstrating that rosiglitazone limits postischemic injury in isolated hearts, suggest an important function
V
for PPARγ in the heart. Yue et al. (Yue et al., 2001) demonstrated that rosiglitazone reduced myocardial infarction and improved contractile dysfunction caused by ischemia/reperfusion injury. Wayman et al. (2002) investigated the effects of various chemically distinct activators of PPARγ and PPARα in a rat model of acute myocardial infarction. Using Northern blot analysis and RT-PCR in samples of rat heart, Wayman et al. (2002) documented the expression of the mRNA for PPARγ (isoform 1 but not isoform 2) as well as PPARδ and PPARα in freshly isolated cardiac myocytes and cardiac fibroblasts and in the left and right ventricles of the heart.
Recent studies in humans(Graf et al., 1997) and animal models(Lapointe et al., 2002) have shown that the expression of myocardial endothelin-1 (ET-1) isincreased during cardiac fibrosis. It is suggested that ET-1 might contribute to cardiac fibroblast proliferation(Piacentini et al., 2000) resulting in cardiac fibrosis (Ammarguellat et al., 2001). In vitro study revealed that Ang II-stimulated cardiomyocyte hypertrophy and fibroblast proliferation is mediated by release of ET-1 from fibroblasts rather than by direct action(Gray et al., 1998; Cheng et al., 2002). AP-1 and GATA2 have been shown to regulate transcription of the ET-1 gene in a cooperative fashion through the GATA and AP1 sites located in the promoter region of ET-1 gene in endothelial cells(Kawana et al., 1995). Mechanical stretch-induced ET-1 gene induction was proposed to be mediated through the AP-1 element of ET-1 gene(Cheng et al., 2001).
研究方法 Materials.
Dulbecco's modified Eagle's medium (DMEM),
fetal calf serum, and tissue culture reagents were from Life Technologies, Inc. A rat ET-1 cDNA probe (accession No. M64711) was obtained as previously described (10). A full length of the ET-1 promoter region (4.4 kb) was fused to the chloramphenicol acetyltransferase (CAT) reporter gene (7). PBLCAT2 (containing CAT reporter gene with its promoter) and PBLCAT3 (containing the CAT gene only) were constructed as previously described (8). 2’,7’-dichlorofluorescin diacetate (DCF-DA) was obtained from Molecular Probes (Eugene, OR, U.S.A.). H2O2 was
purchased from Acros Organics (Pittsburgh, PA, U.S.A.). Rosiglitazone, N-acetyl-cysteine (NAC) and all other reagent-grade chemicals were purchased from the Sigma Chemical Co. (St. Louis, MO, U.S.A.). The plasmid AP-1-Luc containing the firefly luciferase reporter gene driven by a basic promoter element (TATA box)joined to tandem repeats of AP-1 binding element were obtained from Stratagene (La Jolla, CA, U.S.A.).
Culture of Cardiac Fibroblasts.
The investigation was conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and approved by the Institutional AnimalCare and Use Committee of Taipei Medical University. Primary cultures of neonatal rat cardiac fibroblasts were prepared as previously described (5). Briefly, ventricles from 1- to 2-day-old neonatal Sprague-Dawley rats were cut into chunks of approximately 1 mm3 using scissors and subjected to trypsin (0.125%, Gibco) digestion in phosphate-buffered saline (PBS). Dispersed cells were incubated on 100 mm culture dishes for 30 min in 5% CO2 incubator, and
VI
non-myocytes attached to the dishes floor were subsequently incubated with minimum essential medium (MEM) supplemented with 10% fetal calf serum. 2-4 d after seeding, confluent non-myocytes were trypsinized and subcultured. Subconfluent (~70% confluency) cardiac fibroblasts grown in either 60- or 100-mm culture dishes from the second to fourth passage were used for the experiments. Serum-containing medium from these cultured cells was replaced with serum-free medium and exposed to agents as indicated. Cells were then preincubated with trilinolein for 30 min and then with or without Ang II (100 nM) for different incubation times as indicated, followed by harvesting. Cellular viability under all treatmentconditions was determined by cell count, morphology, and trypanblueexclusion. DNA Synthesis
To measure synthesis of new DNA, cells (1 x 105/well) were plated in six-well (35-mm) dishes 24 h before experiments as previously described (6). Cells were incubated with [3H]thymidine (5µ Ci/ml). Following the indicated treatment, cells were harvested by incubation at 4 °C with trichloroacetic acid (5%) followed by solubilization in 0.1 N NaOH. Radioactivity was determined by scintillation counting. Data are presented as the mean ± SEM of 9-12 determinations for three to four different cell preparations and normalized to the untreatedsample x 100 (i.e. percentage of control).
Assay of Intracellular ROS
The level of ROS were measured using a previously described method (7). Prior to the chemical or Ang II treatment, cells were incubated in culture medium containing a fluorescent dye, 2' 7'-dichlorofluorescin diacetate (DCF-DA) (Molecular Probes,
Eugene, OR, U. S. A.) at a concentration of 30µM for one hour to establish a stable intracellular level for the probe. The same concentration of DCF-DA was maintained during either chemical or Ang II treatment. Subsequently, the cells were washed with PBS, removed from Petri dishes by brief trypsinization, and then assessed for their 2’,7’-dichlorofluorescein (DCF) fluorescence intensity. The DCF fluorescence intensity of the cells is an index of intracellular levels of ROS. It was determined by fluorescence spectrophotometry with excitation and emission wavelengths at 475 and 525 nm, respectively. After harvesting, cells were counted in an automatic cell counter (S.ST.II/ZM, Coulter Electronics Ltd., Miami, FL, U.S.A.). The cell number in each sample was counted and used to normalize the DCF fluorescence intensity.
RNA Isolation and Northern Blot Analysis Total RNA was isolated from cells by the guanidine isothiocyanate/phenol chloroform method as previously described (7). The RNA (10 µg/lane) was separated by electrophoresis on a 1% agarose formaldehyde gel and transferred onto a nylon membrane (Nytran, Schleicher & Schuell, Inc, Germany) by a vacuum blotting system (VacuGene XL, Pharmacia, Sweden). Following hybridization with the 32P-labeled ET-1 cDNA probes, the membrane was washed with 0.1x SSC containing 1% SDS at 42°C for 30 min and then exposed to x-ray film at -70°C. Blots of specific mRNA bands were detected by autoradiography and analyzed with a densitometer (Computing Densitometer 300S, Molecular Dynamics). Blots were stripped and reprobed with 18S cDNA probe (obtained from American Type Culture Collection) to control
VII
for loading. The level of expression of ET-1 mRNA was quantitatedand was normalized to the 18S signal.
Transfection and Chloramphenicol Acetyltransferase Assays
For the transient transfections, cells were transfected with different expression vectors by the calcium phosphate method (8). DNA concentration in all samples was adjusted to equal amounts with empty vector pSRα in each experiment. To correct for variability in transfection efficiency, 5 µg of pSV-β‘-galactosidase plasmid DNA was cotransfected in all the experiments. The CAT and β‘-galactosidase assays were performed as previously described (8). The relative CAT activity was corrected by normalizing the respective CAT value to that of β-galactosidase activity. Cotransfected β-galactosidase activity varied by <10% within a given experiment and was not affected by any of the experimental manipulations described. pBLCAT2 (with thymidine kinase promoter) and pBLCAT3 (without promoter) were included in each assay as positive and negative controls.
Western Blot Analysis
Rabbit polyclonal anti-phospho-specific ERK antibodies were purchased from New England Biolabs (Beverly, MA, U.S.A.). Anti-ERK antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, U.S.A.). Western blot analysis was performed as previously described (7).
Luciferase assay
Smooth muscle cells plated on six-well (35-mm) dishes were transfected with the luciferase reporter construct possessing consensus AP-1 binding sites (AP-1-Luc)(Stratagene, La Jolla, CA, U.S.A.). Following incubation for 24 h in serum-freeDMEM, smooth muscle cells were
cultured under various conditions as indicated for a period of 48 h. Smooth muscle cells were assayed for luciferase activity with a luciferase reporter assay kit (Stratagene). As was the case for AP-1 transcriptional activity, the specific firefly luciferase activitiy was normalized for transfection efficiency to its respective
β-galactosidase activity and expressed relative to the control.
Statistical Analysis
Results are expressed as mean + SEM for at least three experiments unless designated otherwise. Statistical analysis was performed using analysis of variance (ANOVA) and Student's t test as appropriate. A value of p<0.05 was considered to be statistically significant.
結果與討論 Results
Ang II-induced proliferation of cardiac fibroblast is mediated via ETA receptor
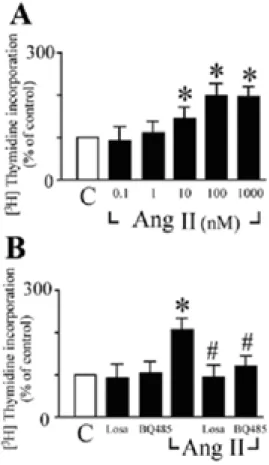
Ang II-stimulated cardiac fibroblast proliferation was assessed by analyzing DNA synthesis with [3H]thymidine incorporation. Ang II increased DNA synthesis in neonatal rat cardiac fibroblasts in a dose-dependent manner (Figure 1A). The maximum level of incorporation was 2.1-fold increase compared with the control. Ang II (100 nM)-stimulated DNA synthesis in cardiac fibroblasts was inhibited by either AT1 receptor antagonist
losartan (1 µM) or ETA receptor antagonist
BQ485 (1 µM) treatment (Figure 1B). Both losartan and BQ485 had no effect on basal [3H]thymidine uptake. These data suggest the possible role of endogenous ET-1 as an autocrine growth factor for the proliferation of cardiac fibroblasts under Ang II stimulation. Ang II-induced ET-1 gene expression in cardiac fibroblasts
VIII
To confirm that Ang II increases ET-1 mRNA levels in cardiac fibroblasts, we performed Northern blot analysis (Figure 2A and B). ET-1 mRNA was induced by Ang II (100 nM) as early as 30 min and then returned to the basal level after 2 h (Figure 2A). When cardiac fibroblasts were treated with Ang II for 30 min, the Ang II-induced ET-1 mRNA expression was dose-dependent with the maximum induction at 100 nM (Figure 2B). To determine whether the Ang II-induced ET-1 expression is regulated at the transcriptional level, an ET-1 promoter construct containing the ET-1 promoter region (–4.4 kb) and the
reporter gene chloramphenicol acetyltransferase (CAT) was constructed and
transiently transfected into cardiac fibroblasts. Cardiac fibroblasts exposed to 24 h of Ang II (100 nM) significantly increased ET-1 promoter activity by 2.6-fold compared with untreated cells (Figure 2C). The effect of AT1
receptor blocker losartan on the Ang II-increased ET-1 promoter activity was also investigated (Figure 2C). Cardiac fibroblasts were pretreated with losartan (1 µM) for one hour and subsequently stimulated with Ang II for 24 h. The Ang II-induced ET-1 gene expression was inhibited by losartan. Ang II dose-dependently increased ET-1 promoter activity. 100 nM of Ang II also gave maximum induction (Figure 2D). These data show that Ang II directly induces ET-1 gene expression in cardiac fibroblasts.
Ang II-induced ET-1 gene expression is mediated via Ras/Raf/ERK signaling pathway
Ang II has been shown to activate ERK, JNK, and p38MAPK in cardiac fibroblasts and the activation of these pathways is redox-sensitive (3). To study whether these
pathways were involved in Ang II-induced ET-1 gene expression in cardiac fibroblasts, we examined the effect of antioxidants on each MAPK pathway and determined the effect of MAPK inhibitors on Ang II-induced ET-1 gene expression. We first confirmed that Ang II increases phosphorylation of ERK1/2, p38MAPK, and JNK in cardiac fibroblasts (Figure 3A, B, and C). Both catalase (350 U/ml) and NAC (10 mM) significantly inhibited Ang II-induced phosphorylation of ERK1/2, p38MAPK, and JNK (Figure 3A, B, and C). These data suggest that ERK1/2, p38MAPK, and JNK are critical components of the redox-sensitive signaling pathways activated by Ang-II in cardiac fibroblasts. We next determined the role of redox-sensitive activation of MAPKs in Ang II-induced ET-1 gene expression. PD98059, a specific inhibitor of MKK-1 (MEK), inhibited augmentation of ET-1 mRNA expression stimulated with Ang II (Figure 3D). SB203580, a specific inhibitor of p38 MAPK, failed to fully inhibit this expression (Figure 3D). Similarly, coincubation with PD98059 also completely abolished Ang II-increased ET-1 promoter activity, but coincubation with SB203580 had no significant effect (Figure 3E). These findings suggest that activation of ERK is a necessary step for ET-1 gene expression induced with Ang II.
To identify the signaling pathway involved in the Ang II-induced ET-1 expression, we also cotransfected cardiac fibroblasts with various dominant negative mutants Ras (RasN17), Raf-1 (Raf301) or a catalytically inactive mutant of ERK2 (mERK), all of which are associated with the Ras/Raf/ERK pathway. Cardiac fibroblasts cotransfected with the empty vector PSRα as control revealed no effect on Ang II-induced ET-1 promoter
IX
activity (Figure 3F). However, cells cotransfected with RasN17, Raf301 or mERK resulted in a significant inhibition in Ang II-induced ET-1 promoter activity. In contrast, cardiac fibroblasts cotransfected with a dominant positive mutant of Ras (RasL61) or MEK1 greatly increased their ET-1 promoter activities. These results further suggest that the Ras/Raf/ERK signaling pathway plays an important role in Ang II-induced ET-1 gene expression in cardiac fibroblasts.
Identification of Ang II-responsive regulatory elements in the ET-1 promoter
The ET-1 promoter contains a number of AP-1 and GATA sites, which can be regulated by multiple activation pathways (10,14). We dissected the Ang II-responsive elements of the ET-1 promoter in cardiac fibroblasts. A series of deletion mutants containing various lengths of the ET-1 promoter region fused to CAT reporter gene were transfected into cardiac fibroblasts and CAT activity was measured in response to Ang II stimulation. Ang II stimulation for 24 hours significantly increased CAT activity by 2.5- and 2.2-fold in -700CAT and -204CAT respectively, both of which contain multiple transcription factor binding sites including GATA (bp -136 to -131) and AP-1 (bp -108 to -102) sites. However, after further truncation of the GATA and subsequent AP-1 site from the 5'-end, the increase of the Ang II-induced ET-1 promoter activity were completely abolished in both -129CAT and -98CAT. It is interesting that deletion of these two sites also resulted in a significant decrease in basal promoter activity (Figure 4A). These findings suggest that the GATA site as well as AP-1 site are necessary for Ang II-stimulated ET-1 gene induction. We further examined whether AP-1 site is essential for the induction
of ET-1 gene by Ang II. In cells transfected with reporter construct -204CAT containing both GATA and AP-1 sites with two-bp mutation in the AP-1 site, the Ang II-induced ET-1 promoter activity was completely abolished. In addition, the basal promoter activity also decreased as compared with control (Figure 4B). These findings suggest that the AP-1 binding element is essential for the induction of ET-1 gene by Ang II.
Rosiglitazone inhibits Ang II-induced proliferation of cardiac fibroblast
We further examined whether rosiglitazone attenuated Ang II-stimulated fibroblast proliferation. Ang II (100 nM)-stimulated incorporation of [3H]thymidine was inhibited by trilinolein (0.1, 1, 10 µM) (Figure 5). These data indicate that rosiglitazone inhibits Ang II-increased proliferation in cardiac fibroblasts.
X
Figure 1. Characteristics of the activation of DNA synthesis by Ang II in cardiac fibroblasts. All experiments were performed with the incorporation of [3H]thymidine into DNA. (A) Effect of Ang II concentration on the DNA synthesis. Cells were incubated with the indicated doses of Ang II for 24 hours and then assayed [3H]thymidine incorporation. (B) Effect of Ang II or ET-1 receptor antagonists on [3H]thymidine incorporation. Cells were preincubated with either losartan (Losa; 1 µM) or BQ485 (1 µM) for one hour followed by an incubation with 100 nM Ang II for 24 hours. Experimental details are given in "Experimental Procedures". [3H]thymidine incorporation is expressed as percentage increase relative to the [3H] content (100%) in their respective control (C). All data are shown as the means + SEM of 9-12 determinations in
three to four different cell preparations. * P < 0.05 vs. control (Student t test); #P<0.05 vs. Ang II alone (ANOVA).
Figure 2. Effect of angiotensin II (Ang II) on endothelin-1 (ET-1) gene expression in neonatal rat cardiac fibroblasts. (A) Time course of Ang II on ET-1 mRNA expression. Cells were incubated with Ang II (100 nM) for the indicated times. (B) Dose-dependent effect of Ang II on ET-1 mRNA expression. Cells were incubated with various doses of Ang II for 30 minutes. (C) Time course of Ang II-increased ET-1 promoter activity. Cardiac fibroblasts were transfected with chimeric ET-1 promoter-CAT fusion genes followed by treatment with Ang II (100 nM) for the time indicated or pretreated with losartan (Losa; 1 µ
M) for one hour followed by Ang II stimulation. (D) Induction of ET-1 promoter activity by different concentration of Ang II. Cells were harvested and CAT activities were measured as described in "Experimental Procedures". C (control), no drugs; CAT2 and CAT3 are shown
XI
as positive and negative control. CAT activities are shown as % incorporation after normalizing to that of β-galactosidase activities. Data are represented as difference relative to control groups. The results are shown as mean + SEM (n = 3 per group). *P<0.05 vs. control (Student t test); #P<0.05 vs. Ang II alone (ANOVA). The experiment was repeated three times with reproducible results.
Figure 3. Ang II increased ET-1 gene expression via
ERK in a redox-sensitive manner. (A) Through (C) Ang II-induced activation of ERK, JNK, and p38MAPK was mediated by ROS-sensitive pathway. Cells were preincubated with either the catalase (350 U/ml), or NAC (10 mM) for 30 minutes and stimulated with Ang II (100 nM) for 30 minutes. Phosphorylation of ERK, JNK, or p38MAPK was detected by Western blotting using anti-phospho-ERK, phospho-JNK, and phospho-p38MAPK antibodies. Both catalase and NAC inhibited Ang II-induced activation of ERK, JNK, or p38MAPK. Phosphorylations of ERK, JNK, or p38MAPK were detected, and densitometric analyses were performed.
The results are shown as mean + SEM (n= 4 per group). *P<0.05 vs. control (Student t test); #P<0.05 vs. Ang II alone (ANOVA). (D) Ang II-induced ET-1 mRNA was attenuated by PD98059 in cardiac fibroblasts. Cardiac fibroblasts were stimulated with Ang II (100 nM) in the presence of PD98059 (PD; 20
µM) or SB203580 (SB; 20 µM), and total RNA was isolated at 30 minutes. (E) Ang II-increased ET-1 promoter activity was inhibited by PD98059 in cardiac fibroblasts. Cardiac fibroblasts were stimulated with Ang II (100 nM) in the presence of PD98059 (PD; 20 µM) or SB203580 (SB; 20 µM), and CAT activity was assayed after 24 h. (F) Ang II-increased ET-1 promoter activity via Ras/Raf/ERK pathway in cardiac fibroblasts. Cells, transfected with either pSRα-empty vector (5 µg), or an expression plasmid encoding the dominant negative mutant
mERK, Raf301, or RasN17 (5 µ g), were
cotransfected with 15 µg of ET-1 promoter-CAT plasmid. Cells cotransfected with ET-1 promoter-CAT plasmid and an expression plasmid encoding MEK1 (5 µg) or RasL61 (5 µg) were used as positive controls. The results are shown as mean + SEM (n=3 per group). *P<0.05 vs. control (Student t test); #P<0.05 vs. Ang II alone (ANOVA).
XII
Figure 4. Identification of Ang II-responsive cis-elements in ET-1 promoter. (A) A series of deletion mutants containing various lengths of ET-1 promoter region were transfected into cardiac fibroblasts. Transfected cells were stimulated with Ang II (100 nM) for 24 h, and CAT activities were measured. Stepwise 5'-deletion constructs were depicted (top). Bars represent mean (+ SEM, n=3 per group). CAT activity of each construct in the presence or absence of Ang II. CAT 2 and CAT 3 were used as positive or negative controls for CAT assay respectively (bottom). (B) Wild type (204 bp) or AP-1 mutant of ET-1 promoter-CAT plasmid was transfected into cardiac fibroblasts. Cells were stimulated with Ang II (100 nM) for 24 h. The mutation of AP-1 strongly abolished the responsiveness to Ang II stimulation. The results are shown as mean + SEM (n=3 per
group). *P<0.05 vs. control (Student t test); #P<0.05 vs. Ang II alone (ANOVA).
Figure 5. Effect of rosiglitazone on Ang II-induced proliferation in cardiac fibroblasts. Effect of rosiglitazone on [3H]thymidine incorporation. Cells were preincubated with different concentration of rosiglitazone (0.1, 1 and 10 µM) for one hour following incubation with 100 nM Ang II for 24 h. Experimental details are given in "Experimental Procedures". Increased in [3H]thymidine incorporation are each expressed relative to the [3H] content (100%) in their respective controls (C). All data are shown as the means + SEM of 9-12 determinations in three to four different cell preparations. * P< 0.05 vs. control; #P<0.05 vs. Ang II alone.
計畫成果自評 :研究內容與原計畫完全相 符、順利達成預期目標、研究成果之學術價 值甚高、適合在學術期刊發表。


![HPSH [ 分子間作用力 - 凡得瓦力 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)