Competitive Acetylide C
-
C Bond Scission vs Formation
of a Quadruply Bridging Carbonyl Ligand. X-ray Crystal
Structures of the Two Pentanuclear Clusters
Cp*
3W
3Ru
2(µ
4-C)(µ
3-CPh)(CO)
9and
Cp*
3W
3Ru
2(µ
3-CCBu
t)(CO)
9Pei-Chiun Su,
†Yun Chi,*
,†Chi-Jung Su,
†Shie-Ming Peng,*
,‡and
Gene-Hsiang Lee
‡Department of Chemistry, National Tsing Hua University, Hsinchu 30043, Taiwan, and Department of Chemistry and Instrumentation Center, National Taiwan University,
Taipei 10764, Taiwan Received December 10, 1996X
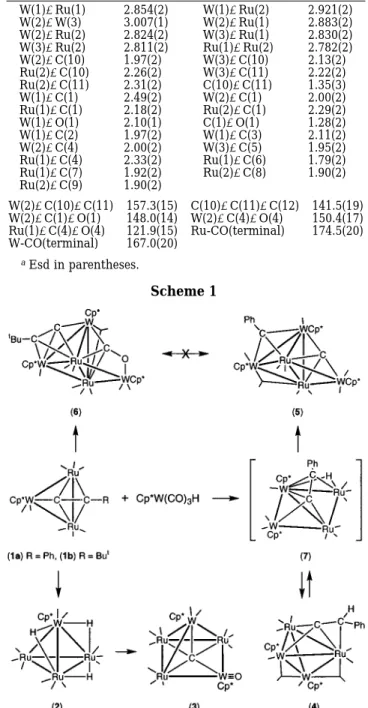
Reactions of Cp*WRu2(CCPh)(CO)8(1a) with excess Cp*W(CO)3H, Cp*)C5Me5, affords a carbido-alkylidyne cluster Cp*3W3Ru2(µ4-C)(µ3-CPh)(CO)9(5) alone with three byproducts, which are identified as hydride cluster Cp*WRu3(µ-H)3(CO)11(2), vinylidene cluster Cp*2W2-Ru2(CCHPh)(CO)9 (4), and a pentanuclear oxo-carbido cluster Cp*2W2(O)Ru3(µ5-C)(CO)11 (3). In contrast, the respective condensation using tert-butyl derivative Cp*WRu2-(CCBut)(CO)8 (1b) gives an acetylide cluster Cp*3W3Ru2(µ3-CCBut)(CO)9 (6). The X-ray structural determinations of 5 and 6 reveals the existence of an edge-bridged tetrahedral core, in which the butterfly crater is occupied by a µ4-carbide in 5 or a quadruply bridging CO ligand in 6. A plausible mechanism leading to the formation of these two cluster compounds is also presented.
Transition metal clusters containing unsaturated alkyne or acetylide fragments have been an area of intense investigation due to their unique physical and chemical properties1,2and their relevance to the trans-formation of small hydrocarbyl intermediates on metal surfaces. Much work has been devoted to the develop-ment of this chemistry in recent years. For example, Carty and co-workers reported the syntheses and crystal structures of phosphido ruthenium clusters containing multisite bound acetylide ligands.3 Adams and co-workers focused on the hydrogenation of alkyne on the layer-segregated, face-shared bioctahedral clusters.4 Other investigations, such as the studies on the ruthe-nium and osmium clusters5 and the heterometallic counterparts,6all provided substantial knowledge about the bonding and reactivity of cluster-bound alkyne and acetylide ligands.
A much less disclosed pattern of reactivity for such complexes is the C-C bond cleavage, which affords two alkylidyne ligands from the coordinated alkynes.7 Like-wise, the related acetylide ligand, which can be
envis-aged as a carbide-alkylidyne in certain polynuclear systems,8 has been observed to convert to the well-separated carbide and alkylidyne ligands.9 In this article, we focus on the irreversible acetylide cleavage through the generation of a Ru2W3mixed-metal cluster, which offers an opportunity to reveal the essence of the acetylide scission vs the competing process involving the generation of the quadruply bridging CO ligand.10 In addition, this study supplements our previous report on the reversible acetylide cleavage between the parent acetylide cluster CpWRu2(CCPh)(CO)8 and the poly-nuclear carbido alkylidyne derivatives CpWRu4(µ5-C)-(µ-CPh)(CO)12and CpWRu5(µ6-C)(µ-CPh)(CO)14, which is also promoted by a cluster building reaction.11
†National Tsing Hua University. ‡National Taiwan University.
XAbstract published in Advance ACS Abstracts, April 1, 1997. (1) (a) Raithby, P. R.; Rosales, M. J. Adv. Inorg. Radiochem. 1985, 29, 169. (b) Bruce, M. I.; Swincer, A. G. Adv. Organomet. Chem. 1983, 22, 59. (c) Kaesz, H. D.; Humphries, A. P. Prog. Inorg. Chem. 1979, 25, 146. (d) Sappa, E.; Tiripicchio, A.; Braunstein, P. Chem. Rev. 1983, 83, 203. (e) Sappa, E.; Tiripicchio, A.; Braunstein, P. Coord. Chem. Rev. 1985, 65, 219. (f) Bruce, M. I. Chem. Rev. 1991, 91, 197.
(2) (a) Nast, R. Coord. Chem. Rev. 1982, 47, 89. (b) Ma, L.; Williams, G. K.; Shapley, J. R. Coord. Chem. Rev. 1993, 128, 261. (c) Manna, J.; John, K. D.; Hopkins, M. D. Adv. Organomet. Chem. 1995, 38, 79. (d) Chi, Y. J. Chin. Chem. Soc. 1992, 39, 591.
(3) (a) Corrigan, J. F.; Taylor, N. J.; Carty, A. J. J. Chem. Soc., Chem. Commun. 1994, 1769. (b) Sun, Y.; Taylor, N. J.; Carty, A. J. Organometallics 1992, 11, 4293.
(4) (a) Adams, R. D.; Barnard, T. S.; Li, Z.; Wu, W.; Yamamoto, J. Organometallics 1994, 13, 2357. (b) Adams, R. D.; Barnard, T. S.; Li, Z.; Wu, W.; Yamamoto, J. J. Am. Chem. Soc. 1994, 116, 9103.
(5) (a) Boyar, E.; Deeming, A. J.; Felix, M. S. B.; Kabir, S. E.; Adatia, T.; Bhusate, R.; McPartlin, M.; Powell, H. R. J. Chem. Soc., Dalton Trans. 1989, 5. (b) Deeming, A. J.; Felix, M. S. B.; Bates, P. A.; Hursthouse, M. B. J. Chem. Soc., Chem. Commun. 1987, 461. (c) Rosenberg, E. Polyhedron 1989, 8, 383. (d) Ma, L.; Rodgers, D. P. S.; Wilson, S. R.; Shapley, J. R. Inorg. Chem. 1991, 30, 3591. (e) Johnson, D. K.; Rukachaisirikul, T.; Sun, Y.; Taylor, N. J.; Canty, A. J.; Carty, A. J. Inorg. Chem. 1993, 32, 5544. (f) Haggitt, J. L.; Johnson, B. F. G.; Blake, A. J.; Parsons, S. J. Chem. Soc., Chem. Commun. 1995, 1263. (g) Paw, W.; Lake, C. H.; Churchill, M. R.; Keister, J. B. Organome-tallics 1995, 14, 3768. (h) Wong, W.-Y.; Chan, S.; Wong, W.-T. J. Chem. Soc., Dalton Trans. 1995, 1497. (i) Koike, M.; Hamilton, D. H.; Wilson, S. R.; Shapley, J. R. Organometallics 1996, 15, 4930. (j) Chi, Y.; Hwang, D.-K. In Comprehensive Organometallics Chemistry II; Wilkin-son, G., Stone, F. G. A., Abel, E. W., Eds.; Pergamon: Oxford, U.K., 1995; Vol. 10, Chapter 3.
(6) (a) Roland, E.; Vahrenkamp, H. J. Mol. Catal. 1983, 21, 233. (b) Shapley, J. R.; McAteer, C. H.; Churchill, M. R.; Biondi, L. V. Organometallics 1984, 3, 1595. (c) Bernhardt, W.; Vahrenkamp, H. J. Organomet. Chem. 1990, 383, 357. (d) Bantel, H.; Powell, A. K.; Vahrenkamp, H. Chem. Ber. 1990, 123, 1607. (e) Adams, K. J.; Barker, J. J.; Charmant, J. P. H.; Ganter, C.; Klatt, G.; Knox, S. A. R.; Orpen, A. G.; Ruile, S. J. Chem. Soc., Dalton Trans. 1994, 477. (f) Adams, R. D.; Barnard, T.; Li, Z.; Wu, W.; Yamamoto, J. H. J. Cluster Sci. 1994, 5, 551. (g) Akita, M.; Hirakawa, H.; Sakaki, K.; Moro-oka, Y. Organometallics 1995, 14, 2775. (h) Adams, H.; Bailey, N. A.; Gill, L. J.; Morris, M. J.; Wildgoose, F. A. J. Chem. Soc., Dalton Trans. 1996, 1437. (i) Norton, D. M.; Eveland, R. W.; Hutchison, J. C.; Stern, C.; Shriver, D. F. Organometallics 1996, 15, 3916.
S0276-7333(96)01041-2 CCC: $14.00 © 1997 American Chemical Society
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
Experimental Section
General Information and Materials. Infrared spectra
were recorded on a Perkin-Elmer 2000 FT-IR spectrometer.
1H and13C NMR spectra were recorded on a Bruker AM-400,
a Varian Gemini-300, or a Varian Unity-400 instrument. 1H
and 13C NMR chemical shifts are quoted with respect to
internal standard TMS. Mass spectra were obtained on a JEOL-HX110 instrument operating in fast atom bombardment (FAB) mode. The acetylide complexes Cp*WRu2(CO)8(CCR),
R ) Ph and Bu
t, were prepared according to literature
procedures.12 All reactions were performed under nitrogen
using deoxygenated solvents dried with an appropriate re-agent. Reactions were monitored by analytical thin-layer chromatography (5735 Kieselgel 60 F254, E. Merck), and the
products were separated on commercially available preparative thin-layer chromatographic plates (Kieselgel 60 F254, E.
Mer-ck). Elemental analyses were carried out at the regional instrumentation Center at National Cheng Kung University, Tainan, Taiwan.
Reaction of 1a with Cp*W(CO)3H. A heptane solution
(80 mL) of acetylide complex Cp*WRu2(CCPh)(CO)8(1a; 100
mg, 0.118 mmol) and Cp*W(CO)3H (135 mg, 0.334 mmol) was
heated to reflux for 8 h, during which the color gradually changed from orange to dark-brown. After removal of the solvent under vacuum, the residue was redissolved in CH2Cl2
and separated by thin-layer chromatography (silica gel, dichlo-romethane-hexane 3:7), producing 35 mg of the starting material Cp*WRu2(CCPh)(CO)8(0.041 mmol, 35%), 2 mg of
orange trihydride complex Cp*WRu3(µ-H)3(CO)11 (2; 0.023
mmol, 2%), 6 mg of orange-red Cp*2W2(O)Ru3(µ5-C)(CO)11(3;
0.0046 mmol, 4%), 3 mg of dark-brown Cp*2W2Ru2
(CCHPh)-(CO)9(4; 0.0025 mmol, 2%), and 22 mg of black Cp*3W3Ru2
-(µ4-C)(µ3-CPh)(CO)9(5; 0.013 mmol, 12%) in the order of their
elution. Crystals of 5 suitable for X-ray diffraction studies were obtained by recrystallization from a layered solution of dichloromethane-methanol at room temperature.
Spectral data of 3: MS (FAB,102Ru,184W) m/z 1275 (M+
). IR (C6H12) ν(CO), 2059 (s), 2022 (vs), 2006 (m), 1990 (s), 1955 (w), 1934 (w), 1802 (vw, br) cm-1 ; IR (KBr) ν(WtO), 928 (s) cm-1;1H NMR (CDCl 3, 294 K) δ 2.14 (s, 15H), 1.72 (s, 15H); 13C NMR (CD 2Cl2, 200 K) δ 441.5 (µ5-C, JW-C)88 and 101 Hz), 223.0 (2CO, br), 212.0 (2CO, JW-C)161 Hz), 205.8 (2CO, br), 201.4 (2CO), 199.6 (2CO, br), 192.2 (1CO), 117.1 (C5Me5),
102.3 (C5Me5), 11.6 (C5Me5), 9.7 (C5Me5). Anal. Calcd for
C32H30O12Ru3W2: C, 30.09; H, 2.37. Found: C, 30.03; H, 2.47.
Spectral data for 4: MS (FAB,102Ru,184W) m/z 1196 (M+);
IR (C6H12) ν(CO), 2049 (s), 2008 (vs), 1990 (m), 1976 (s), 1961 (s), 1920 (w), 1797 (vw, br), 1780 (w) cm-1;1H NMR (CDCl 3, 294 K) δ 7.48 (d, 2H, JH-H )7.4 Hz), 7.27 (t, 2H, JH -H )7.4 Hz), 7.12 (t, 1H, JH-H )7.4 Hz), 6.06 (s, 1H), 2.01 (s, 15H), 1.94 (s, 15H). Anal. Calcd for C37H36O9Ru2W2: C, 37.20; H,
3.04. Found: C, 37.45; H, 3.16.
Spectral data for 5: MS (FAB,102Ru,184W) m/z 1514 (M+
); IR (C6H12) ν(CO), 1994 (w), 1971 (vs), 1936 (s), 1929 (m), 1918 (w), 1909 (w), 1863 (vw, br), 1762 (m) cm-1 ;1H NMR (CDCl 3, 294 K) δ 7.48 (d, 1H, JH-H )7.5 Hz), 7.25-7.09 (m, 3H), 7.00 (t, 1H, JH-H )7.0 Hz), 2.14 (s, 15H), 1.81 (s, 15H), 1.74 (s, 15H);13C NMR (CD 2Cl2, 294 K) CO, δ 251.6 (JW-C )120 Hz), 239.5 (JW-C)170 Hz), 220.1 (JW-C)167 Hz), 218.4 (JW-C )160 Hz), 218.0 (JW-C)160 Hz), 210.0, 209.6, 208.2, 208.0; δ 402.0 (µ4-C, JW-C ) 119 Hz), 280.3 (µ3-CPh, JW-C ) 96 Hz), 160.9 (i-C6H5), 130.1 (CH), 128.7 (CH), 126.3 (CH), 126.0 (CH), 124.8 (CH), 106.6 (C5Me5), 106.0 (C5Me5), 101.0
(C5Me5), 11.3 (C5Me5), 10.8 (C5Me5), 10.2 (C5Me5). Anal. Calcd
for C47H50O9Ru2W3‚
1/
2CHCl3: C, 36.29; H, 3.24. Found: C,
36.00; H, 3.23.
Reaction of 2 with Cp*W(CO)3H. A toluene solution (25
mL) of 2 (11.4 mg, 0.0122 mmol) and Cp*W(CO)3H (26 mg,
0.065 mmol) was brought to reflux under nitrogen. The heating was continued for 4 h, during which (20 mg× 3) of Cp*W(CO)3H was added into the mixture on the hour. After
removal of the solvent in vacuo, the residue was taken up in CH2Cl2and separated by thin-layer chromatography (silica gel,
dichloromethane-hexane 1:5), producing 5.5 mg of orange-red 3 (0.004 mmol, 30%) and 6 mg of unreacted starting material 2 (52%).
Reaction of 4 with Cp*W(CO)3H. A toluene solution (15
mL) of vinylidene complex 4 (9.3 mg, 0.0078 mmol) and Cp*W-(CO)3H (15.7 mg, 0.039 mmol) was heated to reflux for 90 min.
After removal of the solvent in vacuo, the residue was separated by thin-layer chromatography (silica gel, dichlo-romethane-hexane 1:5), producing 8.5 mg of black 5 (0.0056 mmol, 72%).
Reaction of 1b with Cp*W(CO)3H. A heptane solution
(60 mL) of acetylide complex CpWRu2(CCBut)(CO)8(1b; 50 mg,
0.061 mmol) and Cp*W(CO)3H (73 mg, 0.18 mmol) was heated
to reflux for 8 h under nitrogen, during which the color gradually changed from orange to dark-brown. After removal of the solvent in vacuo, the residue was redissolved in CH2Cl2
and separated by thin-layer chromatography (silica gel, dichlo-romethane-hexane 1:3), producing 16 mg of the starting material 1b (0.019 mmol, 32%), and 14 mg of black Cp*3W3
-Ru2(µ3-CCBut)(CO)9 (6; 0.0094 mmol, 15%). Crystals of 6
suitable for X-ray diffraction studies were obtained by recrys-tallization from a layered solution of dichloromethane -methanol at room temperature.
Spectral data for 6: MS (FAB,102Ru,184W) m/z 1494 (M+);
IR (C6H12) ν(CO), 2004 (s), 1972 (vs), 1955 (s), 1930 (vw), 1916 (m), 1871 (vw), 1836 (w, br), 1673 (w, br) cm-1 ; 1H NMR (CDCl3, 294 K) δ 2.14 (s, 15H), 1.99 (s, 15H), 1.98 (s, 15H), 1.51 (s, 9H);13C NMR (CD 2Cl2, 294 K) CO, δ 287.2 (JW-C ) 161 Hz), 285.6 (JW-C )160 Hz), 234.5 (JW -C )184 Hz), 230.1 (JW-C )171 Hz), 219.5 (JW -C )144 Hz), 207.6, 202.5, 201.4, 198.8; δ 174.9 (CCBut, J W-C )134 Hz), 144.1 (CCBu t), 105.8 (C5Me5), 103.6 (C5Me5), 103.3 (C5Me5), 41.5 (CMe3), 37.1 (Me),
12.2 (C5Me5), 11.3 (C5Me5), 9.6 (C5Me5). Anal. Calcd for C46H56Cl2O9Ru2W3: C, 35.02; H, 3.58. Found: C, 35.14; H,
3.57.
X-ray Crystallography. The X-ray diffraction
measure-ments were carried out on a Nonius CAD-4 diffractometer.
(7) (a) Park, J. T.; Shapley, J. R.; Churchill, M. R.; Bueno, C. J. Am. Chem. Soc. 1983, 105, 6182. (b) Gomez-Sal, M. P.; Johnson, B. F. G.; Kamarudin, R. A.; Lewis, J.; Raithby, P. R. J. Chem. Soc., Chem. Commun. 1985, 1622. (c) Chi, Y.; Shapley, J. R. Organometallics 1985, 4, 1900. (d) Eaton, B.; O’Connor, J. M.; Vollhardt, K. P. C. Organo-metallics 1986, 5, 394. (e) Cabrera, E.; Daran, J. C.; Jeannin, Y. J. Chem. Soc., Chem. Commun. 1988, 607. (f) Stone, F. G. A.; Williams, M. L. J. Chem. Soc., Dalton Trans. 1988, 2467. (g) Park, J. T.; Shapley, J. R.; Bueno, C.; Ziller, J. W.; Churchill, M. R. Organometallics 1988, 7, 2307. (h) Johnson, B. F. G.; Lewis, J.; Lunniss, J. A.; Braga, D.; Grepioni, F. J. Chem. Soc., Chem. Commun. 1988, 972. (i) Rumin, R.; Robin, F.; Petillon, F. Y.; Muir, K. W.; Stevenson, I. Organometallics 1991, 10, 2274. (j) Braga, D.; Grepioni, F.; Johnson, B. F. G.; Lewis, J.; Lunniss, J. A. J. Chem. Soc., Dalton Trans. 1991, 2223. (k) Braga, D.; Grepioni, F.; Johnson, B. F. G.; Lewis, J.; Lunniss, J. A. J. Chem. Soc., Dalton Trans. 1992, 1101. (l) Shaposhnikova, A. D.; Drab, M. V.; Kamalov, G. L.; Pasynskii, A. A.; Eremenko, I. L.; Nefedov, S. E.; Struchkov, Y. T.; Yanovsky, A. I. J. Organomet. Chem. 1992, 429, 109. (m) Park, J. T.; Woo, B. W.; Chung, J.-H.; Shim, S. C.; Lee, J.-H.; Lim, S.-S.; Suh, I.-H. Organometallics 1994, 13, 3384. (n) Pasynskii, A. A.; Eremenko, I. L.; Nefedov, S. E.; Kolobkov, B. I.; Shaposhnikova, A. D.; Stadnitchenko, R. A.; Drab, M. V.; Struchkov, Y. T.; Yanovsky, A. I. New J. Chem. 1994, 18, 69. (o) Quenec’h, P.; Rumin, R.; Pe´tillon, F. Y. J. Organomet. Chem. 1994, 479, 93. (p) Adams, H.; Cill, L. J.; Morris, M. J. Organometallics 1996, 15, 4182. (q) Adams, H.; Cill, L. J.; Morris, M. J. J. Chem. Soc., Dalton Trans. 1996, 3909.
(8) (a) Nucciarone, D.; Taylor, N. J.; Carty, A. J. Organometallics 1986, 5, 1179. (b) Blenkiron, P.; Taylor, N. J.; Carty, A. J. J. Chem. Soc., Chem. Commun. 1995, 327.
(9) (a) Wu, C.-H.; Chi, Y.; Peng, S.-M.; Lee, G.-H. J. Chem. Soc., Dalton Trans. 1990, 3025. (b) Chi, Y.; Su, P.-C.; Peng, S.-M.; Lee, G.-H. Organometallics 1995, 14, 5483. (c) Chi, Y.; Chung, C.; Chou, Y.-C.; Su, P.-Y.-C.; Chiang, S.-J.; Peng, S.-M.; Lee, G.-H. Organometallics 1997, 8, 1702.
(10) (a) Horwitz, C. P.; Shriver, D. F. Adv. Organomet. Chem. 1984, 23, 219. (b) Horwitz, C. P.; Shriver, D. F. Organometallics 1984, 3, 756.
(11) Chiang, S.-J.; Chi, Y.; Su, P.-C.; Peng, S.-M.; Lee, G.-H. J. Am. Chem. Soc. 1994, 116, 11181.
(12) Hwang, D.-K.; Chi, Y.; Peng, S.-M.; Lee, G.-H. Organometallics 1990, 9, 2709.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
Lattice parameters were determined from 25 randomly se-lected high-angle reflections. Three standard reflections were monitored every 3600 s. No significant change in intensities, due to crystal decay, was observed over the course of all data collection. Intensities of the diffraction signals were corrected for Lorentz, polarization, and absorption effects (ψ scans). The structure was solved by using the NRCC-SDP-VAX package. All the non-hydrogen atoms had anisotropic temperature factors, and the hydrogen atoms of the organic functional groups were placed at idealized positions with UH)UC+0.1. The crystallographic refinement parameters of complexes 5 and 6 are given in Table 1, while their selective bond distances and angles are presented in Tables 2 and 3, respectively.
Results
Treatment of acetylide cluster Cp*WRu2(CCPh)(CO)8 (1a) with excess Cp*W(CO)3H in refluxing heptane solution for 8 h afforded four cluster complexes in low to moderate yields. These complexes included orange trihydride complex 2 (2%), orange-red, oxo-carbido cluster 3 (4%), dark-brown vinylidene cluster 4 (2%) and
black carbido-alkylidyne cluster 5 (12%) (see Scheme 1). Each cluster complex was separated by thin-layer chromatography on silica gel and was further purified by recrystallization. The first two complexes 2 and 3 were immediately identified by comparing their IR and 1H NMR data with the authentic samples prepared from the reaction of Ru3(CO)12and Cp*W(CO)3H,13and from the condensation of the cluster Cp*WRu3(µ-H)(CO)12or 2 with Cp*W(CO)3H, respectively.14 This reactivity pattern strongly suggests that the oxo-carbido complex 3 is produced by the prior formation of the cluster Cp*WRu3(µ-H)(CO)12or 2, while the latter is generated from a parallel reaction of Cp*W(CO)3H with Ru3(CO)12, and the Ru3(CO)12existed in the solution is most likely produced from the thermally induced decomposition of (13) (a) Chi, Y.; Cheng, C.-Y.; Wang, S.-L. J. Organomet. Chem. 1989, 378, 45. (b) Chen, C.-C.; Chi, Y.; Peng, S.-M.; Lee, G.-H. J. Chem. Soc., Dalton Trans. 1993, 1823.
(14) Su, C.-J.; Su, P.-C.; Chi, Y.; Peng, S.-M.; Lee, G.-H. J. Am. Chem. Soc. 1996, 118, 3289.
Table 1. Crystal Data for the X-ray Diffraction Studies of Complexes 5 and 6a
compound 5 6
formula C47H50O9Ru2W3‚ 1/
2CHCl3 C45H54O9Ru2W3‚CH2Cl2
mol wt 1572.30 1577.52
crystal system monoclinic orthorhombic space group P21/n P212121 a (Å) 12.531(3) 16.949(4) b (Å) 16.970(4) 17.316(4) c (Å) 22.086(6) 32.908(6) β (deg) 99.82(2) volume (Å3) 4628(2) 9658(4) Z 4 8 Dc(g/cm3) 2.256 2.170 F(000) 2972 5984 2θ (max) (deg) 50 50 h,k,l ranges -14 to+14, 0-20, 0-26 0-20, 0-20, 0-39 crystal size, mm 0.10× 0.22 × 0.50 0.30× 0.40 × 0.45 µ(Mo KR) (cm -1) 83.59 79.23 transmission: max, min 1.00, 0.51 1.00, 0.68 no. of unique data 8089 9249 no. of data with
I>2σ(I)
6373 6349
no. of atoms and
params 114, 568 236, 1108 weight modifier, g 0.00005 0.00005 max ∆/σ ratio 0.007 0.082 RF; Rw 0.031; 0.030 0.040; 0.038 GOF 1.61 1.47 D-map, max/min (e/Å3) 1.49/ -1.24 1.42/-1.96
aFeatures common to all determinations: Nonius CAD-4
dif-fractometer, λ(Mo KR))0.7107 Å; minimize function∑(w|Fo -Fc|2), weighting scheme w-1 )σ 2(F o)+|g| Fo 2; GOF )[∑w|Fo -Fc|2/(No - Nv)]1/2 (No, number of observations; Nv, number of variables).
Table 2. Selected Bond Distances (Å) and Bond Angles (deg) of 5a
W(1)-Ru(1) 2.889(1) W(1)-Ru(2) 2.910(1) W(2)-W(3) 3.0560(7) W(2)-Ru(1) 2.742(1) W(2)-Ru(2) 2.873(1) W(3)-Ru(1) 2.887(1) W(3)-Ru(2) 2.767(1) Ru(1)-Ru(2) 2.739(1) W(1)-C(10) 2.037(7) W(3)-C(10) 1.992(7) Ru(1)-C(10) 2.125(8) Ru(2)-C(10) 2.141(7) W(2)-C(11) 2.128(8) W(3)-C(11) 2.104(8) Ru(2)-C(11) 2.120(7) W(1)-C(1) 1.980(9) W(1)-C(2) 1.979(9) W(2)-C(3) 2.094(8) W(2)-C(4) 1.979(9) W(3)-C(5) 1.980(9) Ru(1)-C(6) 1.894(8) Ru(1)-C(7) 1.874(9) Ru(1)-C(3) 2.145(8) Ru(2)-C(8) 1.907(8) Ru(2)-C(9) 1.902(9) W(1)-C(10)-W(6) 172.0(4) Ru(1)-C(10)-Ru(2) 79.9(3) W(2)-C(3)-O(3) 145.2(7) Ru-CO(terminal) 175.7(8) W-CO(terminal) 170.0(8) aEsd in parentheses.
Table 3. Selected Bond Distances (Å) and Bond Angles (deg) of 6
W(1)-Ru(1) 2.854(2) W(1)-Ru(2) 2.921(2) W(2)-W(3) 3.007(1) W(2)-Ru(1) 2.883(2) W(2)-Ru(2) 2.824(2) W(3)-Ru(1) 2.830(2) W(3)-Ru(2) 2.811(2) Ru(1)-Ru(2) 2.782(2) W(2)-C(10) 1.97(2) W(3)-C(10) 2.13(2) Ru(2)-C(10) 2.26(2) W(3)-C(11) 2.22(2) Ru(2)-C(11) 2.31(2) C(10)-C(11) 1.35(3) W(1)-C(1) 2.49(2) W(2)-C(1) 2.00(2) Ru(1)-C(1) 2.18(2) Ru(2)-C(1) 2.29(2) W(1)-O(1) 2.10(1) C(1)-O(1) 1.28(2) W(1)-C(2) 1.97(2) W(1)-C(3) 2.11(2) W(2)-C(4) 2.00(2) W(3)-C(5) 1.95(2) Ru(1)-C(4) 2.33(2) Ru(1)-C(6) 1.79(2) Ru(1)-C(7) 1.92(2) Ru(2)-C(8) 1.90(2) Ru(2)-C(9) 1.90(2) W(2)-C(10)-C(11) 157.3(15) C(10)-C(11)-C(12) 141.5(19) W(2)-C(1)-O(1) 148.0(14) W(2)-C(4)-O(4) 150.4(17) Ru(1)-C(4)-O(4) 121.9(15) Ru-CO(terminal) 174.5(20) W-CO(terminal) 167.0(20)
aEsd in parentheses.
Scheme 1
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
1a. In agreement with this postulation, treatment of 2 with excess Cp*W(CO)3H in toluene affords the expected Ru3W2complex 3 in moderate yield.
The vinylidene complex 4 was established principally from its spectroscopic data. The FAB mass spectrum gave molecular ion M+due to the formula Cp*2W2Ru2-(CCHPh)(CO)9and the peaks corresponding to success-ful loss of nine CO ligands. The IR ν(CO) spectrum shows a pattern similar to that of the structurally characterized W2Os2analogue,15consisting of five Ru
-CO absorptions in the range 2050-1960 cm
-1and three broader peaks at 1920, 1797, and 1780 cm-1, which we attributed to W-bound CO and bridging CO ligands. The presence of vinylidene ligand is evident form the 1H NMR data, which exhibits a singlet at δ 6.06 charac-teristic of a CdCHPh group, in addition to the signals of phenyl and two Cp* ligands.
Complex 5 exhibited three Cp* signals at δ 2.14, 1.81, and 1.74 in the 1H NMR spectrum, suggesting the incorporation of three Cp*W fragments. The13C NMR spectrum showed four Ru-CO resonances between the narrow range δ 210.0 and 208.0 and five W-CO resonances between δ 251.6 and 218.0; the latter were established by their characteristic JW-Ccouplings. In addition, we observed one signal at δ 402.0 and a second one at δ 280.3, which are best assigned to a carbido and an alkylidyne ligand, respectively. In order to confirm this hypothesis, we performed single-crystal X-ray crystallography.
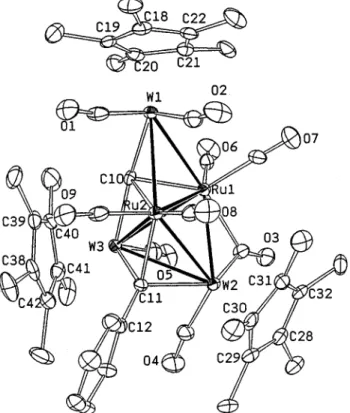
According to the X-ray structural study, complex 5 is as a CH2Cl2solvate. It possesses a pentametallic core of three W atoms and two Ru atoms coordinated by nine CO ligands, one face-bridging alkylidyne ligand and a carbide atom (Figure 1). The metal skeleton consists of an edge-bridged tetrahedral arrangement with a pendant Cp*W(1) unit attached to the Ru2W2 tetrahe-dron. Alternatively, it can be considered to adopt a face-bridged butterfly geometry with the Cp*W(2) fragment coordinated to a Ru2W metal triangle. In this molecule, all metal-metal bonds are normal with the W(2)-W(3) distance being the longest 3.0560(7) Å and the Ru(1) -Ru(2) distance being the shortest 2.7391(11) Å. Other W-Ru distances are within the intermediate range 2.7423(10)-2.9097(10) Å. The variation of metal-metal bond distances is consistent with the fact that the atomic radii of the group 6 metals are slightly greater than that of the group 8 elements. A further shortening of the W(2)-Ru(1) and the W(3)-Ru(2) distances (2.7423-(10) and 2.7674(2.7423-(10) Å) with respect to the rest of W-Ru distances (2.8734(10)-2.9097(10) Å) is probably due to the influence of bridging CO and alkylidyne ligands.
The most important features of 5 are the carbide and alkylidyne ligands which are separated by a distance (3.13(1) Å). The carbide atom C(10) occupied the Ru2W2 butterfly crater; the environment around the carbide atom is analogous to that of the tetranuclear carbide clusters reported in the literature.16 On the other hand, the alkylidyne ligand lies on the adjacent RuW2metal triangle in a virtually symmetrical fashion, W(2)-C(11) ) 2.128(8) Å, W(3)-C(11) ) 2.104(8) Å, and Ru(2) -C(11)) 2.120(7) Å. Its bonding is akin to that of the alkylidyne clusters LWM3(µ3-CR)(CO)11, L )Cp, Cp*; M)Os, Ru; R)H, Ph, OMe.
16e,17 In these alkylidyne
complexes, the face-capping alkylidyne ligand and the edge-bridging CO ligand are all connected to the com-mon LW(CO)2vertex. Moreover, the hexagonal face of phenyl substituent appears to possess some contact with two neighboring Cp* ligands. Consistent with this observation, the13C NMR spectrum at room tempera-ture showed six well-separated signals for the phenyl group, showing the preservation of such highly con-gested conformation even in solution state.
On the contrary, the reaction of tert-butyl derivative 1b and Cp*W(CO)3H led to the formation of acetylide complex 6 (Scheme 1). The1H NMR spectrum showed one tert-butyl group at δ 1.51 and three Cp*W fragments at δ 2.14, 1.99, and 1.98, which agrees with the formula revealed by the FAB mass analysis. In contrast, the 13C NMR spectrum exhibited two high-field signals at
δ 174.9 (JW-C
)134 Hz) and 144.1, which are attributed to the µ3-acetylide ligand12,18 but not the carbide and the alkylidyne as observed in 5.
The structure of 6 was confirmed by X-ray diffraction study. It crystallizes in an orthorhombic space group
P212121with a disordered CHCl3solvent molecule and
(15) Chi, Y.; Wu, C.-H.; Peng, S.-M.; Lee, G.-H. Organometallics 1991, 10, 1676.
(16) (a) Bradley, J. S. Adv. Organomet. Chem. 1982, 22, 1. (b) Harris, S.; Bradley, J. S. Organometallics 1984, 3, 1086. (c) Hriljac, J. A.; Swepston, P. N.; Shriver, D. F. Organometallics 1985, 4, 158. (d) Hriljac, J. A.; Harris, S.; Shriver, D. F. Inorg. Chem. 1988, 27, 816. (e) Chi, Y.; Chuang, S.-H.; Chen, B.-F.; Peng, S.-M.; Lee, G.-H. J. Chem. Soc., Dalton Trans. 1990, 3033. (f) Gong, J.-H.; Tsay, C.-W.; Tu, W.-C.; Chi, Y.; Peng, S.-M.; Lee, G.-H. J. Cluster Sci. 1995, 6, 289.
(17) (a) Busetto, L.; Green, M.; Hesser, B.; Howard, J. A. K.; Jeffery, J. C.; Stone, F. G. A. J. Chem. Soc., Dalton Trans. 1983, 519. (b) Chi, Y.; Lee, G.-H.; Peng, S.-M.; Wu, C.-H. Organometallics 1989, 8, 1574. (c) Gong, J.-H.; Chen, C.-C.; Chi, Y.; Wang, S.-L.; Liao, F.-L. J. Chem. Soc., Dalton Trans. 1993, 1829. (d) Gong, J.-H.; Hwang, D.-K.; Tsay, C.-W.; Chi, Y.; Peng, S.-M.; Lee, G.-H. Organometallics 1994, 13, 1720. (18) (a) Hwang, D.-K.; Chi, Y.; Peng, S.-M.; Lee, G.-H. J. Organomet. Chem. 1990, 389, C7. (b) Chi, Y.; Lee, G.-H.; Peng, S.-M.; Liu, B.-J. Polyhedron 1989, 8, 2003. (c) Carty, A. J.; Cherkas, A. A.; Randall, L. H. Polyhedron 1988, 7, 1045.
Figure 1. Molecular structure of Cp*3W3Ru2(µ4-C)(µ3
-CPh)(CO)9(5) showing the atomic labeling scheme and the
thermal ellipsoids at 30% probability level.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
two crystallographically distinct, but structurally simi-lar, molecules. A prospective view of one of these molecules is depicted in Figure 2. In this compound, the Ru2W2cluster core assumes a closely related edge-bridged tetrahedral skeleton, on which the tert-butyl acetylide resides on the same RuW2face supporting the alkylidyne in 5. The acetylideR-carbon is found to link to W(2) atom via R-bonding, while the C2 vector is bisecting the opposing W(3)-Ru(2) edge. The butterfly cavity is occupied by a µ4-CO ligand with its oxygen atom linking to the Cp*W(CO)2pendent. The formation of µ4-CO ligand is rare in metal clusters, and only a few examples have been reported since it was first docu-mented.19,20 Other important features of 6 involve the relocating of the bridging CO to the W(2)-Ru(1) edge and the removal of terminal CO ligand from the W(2) atom. Such reshuffling of CO ligands is in response to the change of donor capacity from the alkylidyne ligand in 5 (3-electron donor) to the acetylide ligand in 6
(5-electron donor). Finally, the (5-electron counting for both complexes gives 74 valence electrons, which is consistent with the prediction of pentanuclear clusters possessing eight M-M bondings.
Discussion
This work has demonstrated that the formation of the
µ4-CO ligand and the scission of acetylide are two competing processes which can be controlled by chang-ing the substituents on acetylide ligand. We do not understand the actual driving force associated with change of substituents. However, it is obvious that both clusters 5 and 6 are produced through a tetranuclear Ru2W2 intermediate 7 as shown in Scheme 1, which contains a hydride and an acetylide ligand. Evidence in support of this hypothesis is derived from the isolation of the Ru2W2 cluster 4 in trace amount and from the observation of the thermally induced, revers-ible interconversion between the vinylidene and the hydrido-acetylide isomers of its W2Os2 analogue.
15 Moreover, treatment of 4 with excess Cp*W(CO)3H in refluxing toluene afforded the expected pentanuclear complex 5 in high yield, which offers the ultimate confirmation. Unfortunately, we are unable to isolate the corresponding tert-butyl derivatives of 4 or 7, in a finite attempt to illustrate their involvement in the stepwise formation of the acetylide cluster 6.
The generation of the less congested, edge-bridged tetrahedral cluster core also deserves special attention. It can be tracked back to our previous studies on the synthesis of two pentanuclear complexes LWRu4(µ3-H)-(CO)1420eand L2W2Ru3(CO)13, L
) Cp and Cp*. 14 In both cases, the Cp derivatives exhibit the closely packed, trigonal-bipyramidal geometry. However, the Cp* de-rivatives show the formation of the same kind of edge-bridged tetrahedral geometry with one µ4-CO ligand20e or the wingtip-bridged butterfly structure containing one carbide and one oxo ligand generating by C-O bond cleavage.14
On the basis of these experiments, one would instinc-tively expect that the dominant factor in controlling the cluster core arrangement is due to the reduction of interligand repulsion imposed by the Cp* ligand. The coordinative unsaturation generated by changing from the trigonal-bipyramidal to the edge-bridged tetrahedral geometry is thus compensated by forming the µ4-CO ligand as observed in 6. In the presence of ligated acetylide, such as in 5, it would give rise to the carbide and alkylidyne ligands. Clearly, study of the related Cp derivatives of 5 or 6 is vital to the confirmation of this assumption. However, attempts to generate the Cp derivatives via combining CpWRu2(CCR)(CO)8, R)Ph, But, and CpW(CO)
3H under similar conditions gave no stable products at all.
Acknowledgment. We thank the National Science Council of Taiwan, Republic of China for financial support (Grant NSC 85-2113-M007-008).
Supporting Information Available: Tables giving all
bond distances and angles, atomic coordinates, and anisotropic thermal parameters for complexes 5 and 6 (26 pages). Order-ing information is given on any current masthead page. OM9610410
(19) (a) Manassero, M.; Sansoni, M.; Longoni, G.; J. Chem. Soc., Chem. Commun. 1976, 919. (b) Brun, P.; Dawkins, G. M.; Green, M.; Miles, A. D.; Orpen, A. G.; Stone, F. G. A. J. Chem. Soc., Chem. Commun. 1982, 926. (c) Johnson, B. F. G.; Lewis, J.; McPartlin, M.; Pearsall, M.; Sironi, A. J. Chem. Soc., Chem. Commun. 1984, 1089. (d) Horwitz, C. P.; Shriver, D. F. J. Am. Chem. Soc. 1985, 107, 8147. (e) Horwitz, C. P.; Holt, E. M.; Brock, C. P.; Shriver, D. F. J. Am. Chem. Soc. 1985, 107, 8136. (f) Adams, R. D.; Babin, J. E.; Tasi, M. Angew. Chem., Int. Ed. Engl. 1987, 26, 685. (g) Adams, R. D.; Babin, J. E.; Tasi, M. Inorg. Chem. 1988, 27, 2618. (h) Wang, J.; Sabat, M.; Horwitz, C. P.; Shriver, D. F. Inorg. Chem. 1988, 27, 552. (i) Anson, C. E.; Bailey, P. J.; Conole, G.; Johnson, B. F. G.; Lewis, J.; McPartlin, M.; Powell, H. R. J. Chem. Soc., Chem. Commun. 1989, 442.
(20) (a) Chi, Y.; Wu, F.-J.; Liu, B.-J.; Wang, C.-C.; Wang, S.-L. J. Chem. Soc., Chem. Commun. 1989, 873. (b) Chi, Y.; Chuang, S.-H.; Liu, L.-K.; Wen, Y.-S. Organometallics 1991, 10, 2485. (c) Wang, J.-C.; Lin, R.-J.-C.; Chi, Y.; Peng, S.-H.; Lee, G.-H. Organometallics 1993, 12, 4061. (d) Chi, Y.; Su, C.-J.; Farrugia, L. J.; Peng, S.-H.; Lee, G.-H. Organometallics 1994, 13, 4167. (e) Su, C.-J.; Chi, Y.; Peng, S.-M.; Lee, G.-H. Organometallics 1995, 14, 4286.
Figure 2. Molecular structure of Cp*3W3Ru2(µ3-CCBut
)-(CO)9 (6) showing the atomic labeling scheme and the
thermal ellipsoids at 30% probability level.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009