DOI: 10.1002/cbic.201000540
Implantation of Post-translational Tyrosylprotein Sulfation into a
Prokaryotic Expression System
Lu-Yi Lu, Bo-Han Chen, Jennifer Yun-Shin Wu, Chen-Chu Wang, Da-Huang Chen, and Yuh-Shyong Yang*
[a]Tyrosine O-sulfation is a common post-translational modifica-tion of proteins. A previous study estimates that up to approxi-mately 1 % of tyrosine residues in all proteins in an organism are in a sulfated form, and researchers have confirmed tyrosine O-sulfation in about 350 proteins.[1] However, the literature
contains only a few reports in this area since the discovery of protein tyrosine sulfation in 1954.[2]Tyrosine O-sulfation occurs
in the trans-Golgi network, and the targets belong to classes of secretory, plasma membrane, and lysosomal proteins, which reflect their specific intracellular locations.[3]Tyrosylprotein
sul-fotransferases (TPSTs, EC 2.8.2.20) are type-II membrane pro-teins, with a singlea-helical transmembrane segment that ex-poses the catalytic domain in Golgi lumen. They are responsi-ble for catalyzing the transfer of a sulfuryl group from the uni-versal sulfuryl group donor, 3’-phosphoadenosine-5’-phospho-sulfate (PAPS), onto a specific tyrosine residue within target proteins.[4]
Sulfation of tyrosine residues located within specific peptide sequences enhances the strength of protein–protein interac-tions and further mediates numerous crucial physiological events, such as anticoagulation, inflammation, and HIV infec-tion.[1b, 5]The sulfated proteins have great potential for medical
use. An application includes sulfated hirudin, an anticoagulant secreted from the medicinal leech (Hirudo medicinalis), which is a potent thrombin inhibitor.[6] The sulfated antibody helps
defend against HIV infection by binding to the HIV-1 gp120 CCR5-interactive region.[7] However, these recombinant
pro-teins expressed in Escherichia coli or yeast are non-sulfated, due to the absence of TPST gene in these organisms.[3]
Re-searchers have developed a delicate process for the expression of tyrosine-sulfated protein in E. coli, by introducing the amber nonsense codon to encode a sulfotyrosine residue.[8] Despite
the ability to introduce a sulfotyrosine residue during protein translation, the geometric interference of multiple sulfotyrosine clusters, leaky amber suppression, and poor uptake of sulfotyr-osine into expression host are bottlenecks to overcome.[8a]
These days, most sulfated proteins and peptides are available through chemical synthesis and TPST catalysis ; however, the cumbersome procedure, unsatisfactory yield, and the impurity of TPST sources, from either tissue extracts or cell cultures, ob-struct the process.[4b–c, 9]Alternatively, a TPST-catalyzed reaction
provides high specificity for tyrosine-sulfated proteins,
al-though the extremely low catalytic efficiency of TPSTs (the turnover number (kcat) for most known TPSTs is approximately 10 5s 1) might cause low yield and incomplete tyrosine
sulfa-tion.[10]The unavailability of homogeneous and ample
quanti-ties of TPST might also be part of the reason for the lack of re-search activity in this important area. In addition, TPST is labile and difficult to purify by using routine purification methods.[9d]
In this report, we demonstrate an ultra-efficient system for the in vitro synthesis of tyrosine-sulfated proteins. Incorporat-ing this system into a bacterial host produced sulfated tyrosine residue within the specific domain of a target protein. Prokary-otic expression thus provided a constant source of active and homogeneous TPST in large quantities. To prevent interference from the TPST hydrophobic region, we removed the N-terminal transmembrane domain of human TPST isoform 2 (hTPST-2). Topological analysis of the hTPST-2 primary sequence is shown in Figure S1 in the Supporting Information. The NusA fusion tag with hTPST-2 (NusA-hTPST-2) was purified to near homoge-neity (Figure S2). According to previous reports, TPST catalytic efficiency (kcat/Km) is too low (ca. 1 m 1s 1 by using PSGL-1 as
substrate) for analysis by using general spectroscopic methods. Instead, we need a much more sensitive radioactive probe (35S)
to monitor TPST activity.[11]Traditional TPST assay uses
commer-cial PAPS, which are usually contaminated with significant amounts of 3’-phosphoadenosine-5’-phosphate (PAP).[12] PAP is
a potent inhibitor for many sulfotransferases and its presence might be the cause of TPST’s low catalytic efficiency as previ-ously reported.
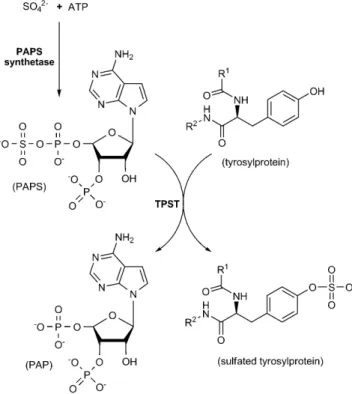
In Scheme 1, we propose to continuously produce fresh PAPS and avoid the drawbacks of instability, contamination, and the expense of commercial PAPS. We prepared PAPS from the reaction catalyzed by PAPS synthetase (PAPSS), a bifunc-tional enzyme composed of ATP sulfurylase and adenosine 5’-phosphosulfate (APS) kinase, in two sequential reactions.[13]
The coupling of human PAPSS isoform 1 (hPAPSS-1) catalysis as delineated in Scheme 1, allowed the continuous generation of in situ PAPS from inorganic sulfate and ATP, and provided a supply of freshly synthesized PAPS to the TPST-catalyzed reac-tion. It is possible to generate PAPS from adequate amounts of hPAPSS-1 (Figure S3). Figure 1 A shows that the productive rate of PSGL-1 sulfation increases dramatically after replacing com-mercial PAPS with PAPSS-generated PAPS. Sulfated PSGL-1 was undetectable by Western blotting when using commercial PAPS as sulfuryl group donor. The kinetics of PSGL-1 sulfation by using PAPSS-generated [35S]PAPS (synthesized from ATP and 35SO
42 ) is shown in Figure 1 B. The apparent Kmvalue obtained
in this study (24mm) is in good agreement with that deter-mined with commercial PAPS as sulfuryl group donor (27mm).[11a]The apparent V
maxobserved in this study, however,
was nearly 45-fold higher than that previously reported (3.2 vs. [a] L.-Y. Lu, B.-H. Chen, J. Y.-S. Wu, C.-C. Wang, D.-H. Chen, Prof. Y.-S. Yang
Department of Biological Science and Technology National Chiao Tung University
75 Po-Ai Street, Hsinchu30050 (Taiwan) Fax: (+ 886) 3-57-29288
E-mail: [email protected]
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.201000540.
0.07 nmol min 1mg 1). We attribute this observation
to the absence of initial PAP inhibition that is so common with commercially available PAPS. PAP might also be a noncompetitive inhibitor for PSGL-1 sulfation in the TPST catalyzed reaction (Scheme 1), and consequently, the use of freshly generated PAPS produced a Km value in agreement with literature
values for commercial PAPS, whereas Vmax increased
(Figure 1 B). Moreover, the recombinant NusA–hTPST-2 remained relatively stable at 30 8C or lower (Fig-ure S4). In addition to the fresh and ample supply of PAPS, other possible reasons for the high catalytic efficiency of PSGL-1 sulfation might be due to a lack of initial PAP, other inhibitor, contamination, and the improved stability of NusA-TPST fusion protein (Fig-ure S4).
We used a similar strategy to produce tyrosine-sul-fated proteins in vivo, by incorporation of suitable genes by using bacterial cultivation. Figure 2 A de-picts the layout of the “built-in” chemoenzymatic ma-chinery. In brief, hPAPSS-1 and hTPST-2 genes were both subcloned into identical expression vector (pA-CYCDuet-1) with separate open-reading frames. The target gene (PSGL-1) was subcloned to another ex-pression vector (pGEX-4T1) with distinct antibiotic re-sistance. By co-expressing the two expression vec-tors, the bacterial colony was simultaneously able to accommodate the genes of hPAPSS-1, hTPST-2, and PSGL-1 (Figure 2 B). Tyrosine-sulfated PSGL-1,
extract-ed from the bacteria containing both plasmids, was detectable by Western blotting (upper panel of Figure 2 C). The control ex-periment (the bacterial host without hTPST-2 gene-containing plasmid) also produced a large amount of GST-fused PSGL-1 protein (lower panel of Figure 2 C) but was free of sulfated GST-PSGL-1 according to the Western blot analysis. Our results strongly indicate that the in vivo chemoenzymatic machinery system was responsible for the highly efficient post-translation-al tyrosine sulfation within the bacteripost-translation-al cell samples. This system can greatly facilitate the development and production of sulfated proteins for therapeutic and medical applications.
In conclusion, we have demonstrated that the chemoenzy-matic machinery for protein tyrosine sulfation is inexpensive, and offers savings in time and effort. The complete system, from production to isolation and detection of sulfated pro-teins, can be integrated with high efficiency and high confi-dence. We were also able to show that protein tyrosine sulfa-tion in Escherichia coli BL21(DE3) provides an excellent niche to specifically produce tyrosine-sulfated products. At present, few literature reports contribute to the understanding of either TPST, or sulfated proteins/peptides at a biochemical level. The successful combination of our PAPS-generating system (by PAPSS), and sulfated-protein production (by TPST) provides a Scheme 1. PAPSS-coupled TPST catalytic reaction. TPST transfers
sulfuryl group from PAPS, which is generated from inorganic sul-fate and ATP in PAPSS catalysis, to the tyrosine residues within specific domain in proteins and peptides.
Figure 1. Characterization of chemoenzymatic machinery for protein tyrosine sulfation. A) Sulfation of 1 with PAPSS-generated PAPS and commercial PAPS. Sulfated PSGL-1 was probed by anti-sulfotyrosine monoclonal antibody (upper panel). The SDS-PAGE of total PSGL-1 stained by Coomassie blue is shown in the lower panel as an internal con-trol. WT and WS were the control experiments, which represented the full catalytic re-action without hTPST-2 and substrate (PSGL-1), respectively. B) Michalis–Menten plot of NusA–hTPST-2 kinetics. The apparent Kmand Vmaxvalues were determined as 24mm and
3.2 nmol min1mg1, respectively. The hTPST-2 catalyzed reactions were determined
under standard condition as described in the Experimental Section.
tool for both fundamental research and industrial applications in protein tyrosine sulfation. Moreover, it would be interesting if we were to implant this tyrosine-sulfation machinery in eukaryotes, which intrinsically possess endogenous TPST and PAPSS, because more substrates and protein-tyrosine sulfation functions would be uncovered.
Experimental Section
The standard PAPSS-coupled TPST catalysis composed of 50 mm MES at pH 6.5, 5 mm b-mercaptoethanol, 4 mm Na2SO4, 1 mm
MgCl2, 1 mm ATP, 20mm PSGL-1 peptide (ATEYEYLDYDFL), 1 mg
hPAPSS-1, 1 U pyrophosphatase, and 4mg hTPST-2 for 45-min incu-bation at 37 8C in a final volume of 20mL. The TPST-catalyzed reac-tion using commercial PAPS was conducted under similar condi-tions except that 20mm PAPS was used to replace the PAPSS system. In vivo protein tyrosine sulfation was demonstrated by co-expressing both plasmids of hPAPSS-1/hTPST-2 (pACYCDuet-1) and PSGL-1 peptide (ATEYEYLDYDFL) (pGEX-4T1) in BL21(DE3) with am-picillin (50mg mL 1
) and chloramphenicol (34mg mL 1
) as antibiot-ics. A single colony was cultivated in LB broth at 37 8C until the ODA600reached to 0.8–1 and then induced with 1 mm IPTG for 16 h
at 20 8C. The cultures were harvested and extracted in lysis buffer (50 mm Tris–HCl at pH 8.0, 150 mm NaCl, and 10 % glycerol) for fur-ther analysis.
Detailed protocols for enzyme cloning, expression, purification, PAPSS assay, TPST kinetics assay, thermal stability, in vivo protein tyrosine sulfation, and immunoblotting are described in the Sup-porting Information.
Acknowledgements
This work was supported by the National Science Council, Taiwan (NSC98-2321-009-001 and 99-2311-B-009-004-MY3).
Keywords: enzyme catalysis · post-translational modifications · protein modifications · sulfation · sulfotransferases
[1] a) P. A. Baeuerle, W. B. Huttner, J. Biol. Chem. 1985, 260, 6434 – 6439; b) C. Seibert, T. P. Sakmar, Biopoly-mers 2008, 90, 459 – 477.
[2] F. R. Bettelheim, J. Am. Chem. Soc. 1954, 76, 2838 – 2839.
[3] K. L. Moore, J. Biol. Chem. 2003, 278, 24243 – 24246.
[4] a) R. W. Lee, W. B. Huttner, Proc. Natl. Acad. Sci. USA 1985, 82, 6143 – 6147; b) R. Beisswanger, D. Corbeil, C. Vannier, C. Thiele, U. Dohrmann, R. Kellner, K. Ashman, C. Niehrs, W. B. Huttner, Proc. Natl. Acad. Sci. USA 1998, 95, 11134 – 11139; c) Y. B. Ouyang, K. L. Moore, J. Biol. Chem. 1998, 273, 24770 – 24774.
[5] a) M. Farzan, T. Mirzabekov, P. Kol-chinsky, R. Wyatt, M. Cayabyab, N. P. Gerard, C. Gerard, J. Sodroski, H. Choe, Cell 1999, 96, 667 – 676; b) J. W. Kehoe, C. R. Bertozzi, Chem. Biol. 2000, 7, R57 – 61; c) S. R. Stone, J. Hofsteenge, Biochemistry 1986, 25, 4622 – 4628.
[6] F. Markwardt, Semin. Thromb. Hemost. 2002, 28, 405 – 414.
[7] H. Choe, W. Li, P. L. Wright, N. Vasilieva, M. Venturi, C. C. Huang, C. Grundner, T. Dorfman, M. B. Zwick, L. Wang, E. S. Rosenberg, P. D. Kwong, D. R. Burton, J. E. Robinson, J. G. Sodroski, M. Farzan, Cell 2003, 114, 161 – 170.
[8] a) C. C. Liu, P. G. Schultz, Nat. Biotechnol. 2006, 24, 1436 – 1440; b) C. C. Liu, H. Choe, M. Farzan, V. V. Smider, P. G. Schultz, Biochemistry 2009, 48, 8891 – 8898.
[9] a) D. Balsved, J. R. Bundgaard, J. W. Sen, Anal. Biochem. 2007, 363, 70 – 76; b) T. Young, L. L. Kiessling, Angew. Chem. 2002, 114, 3599 – 3601; Angew. Chem. Int. Ed. 2002, 41, 3449 – 3451; c) C. Niehrs, W. B. Huttner, EMBO J. 1990, 9, 35 – 42; d) Y. Ouyang, W. S. Lane, K. L. Moore, Proc. Natl. Acad. Sci. USA 1998, 95, 2896 – 2901.
[10] a) W. H. Lin, J. A. Roth, Biochem. Pharmacol. 1990, 40, 629 – 635; b) C. Niehrs, M. Kraft, R. W. Lee, W. B. Huttner, J. Biol. Chem. 1990, 265, 8525 – 8532; c) C. Kasinathan, R. Sundaram, S. William, Gen. Pharmacol. 1995, 26, 577 – 580; d) E. Mishiro, M. Y. Liu, Y. Sakakibara, M. Suiko, M. C. Liu, Biochem. Cell Biol. 2004, 82, 295 – 303.
[11] a) E. Mishiro, Y. Sakakibara, M. C. Liu, M. Suiko, J. Biochem. 2006, 140, 731 – 737; b) F. Monigatti, B. Hekking, H. Steen, Biochim. Biophys. Acta Proteins Proteomics 2006, 1764, 1904 – 1913.
[12] E. S. Lin, Y. S. Yang, Biochem. Biophys. Res. Commun. 2000, 271, 818 – 822.
[13] a) J. P. Girard, E. S. Baekkevold, F. Amalric, FASEB J. 1998, 12, 603 – 612; b) K. V. Venkatachalam, H. Akita, C. A. Strott, J. Biol. Chem. 1998, 273, 19311 – 19320.
Received: September 12, 2010 Published online on December 22, 2010 Figure 2. Implantation of chemoenzymatic machinery in vivo for protein tyrosine sulfation. A) Scheme for in vivo protein tyrosine sulfation. Plasmids that contained hPAPSS-1/hTPST-2 and the target gene (PSGL-1), respectively, were coexpressed in a bacterial host. The gene products were expected to produce a series of reagents for pro-tein tyrosine sulfation in bacteria. The tyrosine residue (Y) within the specific domain of target propro-tein is labeled with asterisk. B) Expression of hPAPSS-1, hTPST-2, and PSGL-1 genes in bacterial host. Lane 1 indicates the PCR products from the bacterium coexpressed with plasmids containing PSGL-1 and hPAPSS-1/hTPST-2 genes. Lane 2 is the control experiment obtained from the bacterium incorporated with both clones of PSGL-1 and hPAPSS-1. C) In vivo production of sulfated PSGL-1. Lane 1 was obtained from bacterium containing PSGL-1 and hPAPSS-1/hTPST-2 genes. Sulfated PSGL-1 was probed by anti-sulfotyrosine monoclonal antibody (upper panel). The SDS-PAGE of total PSGL-1 stained by Coomassie blue was shown in the lower panel as internal control (lower panel). The bac-terium of the control experiment (lane 2) was in the absence of hTPST-2 in the protein sulfation system.