行政院國家科學委員會專題研究計畫 成果報告
以分子動力學研究金奈米管之管徑對水分子行為之影響
計畫類別: 個別型計畫
計畫編號: NSC92-2212-E-110-030-
執行期間: 92 年 08 月 01 日至 93 年 07 月 31 日
執行單位: 國立中山大學機械與機電工程學系(所)
計畫主持人: 朱訓鵬
計畫參與人員: 陳奕瑞、李玟頡
報告類型: 精簡報告
處理方式: 本計畫可公開查詢
中 華 民 國 93 年 8 月 30 日
以分子動力學研究奈米金管尺寸對管內水分子行為之影響
朱訓鵬
中華民國 高雄市
國立中山大學機械研究所
國科會計畫編號:NSC92-2212-E-110-030
摘要 本研究是利用分子動力學探討不同尺寸大小的 奈米金管,對於管內水分子行為的影響。由觀察水分 子的分佈可得知在靠近管壁的水分子會形成兩層類 似殼層的結構,而這種殼層結構在直徑較小的奈米金 管較為明顯。管壁附近的水分子濃度升高,影響了奈 米金管內隨意分佈區域的水分子密度。在較小管徑的 情況下,靠近管壁的高濃度水分子不但降低了隨意分 佈區域內的水分子密度並且減少了水分子之間的氫 鍵數。這些結果都導致奈米金管與水分子之間的平均 交互作用能量會隨著管徑減少而增加。由隨意分佈區 域內的水分子自身擴散係數變化可以得知,水分子在 軸向的擴散能力會隨著奈米金管的尺寸變小而增強。Abstract
This study utilizes molecular dynamics
simulation to investigate the behavior of water
molecules inside Au nanotubes of various
sizes. An observation of the water molecule
distributions reveals that the adsorption of the
water molecules creates two shell-like
formations of water in close vicinity to the Au
nanotube
wall and
these shell-like formations
are found to be more pronounced in nanotubes
of smaller diameter than in larger nanotubes.
The higher concentration of water molecules
near to the nanotube wall influences the
density of the water molecules in the random
distribution region of the Au nanotube. In
smaller Au nanotubes, the
higher
concentration of water molecules in close
vicinity to the wall lowers the density of the
water molecules in the random distribution
region and reduces the number of hydrogen
bonds between a water molecule and its
neighboring water molecules. These results
can be attributed to the increase in the average
interaction energy between the Au nanotube
and the water molecules as the size of the
nanotube decreases.
Finally, an inspection of
the variation in the
self-diffusion coefficients
of the
water molecules in the random
distribution region of the Au nanotube reveals
that water molecules in a smaller Au nanotube
have a greater ability to diffuse along the
longitudinal direction of the nanotube.
關鍵字: 分子動力學模擬、奈米金管、水分子、自 身擴散係數、平行運算 前言 奈米金管在生物科技產業裡具有相當地發 展 潛 力 且 在 近 年 來 引 起 學 術 界 熱 烈 的 討 論 [1-3]。例如:可以利用奈米金管薄膜允許某種擴 散係數的分子通過之特性來作為分子的過濾與 篩選[1-3]。在兩個分子之間的擴散係數比值很大 時,篩選效應就會特別得大。因為擴散係數比值 取決於奈米金管的內徑,所以金管的尺寸是影響 分子篩選過程的重要因素。靠著偵測隨分析濃度
變化的穿越細胞膜電流量,奈米金管同時還能應 用 在 分 子 濃 度 與 超 微 量 離 子 的 化 學 檢 測 上 [2-3]。由以上兩個應用看來,奈米金管與水分子 之間的交互作用是相當的重要,當水分子撞擊管 璧時增加了水分子的摩擦阻力,使得摩擦係數比 自由流動液體的摩擦係數大[2]。所以如果要增 進奈米金管的應用性,就必須清楚地了解奈米金 管壁面與水分子之間的交互作用。 因為分子動力學是作為了解原子尺度下分 子行為很好的方法,再加上原子級的實驗量測具 有相當難度,所以數值模擬是拿來探討奈米管內 分子行為較適當的工具。Takaba 等人採用了分 子動力學去研究當苯、烷基苯以及烷基石腦油精 等分子通過奈米碳管時的動態行為[4]。其研究 的結果指出,分子之間的吸引力會使得分子被吸 入管內。此外,奈米碳管有足夠的伸縮性可以篩 選尺寸大於管徑的分子。Hummer 等人研究水分 子通過碳管時的傳輸行為[5],因為奈米碳管會 阻止管內的水分子與管外的水分子形成氫鍵,造 成管內的水分子平均氫鍵數少於巨觀下的值。通 常來說,管內所形成的一維水分子鏈會影響管內 水分子的動力學行為。除了探討通過管內的傳輸 行為,分子動力學還用來研究奈米管限制對分子 定位與吸附的影響。Gordillo 與 Marti 做了相當 多有關於奈米管限制與溫度對水分子行為影響 的研究[6-8]。結果指出侷限在奈米碳管內的水分 子會破壞氫鍵鍵結的網絡結構,使得平均氫鍵數 會減少,此種限制效應即使在超臨界狀態下也會 發生。除此之外,氫鍵鍵結的網絡結構也會隨著 溫度的升高而被破壞。Brovchenko 等人以分子 動力學來研究環境溫度為 300K、壓力為 1bar 時,奈米孔隙中水分子的性質[9]。研究指出水 與孔隙之間強交互作用力會使得在孔隙表面形 成兩個較為明顯的水殼層。Gallo 等人利用分子 動力學來探討不同水密度與溫度,對在孔徑 2nm 的矽孔隙內水分子行為的影響。水密度與系統溫 度對於氫鍵鍵結網絡的影響以及水分子在矽孔 隙內重新排列的行為都已有很完整的研究內容 [10]。 由以上的介紹可以得知,分子動力學為研究 原子級分子行為的可靠方法。因為水分子是在奈 米金管內溶液的主要成分,所以必須充分了解水 分子在管內的行為表現。故本文即以分子動力學 來探討不同管徑大小的條件下,管內水分子的行 為會受到什麼樣的影響。除此之外,本文還討論 水分子的濃度分佈、平均鄰近氧原子數(nNN)、 平均水分子氫鍵數(nHB)、水分子與奈米金管之 間的交互作用以及水分子軸向的自身擴散係數。 理論說明 圖 1 為水分子在奈米金管內的示意圖。本文 的模擬考慮五種不同大小的奈米金管內徑,分別 為 3nm、6nm、9nm、12nm 以及 15nm。在每一 個模擬尺寸下,奈米管的長度都為三倍的內徑大 小。在軸向的邊界條件設為週期性邊界條件,管 內水分子密度為 0.973g/cm3 ,採用 Scaling 法將 系統的溫度修正在平衡溫度 300K。以多時間步 階法來模擬不同的分子運動,原子軌跡則利用 Verlet 演算法來計算。以 0.1fs 的時間步階來計 算運動較為快速的水分子鍵結與角鍵結,而其他 的運動都用 1fs 來模擬。在模擬最大尺寸的奈米 金管時,所計算的粒子總數為 1035631 個原子。 這個模擬的尺度已經超過傳統個人電腦的運算 能力,故本研究將在 32 個節點的平行運算環境 下執行。 本模擬考慮了各種原子之間的作用力,包含 金原子與金原子之間、水分子與水分子之間、水 分子內部的鍵結與角鍵結以及水分子與金原子 之間的作用力等。本文採用多體緊束法來計算金 原子與金原子之間的作用力[15,16],同一水分子 內的氫氧原子及其它水分子的氫氧原子之間的 作用力都使用 F3C 勢能[17]去描述,水分子彼此 之間的交互作用力則以 Spohr 勢能[18,19]來模 擬。
緊束法為兩個能量的總和,一是狀態 d 軌域 密度的第二動量,一是 Born-Mayer type 的成對 勢能[15,16]。如下:
∑
∑
− − + − − − = j ij j ij r r p A r r q E exp 2 1 exp 1 0 2 / 1 0 2 ζ (1) ζ 為有效期望積分,rij為原子 i 與 j 之間的距 離,r
o是第一鄰近距離。參數 A、p、q 和ξ
都是 由實驗數據所得到,分別代表共價能、晶格常 數、bulk modulus 以及剪力彈性常數。本文緊束 法所使用到的參數都表列在表Ⅰ中[16]。 水分子之間的作用力採用 F3C 勢能去計 算,此勢能不但精確地對應以 X 光與中子衍射所 偵測出來的水結構,還能求得與實驗資料相當吻 合的結構與動力學性質。此勢能的另一個優點為 引入了短截斷範圍,這不但減少了計算上所付出 的時間,同時還允許了分子動力學能在長時間模 擬中計算大量的水分子。F3C 可以表示如下:bond bend vdw els
U
=
U
+
U
+
U
+
U
(2) bondU
和U
bend均為分子內勢能,分別代表水分子 內鍵結能量與角鍵結能量。完整的勢能函數表示 如下:( )
i bond bU
= 1 b N N=∑
OH b K ( 0OH i b −b )2 (3)( )
i bendU
θ
=∑
= θ N N 1 HOH Kθ ( 0HOH iθ θ
− )2 (4) i b 、θ
i、 0OH b 以及θ
0HOH 分別代表水分子第 i 個 的 O-H 鍵長、第 i 個的 H-O-H 鍵角、O-H 的平 衡鍵長以及 H-O-H 的平衡鍵角。 凡得瓦爾勢能乃用來計算分子之間的能量,如 下: )] ( ) / ( 2 ) / ( [ ) ( 0 6 12 0 ij vdw ij ij ij ij ij ij SC ij vdw r A r r r r S r U = ε − ε − (5) SC A 是用來補償短截斷距離所忽略的交互作 用,在 F3C 勢能中已有特定的截斷距離[17]。本 文採用的截斷距離為 10Å 而ASC的值則為 1。 ( ) vdw ij S r 為凡得瓦爾勢能的 truncation shift 函數。 truncation shift 函數通常可以表示如下:( )
( ) (
)
( )
0 c c c c f c df r f r r r r r dr S r r r + − < = > (6)( )
cf r
和rc分別為勢能函數與其截斷距離。 靜電力勢能表示如下: ( ) i j els els ij ij q q U S r r = − ∑
(7) i q 和 j q 為在截斷距離內兩個不同水分子的氫氧原子所帶的部分電荷。Sels( )rij 為 truncation shift
函數,可由(6)式中求得。本文所使用的 F3C 參 數都表列在表Ⅱ中。 本研究使用由 Do 等人[18,19]修正的 Spohr 勢能來計算水分子與金原子之間的作用力。勢能 函數表示如下: ( ) ( ) ( ) 2 1 1 2 2 Au H O Au O Au O Au H Au H Au H Au H U − =U − r − +U − r − +U − r − (8) 上式中的第一項為金原子與氧原子之間的 交互作用,第二與第三項則為金原子與水分子的 兩個氫原子之間的交互作用。勢能函數可以表示 如下: ( ) 0 exp
(
2 ( 1))
2 exp(
( 1))
2( ) Au O O e O e U − r =D − β r−r − −β r−r ⋅S r (9)( )
0exp
(
2
(
2)
)
Au H H eU
−r
=
γ
D
−
β
r
−
r
(10) 2( )
S r
為 shift 函數,表示如下:( )
(
) (
)
(
)
2 2 2 2 2 2 2 3 2 2 1 2 3 on off off on on off off on r r r r r r r S r r r r r r ≤ − + − = < ≤ − (11) on r 和roff為 shift 函數的開始與結束距離,在本文 中分別為 7Å 與 11Å 。此勢能函數所用到的參數 都表列在表Ⅲ中[18,19]。 結果與討論 圖 2 為管內水分子的局部密度分佈圖。為了 表現出奈米管尺寸對水分子定位變化的影響,圖 2 討論了三種不同的直徑,分別為 3nm、9nm 以 及 15nm。縱軸為 reduced 密度(ρlocal/ρbulk),橫 軸為水分子與奈米金管壁之間的距離。每一條曲 線都出現了三個明顯的峰值,第一個明顯的峰值 是由於直接吸附在奈米金管管壁的水層所造成 的。隨著原子間距離的增加,奈米金管與水分子 之間的交互作用跟著變小,第二與第三個峰值也 隨之減少。以第一個與第二個峰值而言,在管徑 3nm 的模擬條件下減少的幅度最大;而在管徑 15nm 的模擬條件下減少的幅度最小。第一個與 第二個峰值都比第三個峰值明顯得許多,此現象 說明了兩個水殼層結構都在奈米金管管壁的附 近形成。密度變化的第三個峰值在不同的管徑 時,幾乎都呈現一樣的量,在第三個峰值以後的 密度只會微小的變動,此現象說明了這個區域的 水分子是隨意且均勻的分佈。相對地,接近第三 個峰值的區域可視為隨意分佈區域與水分子殼 層區域的過渡區域。基於方便的理由,將整個空 間區分為水殼層區域、過渡區域以及隨意分佈區 域。因為水分子的平均密度都一樣(0.973g/cm3 ), 所以靠近管壁的高密度水分子會影響到隨意分 佈區域的密度。密度分佈在隨意分佈區域內與在 水殼層區域內不同。管徑為 15nm 時水密度減少 幅度最大,管徑為 3nm 時則是密度最小的情況。 圖 3 為平均氫鍵數(nHB)與平均鄰近原子數 (nNN)的管內分佈圖。由圖 3 可以更瞭解奈米 金管內水分子局部氫鍵的鍵結情形。因為圖 2 是用來比較不同尺寸的奈米金管管壁附近的水 分子分佈情形,故橫軸為水分子與管壁的距離。 然而圖 3 的目的是表現水殼層區域與隨意分佈 區域內 nHB 以及 nNN的圖形,所以橫軸為水分子 與管中心的距離。因此橫軸的最大值近似於管內 徑,因為水分子不會出現在奈米金管以外的區 域。判斷平均氫鍵數(nHB)的方法則是利用幾何 判斷法[20],當不同水分子的氫氧原子之相對距 離小於 2.4Å[17](F3C 勢能函數,氧氫原子徑向 分佈函數的第一最小值)時,則會形成氫鍵。而 鄰近原子數(nNN)[21]是以氧原子與周遭的氧原 子相距小於 3.4Å[17](F3C 勢能函數,氧氧徑向 分佈函數的第一最小值)時來作為判斷的依據。 圖 3 中的 nHB與 nNN在靠近奈米金管管壁的位置 都有明顯的峰值存在。這些峰值對應的是管壁旁 具有高濃度之第一個水分子殼層(如圖 2)。同樣 地,在第一個水分子殼層左邊的峰值對應的是第 二個水分子殼層。在管徑分別為 3nm、9nm 以 及 15nm 的情況下,nHB與 nNN在隨意分佈區域 內有著明顯的不同。如同圖 2 所示,隨意分佈區 域內的 reduced 密度隨著奈米金管的直徑增大而 增加。由圖 3 可以清楚地看出在管徑最小的情況 中,隨意分佈區域內的水分子會形成最少的氫鍵 數。雖然在此區域內的水分子與金原子並沒有交 互作用,但是 nHB與 nNN的分佈情形是引起水殼 層區域內水分子分佈不同的最可能因素。 圖 4 為平均交互作用能量與奈米金管直徑 的關係圖。平均交互作用能量的定義為第一個水 殼層內水分子之間的作用能量除上與第一個水 殼層水分子作用的金原子總數。由圖 4 可以清楚 地看出平均交互作用能量會隨著管徑的增加而增強。因此可以歸納出小管徑的奈米金管對於水 分子有較強的吸引能力。 因為水殼層內的水分子是吸附在奈米金管 的管壁上,所以當要探討奈米金管尺寸對於水分 子自身擴散係數的影響時,焦點將放在隨意分佈 區域上。自身擴散係數是由以 integrated velocity autocorrelation 為基礎的 Green-Kubo 表示式來求 得。圖 5 為五種不同奈米金管尺寸下,隨意分佈 區域內自身擴散係數與平均 reduced 密度變化關 係圖。隨著管徑的增加,平均 reduced 密度會隨 之變小,此現象同樣地可以由圖 2 看出。除此之 外,由圖 3 可看出 nHB也隨著管徑增加而增加。 由以上結果可以推論,最小尺寸的奈米金管會形 成最少的氫鍵,因此水分子能夠在軸向有更強的 擴散能力。此推論與圖 5 所顯示的軸向自身擴散 係數變化有很好的一致性。 結論 本文研究奈米金管尺寸對其所包含的水分 子性質的影響。結果指出奈米金管與水分子之間 的交互作用受到奈米金管尺寸影響甚巨。交互作 用能量直接影響了奈米金管管壁附近的水分子 分佈,並形成了兩個明顯的水殼層結構。具有高 濃度的水殼層使得隨意分佈區域內的水分子密 度因此降低。較小尺寸之奈米金管在隨意分佈區 域內的氫鍵較為薄弱,這是因為這個區域內的水 分子濃度較低的原因。因此,水分子的擴散能力 會隨著奈米金管的尺寸變小而增強。 參考文獻
1. Yu S., Lee S.B., Kang M., and Martin C.R., Size-Based Protein Separations in Poly(ethylene glycol)-Derivatized Gold Nanotubule Membranes, Nano letters, Vol.1,
pp.495-498, 2001.
2. Wirtz M., Yu S., and Martin C.R., Template synthesized gold nanotube membranes for chemical separations and sensing, Analyst, Vol.127, pp.871-879, 2002.
3. Wirtz M., Parker M., Kobayashi Y., and Martin C.R., Molecular Sieving and Sensing
with Gold Nanotube Membranes, The
Chemical Record, Vol.2, pp.259-267, 2002. 4. Takaba H., Katagiri M., Kubo M., Vetrivel R.,
and Miyamoto A., Molecular design of carbon nanotubes for the separation of molecules, Microporous Materials, Vol.3, pp.449-455, 1995.
5. Hummer G., Rasaiah J.C., and Noworyta J.P., Water conduction through the hydrophobic channel of a carbon nanotube, Nature, Vol.414, pp.188-190, 2001.
6. Gordillo M.C., and Martí J., Hydrogen bond structure of liquid water confined in nanotubes, Chemical Physics Letters, Vol.329, pp.341-345, 2000.
7. Gordillo M.C., and Martí J., Hydrogen bonding in supercritical water confined in carbon nanotubes, Chemical Physics Letters, Vol.341, pp.250-254, 2001.
effects on the static and dynamic properties of liquid water inside nanotubes,Physical Review E, Vol.64, 021504-1~021504-6. M.C, 2001.
9. Brovchenko I.V., Geiger A., and Paschek D., Fluid Phase Equilibria, Vol.183-184, pp.331-339, 2001.
10. Gallo P., Rapinesi M., and Rovere M., Confined water in the low hydration regime, Journal of Chemistry Physics, Vol.117, pp.369~375, 2002.
11. Haile J. M., Molecular Dynamics Simulation (John Wiley & Sons, Canada, 1992).
12. Allen M. P., and Tildesley D. J., Computer Simulation of Liquid (Clarendon Press, Oxford, 1991).
13. Tuckerman M., and Berne B. J., Reversible multiple time scale molecular dynamics, Journal of Chemical Physics, Vol.97, pp.1990-2001, 1992.
14. Frenkel D., and Smit B., Understand Molecular Simulation (Academic Press, San Diego, 1996).
15. Rosato V., Guillope M., and Legrand B., Thermodynamical and structural properties of f.c.c. transition metals using a simple tight-binding model, Philosophical Magazine
A, Vol.59, pp.321-336, 1989.
16. Cleri F., and Rosato V., Tight-binding potentials for transition metals and alloys, Physical Review B, Vol.48, pp.22-33, 1993. 17. Levitt M., Hirshberg M., Sharon R., Laidig K.
E., and Daggett V., Calibration and Testing of a Water Model for Simulation of the Molecular Dynamics of Proteins and Nucleic Acids in Solution, Journal of Physical Chemistry B, Vol.101, pp.5051-5061, 1997. 18. Dou Y., Zhigilei L. V., Winograd N., and
Garrison B. J., Explosive Boiling of Water Films Adjacent to Heated Surfaces: A Microscopic Description Journal of Physical Chemistry A, Vol.105, pp.2748-2755, 2001.
19. Dou Y., Winograd N., Garrison B. J., and Zhigilei L. V., Substrate-Assisted Laser-Initiated Ejection of Proteins Embedded in Water Films, Journal of Physical Chemistry B, Vol.107, pp.2362-2365, 2003.
20. Benjamin I., Theoretical study of the water/1,2-dichloroethane interface: Structure, dynamics, and conformational equilibria at the liquid–liquid interface, Journal of Chemical Physics, Vol.97, pp.1432-1445,
1992.
21. Böcker J., Nazmutdinov R. R., Spohr E., and Heinzinger K., Molecular dynamics simulation studies of the mercury-water interface, Surface Science, Vol.335, pp.372-377, 1995. 圖與表 (a) (b) 圖 1、物理模型示意圖:(a)奈米金管側視圖(b) 含有水分子的奈米金管剖面圖 圖 2、三種不同尺寸的奈米金管內水分子密度分 佈圖 圖 3、平均鄰近原子數(nNN,上曲線)以及平均 氫鍵數 (
n
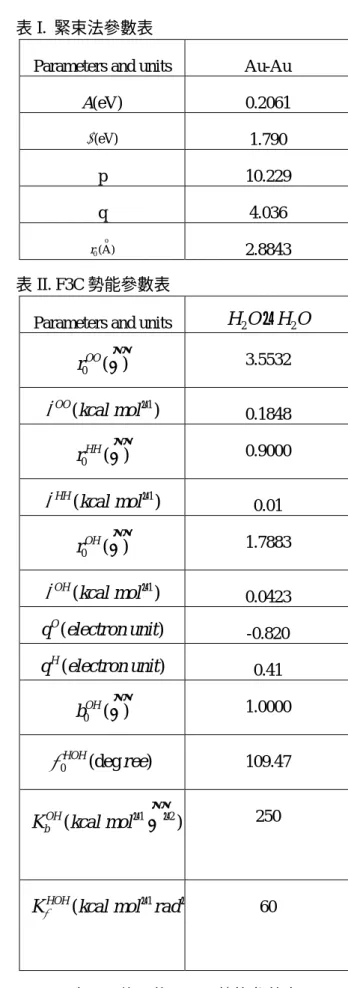
HB,下曲線)與位置的關係圖圖 4、奈米金管管壁與第一個水殼層之間的平均 交互作用能量( 5 10− × eV) 圖 5、在隨意分佈區域內軸向自身擴散係數 ( 2 fs ° Α )以及平均 reduced 密度與管徑大 小關係圖 表 I. 緊束法參數表
Parameters and units Au-Au
A(eV) 0.2061 (eV) ξ 1.790 p 10.229 q 4.036 o 0(A) r 2.8843 表 II. F3C 勢能參數表
Parameters and units
H O
2−
H O
20 ( ) OO r ° Α 3.5532 1 ( ) OO kcal mol
ε
− 0.1848 0 ( ) HH r Α° 0.9000 1 ( ) HH kcal molε
− 0.01 0 ( ) OH r ° Α 1.7883 1 ( ) OH kcal molε
− 0.0423 ( ) O q electron unit -0.820 ( ) H q electron unit 0.41 0 ( ) OH b ° Α 1.0000 0 (deg ) HOH reeθ
109.47 1 2 ( ) OH b K kcal mol ° −Α
− 250 1 ( HOHKθ kcal mol rad− − 60
Parameters and units