國立交通大學
應用化學研究所
碩士論文
無機多面體聚矽氧烷團聯式共聚(4-乙基苯酚)
高分子之表面能、熱性質及自組裝結構研究
Surface Free Energy, Thermal Properties, and
Self-Assembly in Solution State of Poly(4-vinyl phenol)
Tethered to Polyhedral Oligomeric Silsequioxanes
研究生:周幸儀
指導教授:張豐志教授
誌謝
研究所期間,我所獲得的不僅是專業的訓練,更獲得了意想不到的人生經 歷;記憶裡那種痛苦掙扎的感覺漸漸變淡,而那些緊要關頭時讓我突破困境向前 衝的關鍵畫面卻還是一樣鮮明!多次以為要完成碩士學位是個不可能的任務,而 今能走到最後,這都歸功於一路上幫助我的師長朋友們支撐著我走到最後,在此 致上最真誠的感謝。 首先我要感謝指導老師 張豐志教授兩年來的栽培與關懷,謝謝老師在論文 上給予指導與幫助,並提供我良好的研究環境,給我充分的機會與空間,培養我 獨立研究的能力。並感謝口試委員:馬振基教授、邱顯堂教授、王志逢教授及陳 建光教授給予學生研究上寶貴的指導與見解,使學生論文能夠更加完整而嚴謹, 並讓學生對研究有更深的體悟。 感謝廖春雄學長在研究上的指導,並且給予生活上的最大的支持,願意浪費 許多時間心力在這個不成材的學妹身上。感謝呂居樺學長教導我合成的技巧,及 林漢清學長在實驗上的幫忙與協助,讓我順利完成論文;並感謝婉君學姐、倩婷 學姐、世堅學長、筱雯學姐、仁志、徐婕、登揚在這段日子不論是生活上或課業 上的鼓勵,謝謝你們給予我最大的關心與幫助,讓我的研究生涯充滿著歡笑與回 憶,有你們的陪伴與鼓勵,我才能勇敢的面對種種挫折與挑戰。並感謝我的好友: 美惠、怡絜、佩紋、倢如、鴻文、丞以、楷翔、晟瑀、旺樺、彥凱、煜翔、宏鼎、 雁彬、普淨、育廷、瀅太、伯檻…等,謝謝你們總是帶給我滿滿的歡笑,每當我 生氣無助時,你們總是給我最大的精神力量。因為你們的關心和照顧,讓我能順 利完成學業並豐富我的生活。 最後,特別感謝我的家人對我的栽培與支持,在我徬徨無助的時後給我滿滿 的力量,謝謝你們讓我可以無後顧之憂地、恣意的享受自己所選擇的生活。 謹以本文,獻給所有支持與關心我的家人、師長與摯友。 幸儀 2009 年 5 月無機多面體聚矽氧烷團聯式共聚(4-乙基苯酚)高分子之表面能、熱性質及自組裝結構研究
Surface Free Energy, Thermal Properties, and Self-Assembly in Solution State of Poly(4-vinyl phenol) Tethered to Polyhedral Oligomeric Silsequioxanes
研究生:周幸儀 Student : Hsin-Yi Chou
指導教授:張豐志 博士 Advisor : Dr. Feng-Chih Chang
國立交通大學 應用化學研究所
碩士論文
A Thesis
Submitted to Department of Applied Chemistry College of Science
National Chiao Tung University In Partial Fulfillment of the Requirements
For the Degree of Master of Science In Applied Chemistry
May 2009
Hsin-Chu, Taiwan, Republic of China
Outline of Contents
Pages
Acknowledgments
Outline of Contents I
List of Schemes IV
List of Tables VII
List of Figures IX
Abstract (in Chinese) XV
Abstract (in English) XVII
Chapter 1 Introduction to Polyhedral Oligomeric
Silsesquioxane
1-1 A Quick History of Polyhedral Oligomeric Silsesquioxane 1
1-2 Silsesquioxanes and Polyhedral Oligomeric Silsesquioxane 3
1-3 POSS Polymers and Copolymers 5
1-4 Surface Free Energy of POSS Polymer Surface 6
1-4-1 Wettability of POSS Nanostructured Polymer Surfaces 6
1-4-2 Nanostructured Thermosets from Epoxy Resin and an Organic-Inorganic
Amphiphile
7
1-4-3 Structure and Dewetting Behavior of Polyhedral Oligomeric Silsesquioxane
(POSS)-Filled Polystyrene (PS) Thin Films
8
1-5 Thermal Properties of POSS Polymer 9
1-5-1 POSS Reinforced Epoxy Systems 9
1-5-2 Thermal Properties of Poly(norbornyl-POSS) Copolymers 10
1-6 Phase Separation of POSS Polymer 12
1-6-1 An Overview of Self-Organization of Block Copolymers 12
1-6-2 In Dilute Solutions 15
1-6-2-1 Core-Corona Structure of POSS-Poly(ethylene oxide) in Aqueous
Solution
15
1-6-2-2 Vesicle Formation and Its Fractal Distribution by Bola- Amphiphilic
[60]Fullerene
16
1-6-3 Bulk Phases 17
1-6-3-1 Coughlin’s Model 17
1-6-3-2 ABA Triblock Copolymers Containing POSS Pendant Groups 18
Reference 20
Chapter 2
Theory
2-1 Living Free Radical Polymerization 22
2-1-1 Requirements for Living Free Radical Polymerization 22
2-1-2 Peculiarities of Living Free Radical Polymerization 23
2-1-3 Evolution of Living Free Radical Polymerization 26
2-1-4 Nitroxide Mediated Radical Polymerization (NMRP) 29
2-1-5 Anionic Polymerization 32
2-2 Surface Free Energy 34
2-2-1 Interfacial Thermodynamics 34
2-2-2 Contact Angle Equilibrium: Young Equation 36
2-2-3 Determination of Surface Free Energy 39
2-2-4 Surface Free Energy of Polymer 48
Chapter 3
Experimental Section
3-1 Materials 57
3-2 Experimental Part 61
3-2-1 Preparation of POSS-TEMPO Macroinitiator 61
3-2-1-1 Synthesis of N-alkoxyamines (TEMPO-1) 61
3-2-1-2 Synthesis of Hydroxyl-N-alkoxyamines (TEMPO-2) 61
3-2-1-3 Synthesis of Allyl-N-alkoxyamines (TEMPO-3) 61
3-2-1-4 Synthesis of POSS-SiH 62
3-2-1-5 Synthesis of POSS-TEMPO macroinitiator 62
3-2-2 Preparation of POSS-Poly(4-vinyl phenol) (POSS-PVPh) by Nitroxide
Mediated Radical Polymerization
63
3-2-3 Preparation of PMMA-b-P4VP by Anionic Polymerization 63
3-2-4 Preparation of PVPh by Anionic Polymerization 64
3-2-5 Preparation of POSS-PVPh Copolymer and POSS/PVPh Blend Thin Film 64
3-3 Characterizations 66
3-3-1 Fourier Transform Infrared Spectroscopy (FTIR) 66
3-3-2 Mass Spectrometry (MS) 66
3-3-3 Nuclear Magnetic Resonance (NMR) 66
3-3-4 Electron Spectroscopy for Chemical Analysis (ESCA) 66
3-3-5 Gel Permeation Chromatography (GPC) 67
3-3-6 Contact Angle Measurement 67
3-3-7 Atomic Force Microscopy (AFM) 67
3-3-8 Differential Scanning Calorimetry (DSC) 67
3-3-9 Thermogravimetry Analysis (TGA) 68
Reference 69
Chapter 4
Results and Discussion
724-1 Synthesis Characterization 73
4-1-1 Characterization of N-alkoxyamine (TEMPO-1) 73
4-1-2 Characterization of Hydroxyl-N-alkoxyamine (TEMPO-2) 73
4-1-3 Characterization of Allyl-N-alkoxyamine (TEMPO-3) 74
4-1-4 Characterization of POSS-SiH 74
4-1-5 Characterization of POSS-TEMPO Initiator 75
4-1-6 Characterization of POSS-PAS and POSS-PVPh 75
4-1-7 Characterization of PMMA-b-P4VP 76
4-1-8 Characterization of PtBOS and PVPh 76
4-2 Surface Free Energy in PVPh System 78
4-2-1 The Effect of Molecule Weight on Surface Free Energy in PVPh System 78
4-2-2 The Effect of Solvent on Surface Free Energy in POSS-PVPh System 80
4-2-3 The Effect of POSS on Surface Free Energy in PVPh System 81
4-2-4 The Effect of POSS on Surface Free Energy after Thermal Treatment in
POSS-PVPh System
83
4-3 The Effect of POSS on Thermal Properties 85
4-3-1 The Effect of POSS on in POSS-PVPh System 85
4-3-2 The Effect of POSS on Hydrogen Bonding and Thermal Properties in
PVPh/PMMA System
87
4-4 Phase Behavior of POSS-PVPh Copolymers in Solution State 91
4-4-1 The Effect of POSS Nanoparticle on Phase Behavior in POSS-PVPh
Copolymer
4-4-2 Structures of POSS-PVPh/PMMA-b-P4VP Micelles in THF Solution 93
4-4-3 The Effect of Phase Behavior on Thermal Properties in POSS-PVPh
Copolymer
94
Reference 96
Chapter 5
Conclusions
131List of Schemes
Pages
Scheme 3-1 Synthesis of TEMPO-1, TEMPO-2, TEMPO-3, POSS-SiH,
POSS-TEMPO initiator, POSS-PAS and POSS-PVPh.
70
Scheme 3-2 Synthesis of PMMA-b-P4VP copolymer by anionic polymerization. 71
Scheme 3-3 Synthesis of poly(4-vinyl phenol) polymer by anionic polymerization. 71
Scheme 4-1 Schematic illustration of a bilayer vesicle of POSS-PVPh /
PMMA-b-P4VP blemds.
List of Tables
Pages
Table 1-1 Static contact angles and surface free energy of epoxy thermosets
containing POSS-capped PCL.
8
Table 1-2 Elemental compositions of the surfaces of the epoxy thermosets containing
POSS-capped PCL determined by means of XPS.
8
Table 1-3 Static contact angles of water and diiodomethane and surface and
interfacial free energies and spreading coefficient of the PS44k film, CpPOSS/PS44k hybrid films, and the CpPOSS model surface.
9
Table 1-4 DSC results of epoxy glasses. 9
Table 1-5 Summary of thermal characteristics of polynorbornene-POSS copolymers. 10
Table 2-1 Evolution of living free radical polymerization. 28
Table 2-2 Numerical constant for molecular weight dependence of surface free
energy.
48
Table 2-3 Macleod’s exponent for some polymers. 49
Table 4-1 Characterization, root-mean-square surface roughness, advancing contact
angle for water and diiodomethane and surface free energy of POSS-PVPh in different solvents.
101
Table 4-2 Formulation and thermal properties of POSS-PVPh copolymers. 102
Table 4-3 Root-mean-square surface roughness, advancing contact angle for water
and diiodomethane and surface free energy of T8-POSS/PVPh blends.
103
Table 4-4 Root-mean-square surface roughness, advancing contact angle for water
and diiodomethane and surface free energy of Q8M8H -POSS/PVPh
blends.
104
angle for water and diiodomethane and surface free energy of POSS-PVPh copolymers.
Table 4-6 Root-mean-square surface roughness, advancing contact angle for water
and diiodomethane and surface free energy of POSS-PVPh copolymers.
106
Table 4-7 Summary of the glass transition temperatures of PVPh polymer,
POSS-PVPh copolymer, and PS-b-PVPh copolymer in 5 wt% THF solution under different selective (toluene) content.
List of Figures
Pages
Figure 1-1 Plot of the number of POSS publications versus the year in which they were
published.
2
Figure 1-2 Structures of silsesquioxanes. 4
Figure 1-3 POSS nanostructured chemicals. 4
Figure 1-4 AFM topography images of surfaces A (a) and A10 (b). 7
Figure 1-5 Glass transition temperatures Tgs of POSS-PMMA (●) and PMMA (○) as
a function of number-average molecular weight (Mn). The data are the
mean of the three individual experiments.
11
Figure 1-6 Schematic representation of diversity vs. complexity in natural and
synthetic polymers.
14
Figure 1-7 A variety of architectures can be produced by manipulating the ratio of the
volumes of the two blocks and the degree of immiscibility between blocks in solution and bulk states.
14
Figure 1-8 (Left) TEMs of CSSQ-PEO at different concentrations: (a) 0.25 mg/mL, (b)
1.0 mg/mL, and (c) 5.0 mg/mL. (Scale bar: 200 nm). (Right) Schematic representation of CSSQ-PEO aggregates formation at different
concentrations. (a) C≤CAC, (b) C>CAC, and (c) C>>CAC.
16
Figure 1-9 (a) TEM micrographs of dispersed vesicles at various magnifications. (b)
Schematic drawing showing the contacting regions of two vesicles. The double chains are disordered to expose the [60]fullerene moieties to water molecules, producing the hydrophobic force.
17
Figure 1-10 Coughlin model for structure of POSS-copolymer. (a) the TEM image of a
POSS-polybutadiene copolymer (43 wt% POSS) shows raft-like structure.
(b) A model of the 2-D crystallization of the POSS macromers.
Figure 1-11 TEM of thin sections of POSS triblocks prepared with cryomicrotomy at T =
-80 °C to yield samples of thickness ~50 nm. The microtomed sections
were chemically treated with RuO4, an agent selective for POSS. (a) Low
magnification micrograph showing overall morphology, (b)–(c) Higher magnification micrographs revealing cylindrical morphology, (d) Fourier transform of selected area from micrograph (a) revealing symmetry consistent with local hexagonal packing of the cylinders.
19
Figure 2-1 Mechanism of polymerization of styrene by TEMPO- mediated. 31
Figure 2-2 Work of adhesion. 35
Figure 2-3 Work of cohesion. 35
Figure 2-4 Contact angle equilibrium on a smooth, homogeneous, planar, and rigid
surface.
36
Figure 2-5 Advancing contact angle. 37
Figure 2-6 Receding contact angle. 37
Figure 2-7 Zisman plot foe poly(tetrafluoroethylene) (PTFE) using various testing
liquids.
45
Figure 2-8 Process of adhesion force measurement 46
Figure 2-9 Force-distance curve and adhesion force 47
Figure 2-10 Linear additively of surface tension of random copolymers of ethylene oxide
and propylene oxide, and surface-active behavior of blends of poly(ethylene oxide) (PEG 300) and poly(propylene oxide) (PPG 425).
51
Figure 2-11 Surface tension versus composition for ABA block copolymers of ethylene
oxide (A block) and propylene oxide (B block). Degree of polymerization are (1) DP = 16, (2) DP = 30, (3) DP = 56.
Figure 2-12 Surface tension of blends of compatible homopolymers. (1) poly(ethylene oxide) (PEG 300) + poly(propylene oxide) (PPG 425), (2) PPG 2025 + polyepichlorohydrin (PECH 1500), (3) PPG 400 + PECH 2000.
52
Figure 4-1 FTIR spectra of (a) TEMPO-1, (b) TEMPO-2, (c) TEMPO-3, (d) T7-POSS,
(e) POSS-SiH, (f) POSS-TEMPO initiator, (g) POSS-PAS copolymer, and (h) POSS-PVPh copolymer.
108
Figure 4-2 1H-NMR spectra of (a) TEMPO-1, (b) TEMPO-2, (c) TEMPO-3, (d)
T7-POSS, (e) POSS-SiH, (f) POSS-TEMPO initiator, (g) POSS-PAS copolymer, and (h) POSS-PVPh copolymer.
109
Figure 4-3 13C-NMR spectra of (a) TEMPO-1, (b) TEMPO-2, (c) TEMPO-3, (d)
T7-POSS, (e) POSS-SiH, (f) POSS-TEMPO initiator, (g) POSS-PAS copolymer, and (h) POSS-PVPh copolymer.
110
Figure 4-4 EI-MS spectrum of TEMPO-1. 111
Figure 4-5 EI-MS spectrum of TEMPO-2. 111
Figure 4-6 EI-MS spectrum of TEMPO-3. 111
Figure 4-7 FAB-MS spectrum of POSS-SiH. 112
Figure 4-8 FAB-MS spectrum of TEMPO-POSS initiator. 112
Figure 4-9 FTIR spectra of (a) PMMA-b-P4VP, (b) PtBOS, and (c) PVPh. 113
Figure 4-10 1H NMR spectra of (a) PMMA-b-P4VP, (b) PtBOS, and (c) PVPh. 114
Figure 4-11 13C NMR spectra of (a) PMMA-b-P4VP, (b) PtBOS, and (c) PVPh. 115
Figure 4-12 Surface energy of PVPh homopolymers with different molecule weight (■)
at room temperature and (●) after 180 °C thermal treatment process.
116
Figure 4-13 The surface free energies of T8-POSS/PVPh and Q MH
8
8 -POSS/PVPh
blend system.
117
24 hrs thermal treatment.
Figure 4-15 The surface free energies of POSS-PVPh comopolymers with different
molecule weight of PVPh segment (█) at room temperature and (▲) after 180 °C thermal treatment process.
119
Figure 4-16 DSC curves of (a) POSS-PVPh copolymer and (b) PVPh homopolymer
with different molecular weights.
120
Figure 4-17 The glass transition temperature of POSS-PVPh and PVPh as a function of
molecular weight.
121
Figure 4-18 FTIR spectra recorded at room temperature in the 2700-3800 cm-1 region
of PMMA blends with either (a) PVPh or (b) POSS-PVPh with different composition (weight ratio).
122
Figure 4-19 FTIR spectra recorded at room temperature in the 1675-1780 cm-1 region
of PMMA blends with either (a) PVPh or (b) POSS-PVPh with different composition (weight ratio).
123
Figure 4-20 FTIR spectra recorded at room temperature in the 1675-1780 cm-1 region
of PMMA blends with either (a) PVPh or (b) POSS-PVPh under similar composition.
124
Figure 4-21 DSC thermograms of POSS-PVPh or PVPh blemds with PMMA with
different composites (weight ratio).
125
Figure 4-22 Tg vs composition curves based on (a) the Kwei equation for PVPh/PMMA
blends system and (b) POSS-PVPh/PMMA blends system: ( ▲ ) experimental data of POSS-PVPh/ PMMA blends; (■) experimental data of PVPh/PMMA blends.
126
Figure 4-23 Transmission electron micrographs of POSS-PVPh in acetonitrile solution
(a) POSS-PVPh9, (b) POSS-PVPh35, (c) POSS-PVPh120 and (d)
POSS-PVPh264.
Figure 4-24 Transmission electron micrographs of POSS-PVPh120 in different solution
(a) 0.1mL toluene + 4.9mL THF, (b) 0.5mL toluene + 4.5mL THF, (c) 1mL toluene + 4.0mL THF and (d) 2mL toluene + 3.0mL THF.
128
Figure 4-25 Transmission electron micrographs of POSS-PVPh120 in acetonitrile at
different concentrations: (a) 0.5 mg/mL, (b) 1.0 mg/mL, (c) 2.0 mg/mL, (d) 4.0 mg/mL and (e) 8.0 mg/mL.
129
Figure 4-26 Transmission electron micrographs of POSS-PVPh66/ PMMA157-b- P4VP51
at different chains ratios: (a) 1/1 and (b) 2/1.
無機多面體聚矽氧烷團聯式共聚(4-乙基苯酚)
高分子之表面能、熱性質及自組裝結構研究
學生: 周幸儀 指導教授: 張豐志 博士
國立交通大學應用化學研究所 碩士班
摘要
本研究製備出一新型帶有多面體聚矽氧(polyhedral oligomeric silsequioxanes, POSS)之 2,2,6,6-四甲基哌啶氧化物(2,2,6,6-tetramethylpiperidinooxy, TEMPO)起 始劑。以乙醯氧基苯乙烯(acetoxystyrene)為單體,進行氮氧調節自由基聚合反應 (nitroxide-mediated radical polymerization, NMRP)及一水解反應去除保護基,合成 出 無 機 多 面 體 聚 矽 氧 烷 團 聯 式 共 聚(4- 乙 基 苯 酚 ) 高 分 子 (POSS-poly(4-vinyl phenol), POSS-PVPh)。 在本論文中,我們著重於多面體聚矽氧烷團聯式共聚(4-乙基苯酚)高分子的 表面能、熱性質及液態自組裝的研究。我們發現多面體聚矽氧烷的無機核心傾向 於分佈在材料表面,造成高分子薄膜的表面能下降,且經過熱處理過後可得到更 低的表面能性質。 本論文的另一主題是在探討多面體聚矽氧烷奈米粒子的聚(4-乙基苯酚)或聚 (4-乙基苯酚)與聚甲基丙烯酸甲酯(PMMA)混摻系統(POSS-PVPh/PMMA 或 PVPh/PMMA blends),利用傅立葉轉換紅外線光譜(FTIR)發現,帶有多面體聚矽 氧烷奈米粒子的混摻系統有較低的羰基的氫鍵作用力;然而,在相同的混摻比例
下,卻有較高的玻璃轉移溫度,因此我們可以推論多面體聚矽氧烷奈米粒子阻擋 了聚(4-乙基苯酚)分子鏈的運動,造成較高的玻璃轉移溫度。 此外我們也發現在末端帶有多面體聚矽氧烷奈米粒子的聚(4-乙基苯酚)團聯 式共聚合物在溶液(四氫呋喃/甲苯 THF/toluene 或乙腈 acetonitrile)中呈現非常獨 特的自組裝行為;藉由改變四氫呋喃/甲苯的比例或是聚(4-乙基苯酚)高分子鏈段 的長度,可以得到不同的型態。
Surface Free Energy, Thermal Properties, and
Self-Assembly in Solution State of Poly(4-vinyl
phenol) Tethered to Polyhedral Oligomeric
Silsequioxanes
Student: Hsin-Yi Chou Advisor: Dr. Feng-Chih Chang
Institute of Applied Chemistry
National Chiao Tung University
Abstract
Polyhedral oligomeric silsequioxanes-polyacetoxystyrene (POSS-PAS), a novel inorganic-organic polymer, was synthesized through nitroxide-mediated radical polymerization (NMRP) by using a novel macroinitiator, POSS-2,2,6,6-tetramethylpiperidinooxy (POSS-TEMPO). An amphiphilic block copolymer, POSS-PVPh, was obtained from the hydrolysis of POSS-PAS copolymer.
In this dissertation, we focus on the surface free energy, self-assembly in solution and thermal property of POSS-PVPh copolymer. We find that the POSS nanoparticle decrease the surface free energy of POSS-PVPh thin film when it populate on surface and POSS-PVPh polymer thin films possessed extreme low surface free energy after thermal treatment.
The miscibility and specific interaction behavior of POSS-PVPh/PMMA was another important subject in this dissertation. From FTIR results, the POSS-PVPh/PMMA blends possess a lower fraction of hydrogen-bonded carbonyl groups relative to those of PVPh/PMMA blends due to the hinder effect of POSS moiety. However, POSS-PVPh/PMMA blends possess higher Tg than that of the
PVPh/PMMA blends at the same composition. This phenomenon suggests that the POSS nanoparticle hinder the large-scale segmental motions of the polymer chains and results in higher Tg.
For phase behavior in solution, POSS-PVPh copolymer would aggregate in THF/toluene or acetonitrile and self-assembled to well-defined nanostructure. Through changing the composition of THF/toluene mix solution or the length of PVPh segment, the novel morphologies of POSS-PVPh could be observed.
Chapter 1
Introduction to Polyhedral Oligomeric Silsesquioxane
1-1 A Quick History of Polyhedral Oligomeric Silsesquioxane
In 1991, Lichtenhan and the Air Force Research Laboratory received funding from the Air Force Office of Scientific Research for his proposed development of Polyhedral Oligomeric Silsesquioxane (POSS) macromers containing a polymerizable functional group and the subsequent synthesis of a POSS-copolymer [1,2]. The University-Government collaboration between Lichtenhan and Feher rapidly expended to include more academic coaboraors including Laine and Sellinger [3], Mather et al. [4] and others who were all intrigued by the physical and mechanical property improvements imparted by incorporation of these nanostructured materials into polymer systems. In the late nineties not only was government and academic interest growing, but also that of the industrial sector which desired lower costs and larger quantities of the material.
The fall of 1998 marked the start-up of Hybrid Plastics in Fountain Valley, CA, which transitioned the government scale-up facilities to the commercial sector through a cooperative research and development agreement. In addition, the award of a 3 year multimillion dollar NIST Advanced Technology Program grant in 1998 to Hybrid Plastics was critical in both reducing the prices of the POSS feedstocks and macromers ($ 5000-$ 10000 down to $ 50-$ 2000 per pound) and increasing production (< 20 to > 2000 lb/year) to satify the more than 100 companies now investigating how the incorporation of POSS improves material properties for their applications. In the summer of 2003, Hybrid Plastics launched critical agreements with Southern Mississippi State University and the City of Hattiesburg for their
development of a 26000 sq ft production facility and a 1500 sq ft R&D center.
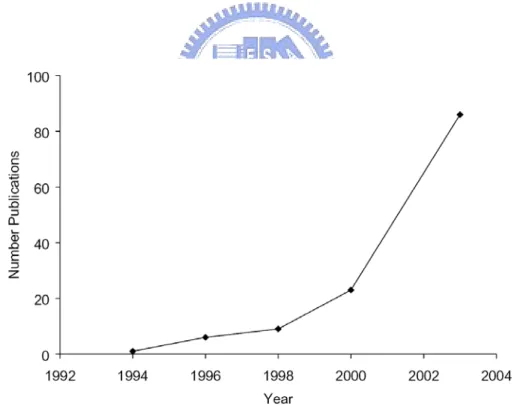
The nearly exponential increase in the number of academic researchers, academic publications (Figure 1-1), government programs, and industrial research efforts on POSS nanostructured chemicals has made it one of the top nanomaterials in the nanoscience/nanotechnology field. Indeed, the versatility of the POSS molecule, the more than one hundred demonstrated compatible polymer systems and the innumerable applications makes it difficult to understand and discern the current and future direction. However, it is clear that at least one concerted effort with a single-minded goal of predicting and controlling structure-property relationships is needed, and pursued by a number of research groups working with POSS nanostructured chemicals.
Figure 1-1. Plot of the number of POSS publications versus the year in which they were published.
1-2 Silsesquioxanes and Polyhedral Oligomeric Silsesquioxane
The term silsesquioxane refers to all structures with the empirical formula RSiO1.5, where the R is hydrogen or any alkyl, alkylene, aryl, arylene, or
organofunctional derivative of alkyl, alkylene, aryl, or arylene groups. The silsesquioxanes include random structures, ladder structures, cage structures, and partial cage structures, as illustrated in Figure 1-2 [5].
In 1995, Baney et al. reviewed the structure, preparation, properties, and applications of silsesquioxanes, especially the ladder-like polysilsesquioxanes shown in Figure 1-2 (structure b). These include poly(phenylsilsesquioxane) (PPSQ) [6,7], poly(methyl silsesquioxane) (PMSQ) [8-10], and poly(hydridosilsesquioxane) (PHSQ) [11,12]. These ladder-like polymers have an outstanding thermal stability and they exhibit oxidative resistance even at temperatures of more than 500 °C. In addition, these ladder-like polymers have good insulating properties and gas permeability.
Therefore, the ladder-like silsesquioxane polymers have a variety of applications in areas such as photoresist coatings [13,14] for electronics and optical devices, interlayer dielectrics and protective coating films [15,16] for semiconductor devices, liquid crystal display elements [17], magnetic recording media [18], optical fiber coatings [19], gas separation membranes [20], binders for ceramics [21]. However, in the past few years, much more attention has been paid to the silsesquioxanes with specific cage structures (Figure 1-2 structure c-f). These Polyhedral Oligomeric Silsesquioxanes have been designated by the abbreviation POSS.
POSS compounds embody a truly hybrid (inorganic-organic) architecture, which contains an inner inorganic framework made up of silicone and oxygen (SiO1.5)x, that
is externally covered by organic substituents. These substituents can be totally hydrocarbon in nature or they can embody a range of polar structures and functional groups. POSS nanostructure chemicals, with sizes of from 1 to 3 nm in diameter, can
Figure 1-2. Structures of silsesquioxanes.
be thought of as the smallest possible particles of silica, as shown in Figure 1-3. They may be viewed as molecular silicas. However, unlike silica, silicones, or fillers, each POSS molecule contains organic substituents on its outer surface that make the POSS nanostructure compatible with polymers, biological systems, or surfaces. Furthermore, these groups can be specially designed to be non-reactive or reactive.
Figure 1-3. POSS nanostructured chemicals.
O ne or more reactive groups for grafting or
polymerization.
Thermally and chemically robust hybrid (organic-inorganic) framework. Si Si O O Si Si Si Si O O O O Si O Si O O O O O R R R R R R R X Nanoscopic size Si-Si distance = 0.5 nm R-R distance = 1.5 nm.
Precise three-dimensional structure for molecular level reinforcement of polymer
segments and coils. Unreactive organic (R)
groups for solubilization and compatibilization.
A variety of POSS nanostructure chemicals have been prepared which contain one or more covalently bonded reactive functionalities that are suitable for polymerization, grafting, surface bonding, or other transformations [22]. Unlike traditional organic compounds, POSS chemicals release no volatile organic components, so they are odorless and environmentally friendly.
1-3 POSS Polymers and Copolymers
The incorporation of POSS derivatives into polymeric materials can lead to dramatic improvements in polymer properties which include, but are not limited to, increases in use temperature, oxidation resistance, surface hardening, and improved mechanical properties, as well as reductions in flammability, heat evolution, and viscosity during processing. These enhancements have been shown to apply to a wide range of thermoplastics and a few thermosetting systems [23]. It is especially convenient that the use of POSS monomers doesn’t require dramatic changes in processing. Monomers are simply mixed and copolymerized. As long as the POSS monomer is soluble in the monomer mixture, it is incorporated in a true molecular dispersion into the copolymer. No phase separation will occur although some aggregation of POSS units bound with polymer will occur. This is a significant advantage over current filler technologies. POSS nanostructures have also shown significant promise for use in catalyst supports and biomedical applications as scaffolds for drug delivery, imaging reagents, and combinatorial drug development.
1-4 Surface Free Energy of POSS Polymer Surface
1-4-1 Wettability of POSS Nanostructured Polymer Surfaces[24,25]
S. Turri and M. Levi prepared polyurethane anionomers containing various amounts (ca. 3-20 %) of a diol functionalized polyhedral oligomeric silsesquioxane (POSS) nanofiller. X-ray diffraction showed the formation of a nanocrystalline structure in all copolymers considered. Static contact angle measurements indicated a significant enhancement of surface hydrophobicity as well as reduction in surface tension components even at the least POSS level (3 %). In particular, the polar component seems very sensitive to the presence of even few POSS percentages. The result is achieved very fast in 3 % POSS-modified sample, and does not significantly improve in the 3-20 % POSS range. This would mean the POSS nanostructures screen the polar groups like urethanes and carboxyls and are preferentially oriented air-side. AFM topography images are shown in Figure 1-4, while numerical results are Ra=94
A°, Rq=132 A° for sample A10 (with 10 wt% POSS present), and Ra=8.5 A°, Rq=11 A°
for sample A (without POSS units present). The presence of POSS macromers therefore seems to increase the surface roughness significantly, although it is considered by both theoretical and experimental evidence that Ra < 100 nm generally
has a limited effect on contact angles and hysteresis. Therefore, surface roughness, although significantly raised, cannot explain the decreased wettability of the POSS nanostructured polyurethane surfaces. This effect is likely related to surface-oriented enrichment of POSS structures bearing low-surface-tension alkyl substituents. Similar results wee found very recently for poly(methyl methacrylate) (PMMA) blended with fluorinated POSS-terminated polymers, [24] and were attributed to coverage of the outermost layer by POSS heads.
Figure 1-4. AFM topography images of surfaces A (a) and A10 (b).
1-4-2
Nanostructured Thermosets from Epoxy Resin and an
Organic-Inorganic Amphiphile [26]
POSS-capped PCL was synthesized via ring-opening polymerization of ε-caprolactone with 3-hydroxypropylheptaphenyl POSS as the initiator. The novel organic-inorganic amphiphile was incorporated into epoxy resin to prepare the organic-inorganic hybrid thermosets. The morphology of the organic-inorganic hybrids was characterized by means of atomic force microscopy (AFM) and transmission electron microscopy (TEM). It is found that the epoxy thermosets displayed a variety of nanostructures depending on the concentration of the POSS-capped PCL. The formation of nanostructures was addressed on the basis of miscibility and phase behavior of the subcomponents (viz. POSS and PCL chains) of the organic-inorganic amphiphile with epoxy after and before curing reaction. It is judged that the nanostructures in the organic-inorganic hybrid composites were formed via a mechanism of self-assembly. The static contact angle measurements show that the organic-inorganic nanocomposites displayed a significant enhancement in surface hydrophobicity as well as reduction in surface free energy. The
improvement in surface properties was ascribed to the enrichment of POSS moiety on the surface of the nanostructured thermosets, which was evidenced by X-ray photoelectron spectroscopy.
Table 1-1. Static contact angles and surface free energy of epoxy thermosets containing POSS-capped PCLa. aH 2O: γL=72.80 mN m -1, d =21.80 L γ mN m-1, p=51.00 L γ mN m-1 (ref 14f). Ethylene glycol: γL =48.3 mN m-1, d =29.3 L γ mN m-1, p =19.0 L γ mN m-1 (ref 14f)
Table 1-2. Elemental compositions of the surfaces of the epoxy thermosets containing POSS-capped PCL determined by means of XPS.
1-4-3 Structure and Dewetting Behavior of Polyhedral Oligomeric
Silsesquioxane (POSS)-Filled Polystyrene (PS) Thin Films [27]
POSS meets increasing interest as a building unit for inorganic-organic hybrid materials. Structural analysis of the cyclopentyl-substituted POSS (CpPOSS) into polystyrene (PS) thin films revealed that CpPOSS segregated to the film surface and crystallized. The segregation of CpPOSS to the surface changes the surface free energy and spreading coefficient of the film. Interfacial structure was also roughened by the segregation of CpPOSS, which can contribute to the inhibition of dewetting by pinning the contact line of the PS film with the substrate. The inhibition of dewetting
can be attributed to the modification of the film surface and interface by the segregation of CpPOSS.
Table 1-3. Static contact angles of water and diiodomethane and surface and interfacial free energies and spreading coefficient of the PS44k film, CpPOSS/PS44k hybrid films, and the CpPOSS model surface.
1-5 Thermal Properties of POSS Polymer
1-5-1 POSS Reinforced Epoxy Systems [28]
Recently, families of mono- and difunctionalized POSS macromers bearing epoxide groups have been developed. This paper presents an investigation of the thermal property enhancements in commonly used model epoxy resins reinforced with monofunctional POSS-epoxy macromers. The glass transitions of these POSS-epoxy nanocomposites were studied using differential scanning calorimetry. They were effective at hindering the molecular motion of the epoxy network junctions. Thus the glass transition temperature, Tg, was observed to increase with increasing
weight fraction of the monofunctional POSS-epoxy.
1-5-2 Thermal Properties of Poly(norbornyl-POSS) Copolymers
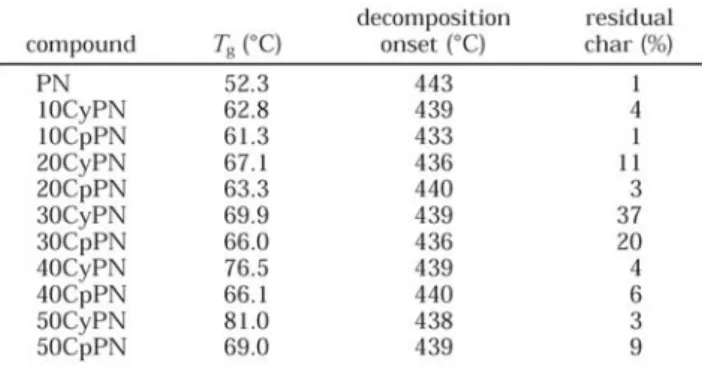
[29]
The mechanical relaxation behavior and microstructure of a series of novel norbornyl-POSS organic-inorganic copolymers have been investigated. Table 1-5 gives a summary of the thermal properties of both CyPOSS and CpPOSS copolymers. The Tg data, as determined from a second heating scan. It can be seen that the glass
transition increases with increasing weight percentage of POSS, with the effect being slightly more pronounced in the CyPOSS copolymers than for the CpPOSS counterparts.
Table 1-5. Summary of thermal characteristics of polynorbornene-POSS copolymers.
1-5-3 The Effect of POSS on Thermal Properties in PMMA System
[30]
The ATRP initiator was synthesized by the reaction of incompletely condensed POSS and trichlorosilanes. The solution polymerization of MMA using these initiators mediated by a copper complex proceeded in a living fashion, thus providing tadpole-shaped polymers with an “inorganic head” of POSS and an “organic tail” of well-defined polymer. DSC studies showed that Tg of the organic/inorganic hybrid
polymers were more and more enhanced compared to those of the model polymers without a POSS moiety, as the polymer tail became shorter or the weight fraction of
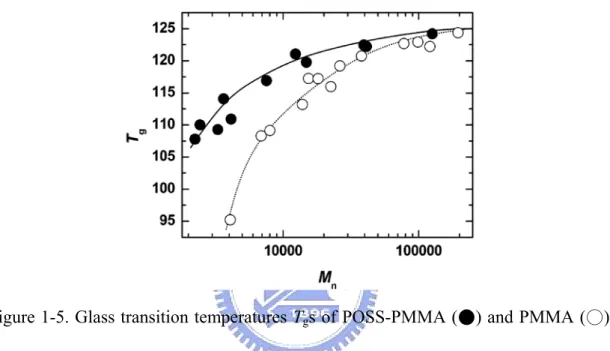
the POSS moiety became larger. Figure 1-5 shows the Tgs of POSS-PMMA and
PMMA as a function of Mn. For both samples, Tg increases with increasing Mn,
leveling off at around 125 °C. This is a well-known behavior of PMMA. Importantly, the Tg value of POSS-PMMA is higher than that of the corresponding PMMA, in
particular, in the lower molar mass region. Namely, the observed Tg enhancement
may be attributed to the high Tg or the low mobility of the POSS segment.
Figure 1-5. Glass transition temperatures Tgs of POSS-PMMA (●) and PMMA (○)
as a function of number-average molecular weight (Mn). The data are the mean of the
1-6 Phase Separation of POSS Polymer
1-6-1 An Overview of Self-Organization of Block Copolymers
Today’s materials require additional processing or modification steps in order to obtain the properties that make them suitable for a particular application. Recently, materials science deals increasingly with nanostructures, i.e. structures with a characteristic dimension of 1-100 nm. This particular range of length scale is called the mesoscopic range, as it is located between the microscopic range of atoms and molecules and the macroscopic range of solids.
Solid-state physics and electronics have entered the field of nanostructures by making use of lithography and etching processes (“top-down” approach), which enable the fabrication of structures no smaller than ~200 nm. Nature, on the other hand, may serve as a model for the building-up of smaller structures. As an alternative to these traditional fabrication pathways, routes that use the self-assembly of low molecular weight oligomeric or polymeric building blocks are attracting increasing attention. By designing these building blocks in such a way that they contain all the necessary information to direct their self-assembly into functional materials, additional processing or modification steps could become superfluous. Individual molecules are integrated into larger functional units and complex structural hierarchies via self-organization (“bottom-up” approach), leading to such advanced materials as wood or bone (Figure 1-6) [31]. It is one of the great challenges in research disciplines like chemistry, physics, or materials science to find ways to structure molecules so as to enable them to produce functional superlattices by self-organization, for example by making use of the self-assembly features of supramolecular chemical devices [32] or block copolymers. [33, 34] Here, we focus on block copolymer type building blocks and discusses their potential for the
development of self-assembled materials.
Block copolymers are macromolecules composed of two or more polymer blocks of chemically different monomers that are linked together by chemical bonds. The resulting chain topologies can be linear, branched, or cyclic. Systematic studies of these materials became possible through developments in polymerization techniques, which made possible the synthesis of well-defined block copolymers with a very small polydispersity. Among the linear block copolymers, the diblock copolymers have been studied in great detail. They can be considered as model systems for more complicated block copolymers, such as block copolymers with more than two components, or block copolymers with other block distributions.
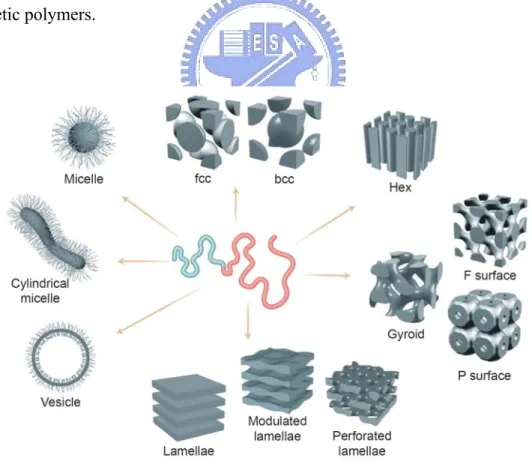
Different microphase separated morphologies of block copolymers occur depending on the relative compositions of the different components and the total molecular weight as expressed by the degree of polymerization. In addition, the aggregation state of the blocks largely influences the morphology, too. The micellar aggregates, lyotropic phases, and solid-state mesophases of diblock copolymers are among the best examined supramolecular systems, which is due to the fact that a simple encoding via the chemical composition and the overall number of repeating units (N) makes it possible to control both the shape and the size of the resulting superstructures. [35, 36] The phases of amphiphilic diblock copolymers which are most commonly observed in solution are spherical and cylindrical micelles and vesicles, and in the solid state bcc-packed spheres (BCC), hexagonally packed cylinders (HEX), lamellae (LAM), and gyroid (Figure 1-7) [37].
Figure 1-6. Schematic representation of diversity vs. complexity in natural and synthetic polymers.
Figure 1-7. A variety of architectures can be produced by manipulating the ratio of the volumes of the two blocks and the degree of immiscibility between blocks in solution and bulk states.
In the following section the self-organization of block copolymers will be described with the help of some examples. They are divided into dilute solutions and bulk phases, each of them being well-suited for the preparation of interesting materials.
1-6-2 In Dilute Solutions
1-6-2-1 Core-Corona Structure of POSS-Poly(ethylene oxide) in
Aqueous Solution [38]
Amphiphilic cubic silsesquioxane-poly(ethylene oxide) (CSSQ-PEO) was
prepared from octakis (dimethylsiloxy)octasilsesquioxane (Q MH
8
8 ) and allyl-PEO
through a hydrosilylation reaction and its aggregation process of CSSQ-PEO in aqueous solution were investigated by fluorescence, dynamic and static light scattering (DLS and SLS), and transmission electron microscopy (TEM). The critical aggregation concentration (CAC) determined by fluorescence measurements was found to be 0.28 mg/mL. Combinations of DLS, SLS, and TEM studies showed the existence of core-corona micelle with hydrophobic CSSQ as the core and hydrophilic PEO as the corona in aqueous solution. A large Rg/Rh ratio (1.46) and the extremely
small value of average chain density (4×10-4 g/cm3) indicate the small hydrophobic CSSQ core was surrounded by the extended PEO coronae. The interconnections between the micellar aggregates lead to the formation of network-like structures at higher concentration.The long PEO segments act as a spacer between the spherical aggregates, which facilitate the formation of a network-like structure at high concentration.
Figure 1-8. (Left) TEMs of CSSQ-PEO at different concentrations: (a) 0.25 mg/mL, (b) 1.0 mg/mL, and (c) 5.0 mg/mL. (Scale bar: 200 nm). (Right) Schematic representation of CSSQ-PEO aggregates formation at different concentrations. (a) C≤ CAC, (b) C>CAC, and (c) C>>CAC.
1-6-2-2 Vesicle Formation and Its Fractal Distribution by Bola-
Amphiphilic [60]Fullerene [39]
A novel amphiphilic [60]fullerene derivative with two ammonium headgroups is synthesized, and its self-organization characteristics in water in the scale ranging from nanometer to micrometer are reported. At the molecular scale, the bola-amphiphilic [60]fullerene forms spherical vesicles. At large scales, most vesicles are not dispersed randomly, but are concentrated in thin walls that are connected three-dimensionally. The connected foam like structure (although the mixture does not foam) produces the large voids whose sizes range from a hundred nanometers to a few micrometers. Only a few vesicles are visible within the voids. At low magnification, only the voids are visible. The novel aggregation modes result from the hydrophobic interaction produced by the [60]fullerene moieties exposed to water molecules by the disordered alkyl tails.
(a) (b)
Figure 1-9. (a) TEM micrographs of dispersed vesicles at various magnifications. (b) Schematic drawing showing the contacting regions of two vesicles. The double chains are disordered to expose the [60]fullerene moieties to water molecules, producing the hydrophobic force.
1-6-3 Bulk Phases
1-6-3-1 Coughlin’s Model [40]
Coughlin et al. have provided a visual model of a POSS-copolymer based on the morphology of POSS polybutadiene and polyethylene copolymers. The hybrid inorganic/organic polymers were made by either ring-opening metathesis copolymerization or single site catalysis, both allowing for good control of POSS incorporation. Physical and mechanical data were taken of the materials and a visual model was proposed (Figure 1-10). This model matches their TEM data, displaying the ‘raft-like’ or lamellae structure of POSS within the polymer matrix. Two-dimensional networks are the highest possible architecture that a POSS copolymer can adopt due to the constraints of the polymer chain, while three-dimensional structures can be expected for most POSS-polymer blends. TGA of
the POSS-polyethylene (12 wt% POSS) showed a 70 °C improvement in the onset of decomposition.
(a) (b)
Figure 1-10. Coughlin model for structure of POSS-copolymer. (a) the TEM image of a POSS-polybutadiene copolymer (43 wt% POSS) shows raft-like structure. (b) A model of the 2-D crystallization of the POSS macromers.
1-6-3-2
ABA Triblock Copolymers Containing POSS Pendant
Groups [41]
The synthesis of ABA triblock copolymers possessing a central rubbery segment of poly(n-butyl acrylate) (pBA) and outer blocks of poly(methacryate-POSS) (p(MA-POSS)) was conducted using a two-step atom transfer radical polymerization (ATRP) strategy. Morphological investigations of p(MA-POSS)6-b-pBA481-b-
p(MA-POSS)6 triblock copolymer thin films prepared with pBA macroinitiator of
higher molar mass (Mn SEC=64,010 g/mol; Mw/Mn= 1.39) by using SAXS and TEM
indicated that no microphase separation was induced during sample preparation; i.e. the resulting morphology and SAXS scattering patterns were completely featureless. However, the morphology of p(MA-POSS)10-b-pBA201-b-p(MA-POSS)10 triblock
copolymer thin films prepared with a difunctional pBA macroinitiator of lower molar mass (Mn SEC= 25,800 g/mol; Mw/Mn=1.20) show remarkably well-defined microphase
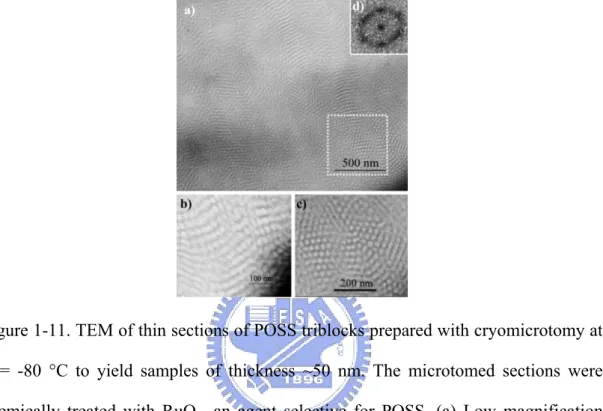
separated structures. (Figure. 1-11) In a relatively low magnification TEM image, well-defined white cylinders of pBA are clearly discerned to be oriented both in and out of the continuous dark phase consists of the p(MA-POSS) block.
Figure 1-11. TEM of thin sections of POSS triblocks prepared with cryomicrotomy at T = -80 °C to yield samples of thickness ~50 nm. The microtomed sections were chemically treated with RuO4, an agent selective for POSS. (a) Low magnification
micrograph showing overall morphology, (b)-(c) Higher magnification micrographs revealing cylindrical morphology, (d) Fourier transform of selected area from micrograph (a) revealing symmetry consistent with local hexagonal packing of the cylinders.
Reference
[1] Lichtenhan, J. D.; Feher, F. J.; Gilman, J. W. Macromolecules 1993, 26, 2141. [2] Lichtenhan, J. D.; Otonari, Y.; Carr, M. J. Macromolecules 1995, 28, 8435. [3] Laine, R. M.; Sellinger, A. Macromolecules 1996, 29, 2327.
[4] Mather, P. T.; Haddad, T. S.; Oviatt, H. W.; Schwab, J. J.; Chaffee, K. P.; Lichtenhan, J. D. Polym Prepr 1998, 39, 611.
[5] Baney, R. H.; Itoh, M.; Sakakibara, A.; Suzuki, T.; Chem. Rev. 1995, 95, 1409. [6] Zhang, X.; Shi, L.; Chin, J. Polym. Sci. 1987, 5, 197.
[7] Brown, Jr. J. F. J. Polym. Sci. C 1987, 1, 83.
[8] Xie, Z.; He, Z.; Dai, D.; Zhang, Chin. R. J. Polym. Sci. 1989, 7, 183.
[9] Maciel, G. E.; Sullivan, M. J.; Sindorf, D. W. Macromolecules 1981, 14, 1607. [10] Engelhavdt, G.; Jancke, H.; Lippmaa, E.; Samoson, A. J. Organomet. Chem.
1981, 210, 295.
[11] Frye, C. L.; Collins, W. T. J. Am. Chem. Soc. 1970, 92, 5586.
[12] Belot, V.; Corriu, R.; Leclerq, D.; Mutin, P. H.; Vioux, A. Chem. Mater. 1991, 3, 127.
[13] Adachi, H.; Hayashi, O.; Okahashi, K. Japanese Patent Kokoku-H- 2-15863. [14] Adachi, H.; Hayashi, O.; Okahashi, K. Japanese Patent Kokai-S-60-108841. [15] Adachi, E.; Aiba, Y.; Adachi, H. Japanese Patent Kokai-H-2-277255. [16] Aiba, Y.; Adachi, E.; Adachi, H. Japanese Patent Kokai-H-3-6845. [17] Shoji, F.; Sudo, R.; Watanabe, T. Japanese Patent Kokai-S-56-146120. [18] Imai, E.; Takeno, H. Japanese Patent Kokai-S-59-129939.
[19] Mishima, T.; Nishimoto, H. Japanese Patent Kokai-H-4-247406. [20] Saito, Y.; Tsuchiya, M.; Itoh, Y. Japanese Patent Kokai-S-58-14928. [21] Mine, T.; Komasaki, S. Japanese Patent Kokai-S-60-210570.
[23] Ellsworth, M. W.; Gin, D. L. Polym. News 24, 331.
[24] Turri, S.; Levi, M. Macromol. Rapid Commun. 2005, 26, 1233. [25] Turri, S.; Levi, M. Macromolecules 2005, 38, 5569.
[26] Ni; Y.; Zheng, S. Macromolecules 2007, 40, 7009.
[27] Hosaka, N.; Torikai, N.; Otsuka, H.; Takahara, A. Langmuir 2007, 23, 902. [28] Lee, A.; Lichtenhan, J. D. Macromolecules 1998, 31, 4970.
[29] Mather, P. T.; Jeon, H. G.; Romo-Uribe, A. Macromolecules 1999, 32, 1194. [30] Ohno, K.; Sugiyama, S.; Koh, K.; Tsujii, Y.; Fukuda, T.; Yamahiro, M.; Oikawa,
H.; Yamamoto, Y.; Ootake, N.; Watanabe, K. Macromolecules 2004, 37, 8517. [31] Rodríguez-Hernández, J.; Chécot, F.; Gnanou, Y.; Lecommandoux, S. Prog.
Polym. Sci. 2005, 30, 691.
[32] Elemans, J. A. A. W.; Rowan, A. E.; Nolte, R. J. M. J. Mater. Chem. 2003, 13, 2661.
[33] Förster, S.; Plantenberg, T. Angew. Chem. Int. Ed. 2002, 41, 688. [34] Park, C.; Yoon, J.; Thomas, E. L. Polymer 2003, 44, 6725.
[35] Bates, F. S.; Fredrickson, G. H. Annu. Rev. Phys. Chem. 1990, 41, 525. [36] Förster, S.; Antonietti, M. Adv. Mater. 1998, 10, 195.
[37] Bucknall, D. G.; Anderson, H. L. Science 2003, 302, 1904.
[38] Mya, Y. K.; Li, X.; Chen, L.; Ni, X.; Li, J.; He, C. J. Phys. Chem. B 2005, 109, 9455.
[39] Sano, M.; Oishi, K.; Ishi-I, T.; Shinkai, S. Langmuir 2000, 16, 3773.
[40] Phillips, S. H.; Haddad, T. S.; Tomczak, S. J.; Opin, C. Solid State Mater. Sci.
2004, 8, 21.
[41] Pyun, J.; Matyjaszewski, K.; Wu, J.; Kim, G. M.; Chun, S. B.;Mather, P. T. Polymer 2003, 44, 2739.
Chapter 2
Theory
2-1 Living Free Radical Polymerization
2-1-1 Requirements for Living Free Radical Polymerization
Living free radical polymerization was first defined by Szwarc [1] as a chain growth process without chain break reactions transfer ant termination. Such a polymerization provides end-group control and enables the synthesis of block copolymers by sequential monomer addition. However, it does not necessarily provide for molecular weight control and narrow molecular weight distribution (MwD or PDI). Additional prerequisites to achieve these goals are that the initiator should be consumed at early stages of polymerization and that exchange between species of various reactivities is fast in comparison with propagation [2-4]. If these additional criteria are met, a controlled polymerization results. Polymerization can also be defined as controlled of side reaction occur, but only to an extent which does not considerably disturb the control of the molecular structure of the polymer chain.
Recently, many new “living” polymerization systems, such as carbocationic, ring-opening metathesis, group transfer, and radical polymerization, have been developed. The term ‘living’ (with quotation marks) indicates synthesis of well-defined polymers under conditions in which chain breaking reaction undoubted occur, as in radical polymerization, but polymerization systems which lead to well-defined polymers but are not completely free of termination or transfer, as are radical polymerization, should be named controlled/living may also described the essence of these systems [5].
Ideally, controlled/living systems lead to polymers with degrees of polymerization predetermined by the ratio of the concentrations of consumed monomers to the introduced initiator DPn =Δ[M]/[I]0, polydispersities close to Poission distribution (DPw/DPn =1+1/DPn), and with all chains end-functionalized. Experimentally, the best way to evaluate such system is to follow the kinetics of polymerization and the evolution of molecular weights, polydispersities and functionalities with conversion. Well-defined systems should provide:
(a) Linear kinetics plots in semilogarithmic coordinates (ln
(
[M]0/[M])
vs time), if the reaction is first order in monomer concentration; acceleration on such plots may indicate slow initiation whereas deceleration may indicate termination or deactivation of the catalyst.(b) Linear evolution of molecular weights with conversion; molecular weights lower than predicted by Δ[M]/[I]0 ratios indicate transfer, molecular weights higher than predicted by Δ[M]/[I]0 ratios indicate inefficient initiation or chain coupling (at most, twice higher than predicted molecular weights can be formed by bimolecular radical coupling).
(c) Polydispersities should decrease with conversion for systems with slow initiation and slow exchange; polydispersites increase with conversion when the contribution of chain breaking reactions becomes significant.
(d) End functionalities are not affected by slow initiation and exchange but they are reduced when chain breaking reactions become important.
2-1-2 Peculiarities of Living Free Radical Polymerization
Conventional radical polymerizations can never be controlled due to slow initiation and unavoidable termination. Fortunately, the concept of exchange between active and dormant species has been very helpful in converting ill-defined radical
systems to controlled polymerizations.
(a) Suppression of chain transfer and termination:
Chain transfer is not the main obstacle for the synthesis of well-defined polymers by a radical mechanism. Monomer transfer coefficients are usually below
4 10 / p < −
trm k
k . This means that half of the chains participate in transfer at the stage of
10000 =
n
DP but less than 10 % of chains at DPn =1000. Thus, transfer to monomer should not interfere with the synthesis of polymers with DPn <1000. Of course, transfer to solvent, polymer and transfer agents will operate as usual, but the proper choice of reaction conditions should lead to well-defined polymers, especially at lower ranges of molecular weights.
In contrast, termination is impossible to avoid in homogeneous radical polymerization. Two radicals will recombine or disproportionate with nearly diffusion controlled rates. Fortunately, propagation is first order, whereas termination is second order with respect to the concentration of growing radicals. Thus, the contribution of termination decreases when the concentration of propagating radicals is reduced. In conventional radical polymerization, a sufficiently low concentration of radical is attained by slow initiation. However, as discussed above, slow concentration of growing radicals must be achieved by other means.
(b) Exchange between active and dormant species:
Thus, a requirement for controlled radical system is that initiation should be completed at low monomer conversions; the concentration of propagating radicals should be low, but the total concentration of growing chains should be much higher. These requirements can be met through dynamic equilibration of minute amounts (ppm) of propagating radicals with some form of dormant chains. Three general equilibrium systems are shown schematically below:
System Ⅰ P ⋅ X+ ⋅ n kd ka P X n − System Ⅱ Z Pn⋅+ kd ka
{
P − Z}
⋅ n System Ⅲ P P X m n⋅+ kexch X Pn − + Pm⋅The first system is based on the deactivation of growing radical with relatively stable (persistent) radicals, which may also be inorganic or organometallic species.
The deactivation in the second system involves species with even numbers of electrons forming stable (persistent) radicals. Z may be a nonpolymerizable vinyl monomer, or inorganic, or organometallic species, etc. In both systems, the equilibriums are very strongly shifted towards the dormant species to reduce the concentration of growing radicals.
The last system employs the so-called degenerative transfer process which has equilibrium constant of K=1. In this case, the protected group is never spontaneously cleaved but rather transferred in bimolecular process from the dominating dormant species carrying the protecting group X to a minute amount of propagating radicals. If exchange is faster than propagation, well-defined polymers can be prepared. This system can employ conventional initiators such as AIBN, benzoyl peroxide (BPO), etc, provided that their concentration is smaller than that of the transfer agent (R-X).
In these systems, the prerequisite for the synthesis of controlled polymers is a sufficiently fast exchange process. Polydispersities decrease with conversion and are
lower systems with higher concentration of deactivator, lower kp/kd ratios and
lower concentration of chains.
The thermodynamics and the dynamics of exchange in these reactions are the most important parameters affecting the control of radical polymerizations. Therefore,
structures and reactivities of persistent radicals and catalyst, which may accelerate exchange, are among the most important features of these systems.
(c) Contribution of thermal self-initiation:
In the controlled polymerization of styrene and substituted styrenes, the thermal self-initiated polymerization of styrene occurs simultaneously with the controlled process (system ⅠⅡⅢ). The contribution of thermal self-initiation to the overall rate and to the total number of polymer chains in the system should be carefully assessed.
The rate of self-initiation has been studied and the rates of generation of new radicals are known at various temperatures [6]. The so-called Mayo dimmer [7], which is an intermediate in this process, is additionally responsible for the reduction of molecular weights since it has a very large transfer coefficient [8]. The overall rate of self-initiated polymerization, however, should not be confused with the rate of radical generation. For example, approximately 1 M/hour of styrene is consumed at 130 °C in bulk polymerization, but less than 10-3 M/hour of radicals are generated. Thus, in ten hours less than 10-2 M radicals are generated. Usually this number may even be lower because the rate of self-initiation is second or third order in respect to monomers [6].
2-1-3 Evolution of Living Free Radical Polymerization
Table 2-1 illustrates development of fundamental concepts in organic radical processes, some concepts in living polymerization and advanced in radical polymerization which all have contributed to controlled/living free radical polymerization and without which the present understanding and control of radical reactions would possible.
difficult to control due to termination reactions which could not be avoided. Probably the first example of successful addition of halogenated compounds to alkenes via radical intermediates (Atom transfer radical addition, ATRA) was provided by Kharach [9] under photochemical condition. This atom transfer radical process was subsequently converted to metal catalyzed reactions by Minisci [10], Vofsi [11] and others during the 1960s. At approximately the same time, various nitroxides were prepared as stable radicals but without any application to organic synthesis at that time [12].
These reactions have found many synthesis applications as thoroughly summarized by Bellus in 1985 [13]. At that time, it was not clear how these reactions occurred until Fischer [14] provided the explanation based on the persistent radical effect for the observed unexpectedly high chemoselectivities. Numerous systems based on atom transfer reactions were used for structure-reactivity comparisons and for refined organic/bioorganic synthesis [15] with many organometallic catalysts [16]. Atom transfer radical reactions gained increasing importance in organic synthesis because of their high chemo-, region-, and stereoselectivity and tolerance to many useful functional groups [17]. Future progress in this area requires additional developments of new catalysis which could increase the rates and selectivity of the addition, elimination and rearrangement reactions.
Table 2-1. Evolution of living free radical polymerization.
Organic Synthesis Living Polymerization Controlled Radical
Polymerization (CRP) 1940’s Kharash
(1st ATRA (hv))
1950’s Szwarc (Living Anionic, Block
Copolymer) 1960’s Minisci (CuX/RX/olefin) Rosantsev (Nitroxide) 1970’s Matyjaszewski (A*⇔D in CROP) Borsig (Ar2CH, Ar3C) Minoura (Cr(Ⅱ)Acetate/MMA) 1980’s Fisher (Persistent Radical Effect) Curran (RI, RSn, RSeAr) Kochi (Organocobalt) Kennedy (Inifers) Matyjaszewski, Sawamoto (“living” Carbocationic) DuPont: Quirk (GTP) Novak, Grubbs (Living ROMP) Otsu (1st CRP, Iniferters) Solomon/Rizzardo (TEMPO) Rizzardo (RAFT) 1990’s Various Mt in ATRA Stereo-control….
Matyjaszewski (Controlled & Ranking) Muller (Exchange Reactions) George (TEMPO/Styrene) Matyjaszeski (ATRP) Sawamoto
(Various Nitroxides & Mt/RX in ATRP)
In this section we introduce two polymerizations; nitroxide mediated radical polymerization and anionic polymerization, which are utilized in this thesis.
2-1-4 Nitroxide Mediated Radical Polymerization (NMRP)
In 1993, Georges et al. [18] reported on the controlled radical polymerization of styrene initiated by benzoyl peroxide (BPO) and mediated by 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) as a stable radical together as shown in Figure 2-1 with a conventional radical initiator such as BPO to prepare polystyrene with molecular weights approaching Mn≒100000 and relatively low polydispersities,
Mw/Mn = 1.2 to 1.4. In fact, TEMPO alone can also be used as a moderator as styrene
self-initiates [19]; however, control of the polymerization is reduced because of competing some side reactions. The current knowledge of nitroxide-mediated radical polymerization can be summarized as follows. Firstly controlled polymerization in the presence of TEMPO is limited to styrene and styrene copolymers in the temperature range of 110-140 °C. Molecular weight up to Mn ≒ 50000 are well controlled and
predetermined by the ratio of reacted monomer to the initial concentration of TEMPO or its adducts.
Secondly, polymerization of styrene with TEMPO and some other nitroxide as the counter radical is very slow and independent of the concentration of alkoxyamines present in the system [20]. The rate of polymerization is very similar to the rate of styrene thermal polymerization [21]. This indicates that self-initiation controls the polymerization rate but that generated radicals must exchange with dormant chains that are reversibly homolytically cleaved.
A third thing we know about nitroxide-mediated radical polymerization is that styrene is a unique monomer for TEMPO-mediated polymerization, because not only does it self-initiate, but it also polymerizes more slowly than acrylaies and
methacrylaies, enabling the preparation of polymers with low polydispersities. In contrast an increase in polydispersity is observed at higher conversion and higher molecular weights: this increase has been ascribed, at least in part, to self-initiation and the decomposition of alkoxyamines [22]. Fourthly, TEMPO-mediated systems allow the preparation of some block and random copolymers (although at present styrene must be a part of both blocks [23] as well as of branched and hyperbranched polymers [24].
To summarize, TEMPO-mediated systems are currently limited to styrene and copolymers of styrene but it is expected that, by using other nitroxides, these systems can be successfully extended to (meth)acrylates and other monomers. The slow polymerization rate can be increased by using some additives, acids and salts for example [25], although the higher rate is often accompanied by an increase in polydispersity.
I I 2I I + I I + O N I O N n TEMPO N O + I I O n styrene N Initiation Polymerization n
2-1-5 Anionic Polymerization
Anionic polymerization is a method of making polymers from small molecules containing carbon-carbon double bonds. It is a type of vinyl polymerization. In anionic polymerization, the process is begun by an initiator. In this case, the initiator is an anion; that is, an ion with a negative electrical charge. There are a lot of different initiators used in anionic vinyl polymerization, but the most often used is an unassuming little molecule called butyl lithium.
A little bit of the butyl lithium will always fall apart. Not a lot, but some. If falls apart to form a positive lithium cation and a negative butyl anion. We call an anion like this where the negative charge is on a carbon atom a carbanion. A pair of election from the butyl anion will be donated to one of the double bond carbon atoms of the monomer. Now this carbon atom already has eight elections in its outer shell which it shares with the atoms to which it is bonded, so one pair of these electrons, specifically a pair on the carbon-carbon double bond, will leave the carbon atom, and settle on the other carbon atom of the carbon-carbon double bond. This forms a new carbanion, with the negative charge resting on that carbon. The process in which the butyl lithium falls apart, and the butyl anion reacts with a monomer molecule is called initiation. The carbanion now reacts with another monomer molecule in just the same manner as the initiator reacted with the first monomer molecule; another carbanion is generated. This keeps happening, and each time another monomer is added to the growing chain, a new anion is generated allowing another monomer to be added. In this way the polymer chain grows. This adding of monomer after monomer is called propagation.
Termination of propagation carbanion by combination with the counterion occurs in only a few instances, such as in electroinitiated polymerization when the contents of the anode and cathode chambers are mixed and in initiation by ionizing
radiation. Termination by combination of the anion with a metal counterion does not take place. Many anionic polymerizations, especially of nonpolar monomers such as styrene and 1,3-butadiene, take place under conditions in which there are no effective termination reactions. Propagation occurs with complete consumption of monomer to form living polymers. The propagating anionic centers remain intact because transfer of proton or other positive species from the solvent does not occur. Living polymers are produced as long as one employs solvents, such as benzene, tetrahydrofuran, and 1,2-dimethoxyethane, which are relatively inactive in chain transfer with carbanions. The polymerization of styrene by amide ion in liquid ammonia, one of the first anionic systems to be studied in detail, is one of the few anionic polymerizations where chain transfer to solvent is extensive. [26]
The nonterminating character of living anionic polymerization is apparent in several different ways. Many of the propagating carbanions are colored. If a reaction system is highly purified so that impurities are absent, the color of the carbanions is observed to persist throughout the polymerization and does not disappear or change at 100 % conversion. Further, after 100 % conversion is reached, additional polymerization can be effected by adding more monomer, either the same monomer or a different monomer. The added monomer is also polymerized quantitatively and the molecular weight of the living polymer is increased.
2-2 Surface Free Energy
2-2-1 Interfacial Thermodynamics
The interface (surface) is a region of finite thickness (usually less than 0.1 μm) in which the composition and energy very continuously from one bulk phase to other. The pressure (force field) in the interfacial zone is therefore nonhomogeneous, having a gradient perpendicular to the interfacial boundary. In contrast, the pressure in a bulk phase is homogeneous and isotropic. Consequently, no net energy is expended in reversibly transporting the matter within a bulk phase. However, a net energy is required to create an interface by transporting from the bulk phase to the interfacial zone. The reversible work require to create a unit surface area is the surface free energy, that is,
n P T A G , , ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ ∂ ∂ = γ (2.1)
where γ is the surface free energy, G the Gibbs free energy of the total system, A the interfacial area, T the temperature, P the pressure, and n the total number of moles of matter in the system.
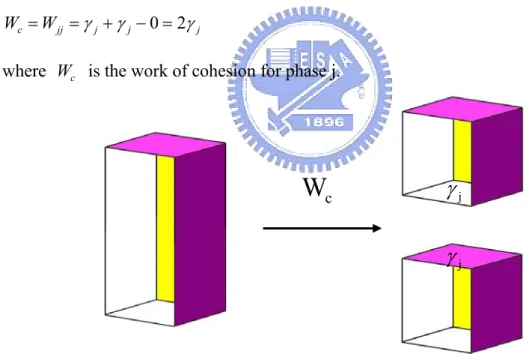
The work requires separating reversibly the interface between two bulk phases α and β form their equilibrium separation to infinity is the work of adhesion.
a
W =Wαβ =γα +γβ −γαβ (2.2)
Where Wa is the work of adhesion, γα the surface free energy of phase α , γβ the surface free energy of phase β, and γαβ the interfacial energy between phase α and β (Figure 2-2).