Cardofluorene Units: Synthesis and Characterization
CHING-HSIN CHEN, CHING-FONG SHUDepartment of Applied Chemistry, National Chiao Tung University, Hsin-Chu, Taiwan, 30035
Received 8 March 2004; accepted 8 March 2004 DOI: 10.1002/pola.20179
Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: We have synthesized aromatic polyquinolines containing both spirobiflu-orene and cardofluspirobiflu-orene moieties in the main chain by applying acid-catalyzed Fried-la¨nder quinoline synthesis. The incorporation of these rigid nonplanar structures into the polymer backbone, which restricts segmental mobility, significantly increases both the glass-transition temperature and thermal stability, while providing enhanced sol-ubility as a result of a decrease in the degree of molecular packing and crystallinity. We have also examined the optical and electrochemical properties of these polyquinolines. The low-lying lowest unoccupied molecular orbital energy level and quasireversible electrochemical reduction of these polyquinolines suggest their potential for use as electron-injecting/transporting materials in polymer light-emitting diodes.© 2004 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 42: 3314 –3322, 2004
Keywords: electrochemistry; fluorescence; light-emitting diodes (LED); polyquino-lines; solution properties; thermal properties
INTRODUCTION
Polyquinolines, a class of high-performance poly-mer materials developed by Stille1 in the 1970s,
are thermally stable and mechanically strong. Be-cause of their excellent optical and electronic properties,2,3 these polymers have recently been
explored extensively as potentially useful materi-als in electronic and photonic applications, such as organic light-emitting diodes,4 –12 thin-film
transistors,13photovoltaic cells,14nonlinear
opti-cal devices,15–21 and optical sensors.22–24
Although rigid-rod polyquinolines exhibit high strength and possess excellent thermal proper-ties, they have limited solubility in organic sol-vents, and this makes fabrication difficult.25,26In
attempts to enhance the solubility of
polyquino-lines, long alkyl side chains,27,28 flexible ether linkages,29 cardo units,30,31 and spiro moieties32 have all been incorporated into the polymer back-bone. Generally, phenylated polyquinolines are synthesized by acid-catalyzed Friedla¨nder con-densation reactions of bis(o-aminoketone)s with bisacetyl monomers.33–35 This synthetic route
provides a great degree of flexibility for introduc-ing various structural features into the backbone of the polymers. In this article, we report the synthesis of polyquinolines that contain both spi-robifluorene and cardofluorene moieties in the polymer main chain, which we have prepared with acid-catalyzed Friedla¨nder quinoline syn-thesis. The bifluorene rings in the spiro segment are orthogonally arranged and connected via a common tetracoordinated carbon atom.36 –39 The
pendent fluorenyl loop in the cardo segment lies perpendicular to the planar aromatic back-bone.30,40,41 We expected that these structural
features would reduce the probability of
inter-Correspondence to: C.-F. Shu (E-mail: [email protected])
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 42, 3314 –3322 (2004) © 2004 Wiley Periodicals, Inc.
chain interactions and prevent the close packing of polymer chains, and this would impart good solubility to the polymers. Additionally, we ex-pected the incorporation of these rigid nonplanar structures into the polymer backbone to restrict segmental mobility and result in a significant im-provement in both the glass-transition tempera-ture (Tg) and thermal stability. Furthermore, the
sp3-hybridized carbon atoms that are present at the spiro and cardo centers connect the conju-gated moieties via a-bonded network and serve as spacers and conjugation interrupts and thus effectively control the conjugation length of the polymers.32,42We have studied the basic proper-ties of these new polyquinolines, such as their thermal properties and solubility. Because of the electron-deficient nature of the quinoline ring,43 polyquinolines and their copolymers have been demonstrated to be electron-injecting/transport-ing materials in organic light-emittelectron-injecting/transport-ing diode de-vices.4 – 8 In light of these observations, we have also explored the optical and electrochemical properties of these new polyquinolines.
EXPERIMENTAL
Materials
Monomers 532 and 644 and compound 732 were prepared as described in the literature. m-Cresol was purified by distillation under reduced pres-sure. Tetrabutylammonium hexafluorophosphate was purified by recrystallization from ethyl ace-tate and dried in vacuo at 60 °C. All other chem-icals were used as received unless otherwise stated.
Characterization
1
H and13C NMR spectra were recorded on a Var-ian Unity 300-MHz spectrometer or a Bruker-DRX 300-MHz spectrometer with CDCl3 as the
solvent. Mass spectra were obtained on a JEOL JMS-SX/SX 102A mass spectrometer. Size exclu-sion chromatography was performed on a Waters chromatography unit interfaced with a Waters 410 differential refractometer, with three 5-m Waters Styragel columns (300 ⫻ 7.8 mm) con-nected in series in order of decreasing pore size (104, 103, and 102Å); tetrahydrofuran (THF) was the eluent, and standard polystyrene samples were used for calibration. Differential scanning
calorimetry (DSC) was performed on a Seiko SSC 5200 DSC unit at heating and cooling rates of 20 and 40 °C min⫺1, respectively. The samples were scanned from 30 to 500 °C, cooled to 30 °C, and scanned for a second time from 30 to 500 °C. Tg’s
were determined from the second heating scan. Thermogravimetric analysis (TGA) was carried out on a Dupont TGA 2950 instrument. The ther-mal stability of the samples was determined in nitrogen through the measurement of the weight loss at a heating rate of 10 °C min⫺1. The ultra-violet–visible (UV–vis) spectra were measured with an HP 8453 diode-array spectrophotometer. The photoluminescence (PL) spectra were ob-tained on a Hitachi F-4500 luminescence spec-trometer. Cyclic voltammetry measurements of the polymer films were performed on a BAS 100 B/W electrochemical analyzer in acetonitrile, with 0.1 M tetrabutylammonium hexafluorophos-phate as the supporting electrolyte, at a scanning rate of 100 mV s⫺1. The potentials were measured against an Ag/Ag⫹(0.01 M AgNO3) reference
elec-trode with ferrocene as the internal standard. The onset potentials were determined from the inter-section of two tangents drawn at the rising cur-rent and background curcur-rent of the cyclic voltam-mogram.
9,9-Bis(4-acetylphenyl)fluorene (1)
A mixture of fluorene (2.00 g, 12.0 mmol), 4-flu-oroacetophenone (4.15 g, 30.1 mmol), K2CO3(6.64
g, 48.1 mmol), [18]crown-6 (1.59 g, 6.02 mmol), benzene (5.0 mL), and N,N-dimethylformamide (DMF; 10 mL) was heated at 150 °C. The water that formed during the reaction was removed by azeotropic distillation and collected in a Dean– Stark trap. After 3 h, the remaining benzene in the reaction mixture was removed by distillation. The mixture was then heated under reflux for 20 h, cooled, and poured into water (300 mL). The precipitated product was purified by column chro-matography, eluting with hexane/ethyl acetate (3: 1); this was followed by recrystallization from ethyl acetate to yield 1 (3.05 g, 63.0%).
1 H NMR (CDCl3,␦): 7.79 (d, J ⫽ 10.5 Hz, 4 H), 7.76 (d, J⫽ 7.2 Hz, 2 H), 7.36 (ddd, J ⫽ 7.2, 7.2, and 1.2 Hz, 2 H), 7.33–7.27 (m, 4 H), 7.23 (d, J ⫽ 9.3 Hz, 4 H), 2.51 (s, 6 H).13 C NMR (CDCl3,␦): 197.5, 150.7, 149.6, 140.2, 135.8, 128.5, 128.2, 128.1, 125.9, 120.5, 65.5, 26.5. High-resolution mass spectrometry (HRMS): [M⫹] calcd. for C29H22O2, 402.1620; found, 402.1624. ELEM. ANAL.
Calcd. for C29H22O2: C, 86.54%; H, 5.51%. Found:
C, 86.42%; H, 5.71%.
9,9-Bis(4-nitrophenyl)fluorene (2)
A mixture of fluorene (3.00 g, 18.0 mmol), 4-fluo-ronitrobenzene (5.68 g, 41.1 mmol), K2CO3(5.98
g, 43.4 mmol), [18]crown-6 (2.39 g, 9.09 mmol), benzene (5.0 mL), and DMF (15 mL) was heated at 150 °C. The water that formed during the re-action was removed by azeotropic distillation and collected in a Dean–Stark trap. After 3 h, the remaining benzene in the reaction mixture was removed by distillation. The mixture was then heated under reflux for 3 h, cooled, and poured into water (300 mL). The precipitate was washed with water and recrystallized from ethyl acetate to give 2 (6.65 g, 90.3%). 1 H NMR (CDCl3,␦): 8.09 (d, J ⫽ 9.0 Hz, 4 H), 7.81 (d, J ⫽ 7.5 Hz, 2 H), 7.46–7.40 (m, 2 H), 7.34 –7.29 (m, 8 H). 13C NMR (CDCl3, ␦): 152.3, 148.6, 147.1, 140.3, 128.9, 128.8, 128.6, 125.9, 123.9, 121.0, 65.4. HRMS: [M⫹] calcd. for C25H16N2O4, 408.1110; found, 408.1113. ELEM.
ANAL. Calcd. for C25H16N2O4: C, 73.52%; H,
3.95%; N, 6.86%. Found: C, 73.46%; H, 4.32%; N, 6.80%.
9,9-Bis[5-(3-phenyl-2,1-benzisoxazolyl)]fluorene (3)
Benzyl cyanide (1.63 mL, 14.7 mmol) was added dropwise to an ice-cooled mixture of sodium hy-droxide (2.83 g, 70.8 mmol), methanol (12 mL), and THF (18 mL). After 5 min of stirring, com-pound 2 (3.00 g, 7.35 mmol) was added slowly, and then the mixture was heated at 80 °C for 10 h. Methanol (20 mL) was added to the reaction mixture, which was then cooled in an ice bath. The resulting precipitate was isolated by filtra-tion and washed with a cold mixture of methanol (10 mL) and H2O (40 mL). The crude product was
recrystallized from ethyl acetate to afford com-pound 3 (2.88 g, 71.0%). 1 H NMR (CDCl3,␦): 7.85 (d, J ⫽ 7.2 Hz, 2 H), 7.78 –7.75 (m, 4 H), 7.57–7.54 (m, 4 H), 7.51 (dd, J ⫽ 6.8, 0.9 Hz, 2 H), 7.47–7.41 (m, 8 H), 7.37–7.30 (m, 4 H).13C NMR (CDCl3,␦): 164.4, 157.1, 149.1, 140.6, 140.3, 132.5, 130.2, 129.2, 128.4, 128.2, 128.0, 126.4, 125.7, 120.8, 117.9, 116.0, 114.2, 65.0. HRMS: [M⫹] calcd. for C39H24N2O2,
552.1838; found, 552.1842. ELEM. ANAL. Calcd. for
C39H24N2O2: C, 84.76%; H, 4.38%; N, 5.07%.
Found: C, 84.62%; H, 4.60%; N, 5.01%.
9,9-Bis(4-amino-3-benzoylphenyl)fluorene (4)
Iron powder (0.75 g, 13.4 mmol) and water (0.5 mL) were added to a stirred suspension of com-pound 3 (1.00 g, 1.81 mmol) in acetic acid (AcOH; 20 mL) at 95 °C. The reaction mixture was heated at 95 °C for 1 h, cooled, and filtered to remove the iron powder. The filtrate was poured into water (100 mL). The resulting precipitate was collected by filtration and recrystallized from methylene chloride (CH2Cl2) to give 4 (910 mg, 90.4%). 1 H NMR (CDCl3,␦): 7.61 (d, J ⫽ 7.3 Hz, 2 H), 7.43–7.39 (m, 6 H), 7.30 (d, J ⫽ 7.3 Hz, 4 H), 7.26 –7.20 (m, 6 H), 7.17 (m, 2 H), 7.10 (dd, J ⫽ 8.6, 2.3 Hz, 2 H), 6.56 (d, J ⫽ 8.6 Hz, 2 H).13C NMR (CDCl3,␦): 198.6, 151.1, 149.5, 139.9, 139.5, 134.1, 133.9, 132.4, 131.2, 129.3, 127.9, 127.6, 127.5, 125.6, 120.2, 117.7, 117.1, 63.3. HRMS: [M⫹] calcd. for C39H28N2O2, 556.2151; found, 556.2146. ELEM. ANAL. Calcd. for C39H28N2O2: C,
84.15%; H, 5.07%; N, 5.03%. Found: C, 84.14%; H, 5.36%; N, 5.01%.
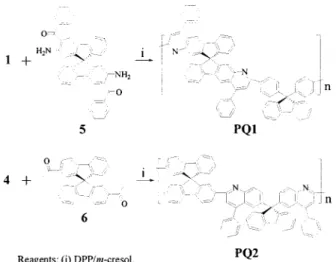
Polyquinoline PQ1
A mixture of bisacetyl monomer 1 (200 mg, 497 mol), bis(o-aminoketone) monomer 5 (275 mg, 497mol), diphenyl phosphate (DPP; 3.00 g, 12.0 mmol), and freshly distilled m-cresol (0.6 mL) was flushed with nitrogen while stirring for about 20 min and then was heated at 140 °C for 72 h under a nitrogen atmosphere. After cooling, the result-ing viscous solution was diluted with chloroform (CHCl3; 2.0 mL) and added dropwise to an agi-tated solution of 10% (v/v) triethylamine in meth-anol (110 mL). The collected polymer was purified by being precipitated three times from CHCl3(2.0
mL) into a 10% (v/v) triethylamine solution in methanol (150 mL). With a Soxhlet extractor, the polymer was then extracted continuously with a 10% (v/v) triethylamine solution in methanol for 24 h. The solid was then dried at 100 °C in vacuo to give PQ1 (412 mg, 93.6%). 1H NMR (CDCl 3,␦): 8.25 (s, 2 H), 7.81–7.51 (m, 22 H), 7.30 –7.25 (m, 6 H), 7.18 –7.13 (m, 8 H), 6.82 (d, J ⫽ 6.8 Hz, 2 H). 13C NMR (CDCl 3, ␦): 155.8, 151.9, 150.7, 149.5, 149.1, 149.0, 146.8, 140.7, 140.3, 140.1, 138.7, 137.9, 129.6, 128.9, 128.7, 128.4, 128.2, 127.7, 127.5, 127.2, 126.1, 125.9, 125.1, 124.6, 120.9, 120.1, 119.0, 115.7, 65.2, 65.0.
Polyquinoline PQ2
With the procedure described for PQ1, the poly-merization of monomers 4 and 6 gave PQ2 (94.9% yield). 1 H NMR (CDCl3,␦): 8.31 (d, J ⫽ 7.4 Hz, 2 H), 7.97 (d, J⫽ 6.8 Hz, 4 H), 7.87 (d, J ⫽ 6.7 Hz, 2 H), 7.61–7.47 (m, 10 H), 7.31–7.09 (m, 20 H), 6.73 (d, J ⫽ 6.9 Hz, 2 H). 13C NMR (CDCl3, ␦): 156.6, 150.3, 149.5, 148.9, 148.7, 147.9, 143.3, 143.1, 141.2, 140.1, 139.5, 137.6, 130.4, 130.0, 129.4, 128.2, 127.9, 127.7, 126.0, 124.9, 124.2, 123.3, 120.7, 120.6, 120.4, 119.4, 66.1, 65.3.
Synthesis of Model Compound 8
A mixture of 7 (30 mg, 81 mol), 4-tert-butylac-etophenone (17 mg, 98mol), DPP (146 mg, 590 mol), and freshly distilled m-cresol (0.1 mL) was flushed with nitrogen while stirring at 65 °C for about 30 min and then was heated under nitrogen at 140 °C for 12 h. After cooling, the reaction mixture was added to water (15 mL) and ex-tracted with ethyl acetate. The combined organic phases were dried (MgSO4) and evaporated, and
then the residue was purified by column chroma-tography (30:1 hexane/ethyl acetate) to afford compound 8 (37 mg, 89%). 1 H NMR (CDCl3,␦): 8.17–8.12 (m, 4 H), 7.79 (s, 1 H), 7.71–7.54 (m, 8 H), 7.37–7.32 (m, 3 H), 2.18 –2.01 (m, 4 H), 1.38 (s, 9 H), 0.80 – 0.67 (m, 10 H). 13C NMR (CDCl3, ␦): 156.2, 153.2, 152.5, 151.3, 149.2, 149.1, 140.6, 140.1, 139.0, 137.0, 129.7, 128.7, 128.4, 128.3, 127.3, 127.1, 125.9, 125.5, 123.5, 123.2, 120.6, 119.0, 115.0, 55.3, 43.6, 34.8, 31.4, 31.2, 17.4, 14.5. HRMS: [M⫹] calcd. for C39H39N, 509.3082; found, 509.3097.
RESULTS AND DISCUSSION
Synthesis
Scheme 1 illustrates the synthetic route used to prepare the fluorene-based bisacetyl (1) and bis(o-aminoketone) (4) cardo monomers. Starting from fluorene, which contains an activated methylene group at the C-9 position, our synthetic strategy is based on the nucleophilic aromatic substitution of 4-fluoroarenes with the fluorenyl anion gener-ated by K2CO3in DMF. This approach is a simple and direct method for adding 4-acetylphenyl and 4-nitrophenyl functionalities to the C-9 position of fluorene for the preparation of 1 and 2, respec-tively; in comparison, previously reported meth-ods for the synthesis of these compounds require
Scheme 1
Scheme 2
Table 1. Molecular Weights and Thermal Properties of Polyquinolines PQ1 and PQ2
Polymer Mn⫻ 104a Mw⫻ 104a Tg (°C)b TGA (°C)c 5% 10% PQ1 1.0 1.4 440 619 689 PQ2 1.1 2.5 —d 576 596
aThe molecular weights were determined by GPC in THF by comparison with polystyrene standards.
bThe value of T
gwas determined by DSC at heating rate of 20 °C min⫺1under a nitrogen atmosphere.
cThe temperatures at which 5 and 10% weight losses oc-curred were determined at a heating rate of 20 °C min⫺1 under a nitrogen atmosphere.
several synthetic steps.30,45,46The conversion of 2
into bis(o-aminoketone) monomer 4 followed a lit-erature procedure.30The reaction of compound 2
with benzyl cyanide in the presence of a base yielded bisbenzisoxazole 3, which was then trans-formed into monomer 4 by hydrogenation with iron powder in AcOH. The structures of com-pounds 1–4 were verified by 1H and 13C NMR
spectroscopy, mass spectrometry, and elemental analysis. The other two monomers, bisacetyl 5 and bis(o-aminoketone) 6, containing spirobiflu-orene skeletons, were prepared as reported previ-ously.32
Polyquinolines PQ1 and PQ2 were synthesized (Scheme 2) with a condensation based on the acid-catalyzed Friedla¨nder reaction.17,18An equimolar
mixture of the appropriate bisacetyl monomer and the bis(o-aminoketone) monomer was reacted in an acidic medium of DPP and m-cresol at 140 °C for 72 h under nitrogen. The resulting viscous polymer solution was diluted with CHCl3and pre-cipitated into methanol containing 10% triethyl-amine; this was followed by Soxhlet extraction with the same solution to afford the polyquino-lines in high yields. The structures of PQ1 and PQ2 were characterized by1H and13C NMR
spec-troscopy. The molecular weights of these poly-mers were determined by gel permeation chroma-tography (GPC) with THF as the eluent and with calibration against polystyrene standards; Table 1 lists the results. In the case of PQ1, the molec-ular weights are probably higher than the values listed because it is only partially soluble in THF; the insoluble portion corresponds to a higher mo-lecular weight material.
Properties of the Polyquinolines
We investigated the thermal properties of the polyquinolines by DSC and TGA; the results are tabulated in Table 1. DSC was performed from 30 to 500 °C. In repeated heating and cooling DSC cycles for PQ2, we did not detect any possible phase-transition signals. This observation proba-bly results from the stiffness of this polymer’s chains. As presented in Figure 1, we observe a distinct Tgfor PQ1 at 440 °C, which is higher than that of the polyquinoline, PQ3 (Tg ⫽ 390 °C), which contains only fluorene cardo units:30
In general, the presence of bulky fluorene cardo groups, which restrict the rotation of the polymer chain, leads to polymers that have highly rigid backbones. The incorporation of rigid 9,9 ⬘-spiro-bifluorene units into the polymer backbone fur-ther increases the chain stiffness and results in a higher Tg. As evidenced from TGA, both PQ1 and PQ2 have excellent thermal stability. The 5 and 10% weight-loss temperatures in nitrogen were 576 – 619 and 596 – 689 °C, respectively, and the char yields greater than 80% in nitrogen at 900 °C. These results reflect the high thermal
stabil-Figure 1. DSC data for PQ1 recorded under a nitro-gen atmosphere.
Table 2. Solubility of Polyquinolines PQ1 and PQ2a
Polymer
Solvent
CH2Cl2 CHCl3 PhCl Py THF m-Cresol DMF NMP HCOOH AcOH CF3COOH
PQ1 ⫺ ⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫹ ⫺ ⫹ ⫺ ⫺ ⫺ ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹
PQ2 ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫺ ⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹
ity of the cardofluorene and spirobifluorene units present in the polymer backbones.
Like most polyquinolines, PQ1 and PQ2 are soluble in protic acid solvents such as formic acid (HCOOH), AcOH, and trifluoroacetic acid (CF3COOH). The solubility of these
polyquino-lines was also tested in a variety of aprotic or-ganic solvents; the results are summarized in Ta-ble 2. The polyquinolines have improved solubil-ity in common organic solvents such as CHCl3,
pyridine (Py), and N-methyl-2-pyrrolidinone
(NMP). PQ1 is only partially soluble in CH2Cl2,
but PQ2 is soluble. PQ2 is soluble in THF and
m-cresol, but PQ1 is soluble in them only at
ele-vated temperatures. The good solubility of these aromatic polyquinolines is significant in that it does not arise from the presence of flexible ether linkages or long alkyl pendent groups; rather,
their solubility appears to result from the rigid three-dimensional structures of the spirobiflu-orene and cardofluspirobiflu-orene moieties in the polymer backbones. These structures hinder close packing and prevent strong interpolymer interactions, and this leads to an enhancement in solubility.
Figure 2 presents the absorption and emission spectra of the polyquinolines; these spectral prop-erties are summarized in Table 3. In CHCl3
solu-tions, PQ1 and PQ2 have similar lowest energy transitions, which we attribute to their –* transitions, with maximum wavelengths (max) of 374 and 352 nm, respectively. The emission max-ima are 389 nm (408 nm, shoulder) for the former and 391 nm for the latter. The shoulder at 408 nm is associated with a vibronic fine structure, which indicates that the polymer has a rigid and well-defined backbone.42 The PL quantum yields of
PQ1 and PQ2 in CHCl3solutions were estimated to be 0.27 and 0.24, respectively, with respect to the standard 9,10-diphenylanthracene (ca. 5 ⫻ 10⫺6M solution in cyclohexane, with a
fluores-cence quantum yield of 0.90).47 The absorption spectra of the solutions and their corresponding films are nearly identical, whereas the emission from films is redshifted with respect to that ob-tained from dilute solutions. This most likely re-sults from either intermolecularly interacting species, namely aggregates and excimers, or a change in the polymer conformation in the solid state.48,49 The redshift of the emission spectrum in the thin film has also been seen for other rigid-rod polyquinolines.27,32
To demonstrate that the sp3-hybridized carbon
atoms at the centers of both the spiro and cardo moieties could serve as conjugation interrupts controlling the conjugation length of the poly-mers, we synthesized model compound 8 (Scheme 3) to act as the repetitive conjugated unit of poly-mer PQ1. As shown in Figure 3, the UV–vis
ab-Figure 2. UV–vis absorption and PL spectra of (a) PQ1 and (b) PQ2 recorded in CHCl3 solutions and in the solid state.
Table 3. Optical Properties of Polyquinolines PQ1 and PQ2
Polymer
Absorption max(nm)
Photoluminescence max(nm)
Solutiona Filmb Solutiona Filmb
PQ1 374 377 389, 408 (sh) 419, 439 (sh)
PQ2 352 359 391 420, 442 (sh)
aIn CHCl 3.
bFormed via spin coating from their CHCl
sorption and PL spectra of PQ1 in CHCl3closely
resemble those obtained from compound 8, and this indicates that the effective conjugation length of PQ1 is controlled well by the sp3 -hybrid-ized carbon atoms and is nearly identical to that of compound 8.
The electrochemical behavior of the polyquino-lines was investigated with cyclic voltammetry, with ferrocene as the internal standard. The cy-clic voltammograms of PQ1 and PQ2 are dis-played in Figure 4, and the data are tabulated in Table 4. Upon cathodic sweeps, PQ1 and PQ2 exhibit quasireversible reductions with onsets at about⫺2.10 and ⫺2.13 V, respectively; the oxida-tion processes of these polymers are irreversible, with onsets at about 1.10 and 1.13 V, respectively. On the basis of the onset potentials of the oxida-tion and reducoxida-tion processes, we estimated the highest orbital molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) en-ergy levels of the polyquinolines with respect to the energy level of the ferrocene (FOC) reference (4.8 eV below the vacuum level);50 Table 4 sum-marizes these values. The HOMO–LUMO energy gaps estimated from the oxidation and reduction potentials are in close agreement with those
de-termined from the edges of the electronic absorp-tion bands. The low-lying LUMO energy levels, which originate from the electron-deficient nature of the quinoline ring, are similar to those of typ-ical quinoline-containing polymers.2,5 From the
quasireversible reduction and high electron affin-ity, we believe that PQ1 and PQ2 have potential for use as electron-injecting/transporting materi-als in polymer light-emitting diodes.
CONCLUSIONS
We have reported a direct and simple method for the synthesis of bis(o-aminoketone) and bisacetyl monomers containing cardo fluorenyl groups. The cardo-type bis(o-aminoketone) monomer, A–A (or bisacetyl, B–B), is in turn subjected to polycon-densation with a bisacetyl monomer, B–B [or bis(o-aminoketone), A–A], possessing a spirobiflu-orene linkage to furnish polyquinolines with both spirobifluorene and cardofluorene moieties in their main chains. As a result of the incorporation of these rigid, nonplanar structures into the poly-mer backbone, the polyquinolines possess en-hanced thermal stability and improved solubility with respect to regular polyquinolines. These new polymers have similar lowest energy transitions, which we attribute to their–* transitions, with values of max in the range 352–374 nm; their
polymer films exhibit blue emissions. We have also synthesized a model compound to demon-strate that the sp3-hybridized carbon atoms at the centers of the spiro and cardo moieties serve as a
Figure 3. UV–vis absorption and PL spectra of model compound 8 and PQ1 recorded in CHCl3.
Scheme 3
Figure 4. Cyclic voltammograms recorded in CH3CN for PQ1 and PQ2 films coated on a glassy carbon elec-trode.
conjugation interrupts that effectively control the conjugation length of the polymers. The electro-chemical behavior of these polymers has been in-vestigated with cyclic voltammetry, which indi-cates that the polyquinolines have potential to be used as electron-injecting/transporting materials in polymer light-emitting diodes.
The authors thank the National Science Council of the Republic of China for its financial support.
REFERENCES AND NOTES
1. Stille, J. K. Macromolecules 1981, 14, 870. 2. Agrawal, A. K.; Jenekhe, S. A. Chem Mater 1996, 8,
579.
3. Zhang, X.; Shetty, A. S.; Jenekhe, S. A. Macromol-ecules 1999, 32, 7422.
4. Parker, I. D.; Pei, Q.; Marrocco, M. Appl Phys Lett 1994, 65, 1272.
5. Jenekhe, S. A.; Zhang, X.; Chen, X. L. Chem Mater 1997, 9, 409.
6. Kim, J. L.; Kim, J. K.; Cho, H. N.; Kim, D. Y.; Kim, C. Y.; Hong, S. I. Macromolecules 2000, 33, 5880. 7. Liu, Y. Q.; Ma, H.; Jen, A. K. Y. J Mater Chem
2001, 11, 1800.
8. Zhan, X. W.; Liu, Y. Q.; Wu, X.; Wang, S. A.; Zhu, D. B. Macromolecules 2002, 35, 2529.
9. Akcelrud, L. Prog Polym Sci 2003, 28, 875. 10. Kulkarni, A. P.; Jenekhe, S. A. Macromolecules
2003, 36, 5285.
11. Alam, M. M.; Tonzola, C. J.; Jenekhe, S. A. Macro-molecules 2003, 36, 6577.
12. Zhu, Y.; Alam, M. M.; Jenekhe, S. A. Macromole-cules 2003, 36, 8958.
13. Babel, A.; Jenekhe, S. A. J Am Chem Soc 2003, 125, 13656.
14. Alam, M. M.; Jenekh, S. A. J Phys Chem B 2001, 105, 2479.
15. Agrawal, A. K.; Jenekhe, S. A.; Vanherzeele, H.; Meth, J. S. Chem Mater 1991, 3, 765.
16. Agrawal, A. K.; Jenekhe, S. A. Macromolecules 1993, 26, 895.
17. Jen, A. K.-Y.; Wu, X.-M.; Ma, H. Chem Mater 1998, 10, 471.
18. Ma, H.; Jen, A. K.-Y.; Wu, J.; Wu, X.; Liu, S.; Shu, C.-F.; Dalton, L. R.; Marder, S. R.; Thayumanavan, S. Chem Mater 1999, 11, 2218.
19. Stenger-Smith, J. D.; Zarras, P.; Hollins, R. A.; Chafin, A. P.; Merwin, L. H.; Yee, R.; Lindsay, G. A.; Herman, W. N.; Gratz, R. F.; Nickel, E. G. J Polym Sci Part A: Polym Chem 2000, 38, 2824.
20. Gubbelmans, E.; Van den Broeck, K.; Verbiest, T.; Van Beylen, M.; Persoons, A.; Samyn, C. Eur Polym J 2003, 39, 969.
21. Zhan, X. W.; Liu, Y. Q.; Zhu, D. B.; Xu, G.; Liu, X. C.; Ye, P. X. Appl Phys A 2003, 77, 375. 22. Lee, T. S.; Yang, C.; Kim, J. L.; Lee, J. K.; Park,
W. H.; Won, Y. J Polym Sci Part A: Polym Chem 2002, 40, 1831.
23. Tong, H.; Wang, L. X.; Jing, X. B.; Wang, F. Mac-romolecules 2002, 35, 7169.
24. Tong, H.; Wang, L. X.; Jing, X. B.; Wang, F. Mac-romol Rapid Commun 2002, 23 877.
25. Sutherlin, D. M.; Stille, J. K. Macromolecules 1985, 18, 2669.
26. Agrawal, A. K.; Jenekhe, S. A. Chem Mater 1992, 4, 95.
27. Tonzola, C. J.; Alam, M. M.; Jenekhe, S. A. Adv Mater 2002, 14, 1086.
28. Kru¨ ger, H.; Janietz, S.; Sainova, D.; Wedel, A. Mac-romol Chem Phys 2003, 204 1607.
29. Sutherlin, D. M.; Stille, J. K. Macromolecules 1985, 18, 2669.
30. Stille, J. K.; Harris, R. M.; Padaki, S. M. Macro-molecules 1981, 14, 486.
31. Concilio, S.; Pfister, P. M.; Tirelli, N.; Kocher, C.; Suter, U. W. Macromolecules 2001, 34, 3607. 32. Chiang, C.-L.; Shu, C.-F. Chem Mater 2002, 14,
682.
33. Imai, Y.; Johnson, E. P.; Katto, T.; Kurihara, M.; Stille, J. K. J Polym Sci Polym Chem Ed 1975, 13, 2233.
Table 4. Electrochemical Properties of Polyquinolines PQ1 and PQ2 Polymer Eonset
ox(V)a E onset
red(V)a HOMO (eV)b LUMO (eV)c E g el(eV)d E g opt(eV)e PQ1 1.00 ⫺2.10 ⫺5.80 ⫺2.70 3.10 3.11 PQ2 1.13 ⫺2.13 ⫺5.93 ⫺2.67 3.26 3.24
aPotential values determined versus Fc/Fc⫹. bDetermined from the onset oxidation potential. cDetermined from the onset reduction potential.
dElectrochemical band gap, estimated with the equation E g el⫽ E onset ox⫺ E onset red. eOptical band gap, calculated from the absorption edge of the UV–vis spectrum.
34. Wolfe, J. F.; Stille, J. K. Macromolecules 1976, 9, 489.
35. Norris, S. O.; Stille, J. K. Macromolecules 1976, 9, 496.
36. Wu, R.; Schumm, J. S.; Pearson, D. L.; Tour, J. M. J Org Chem 1996, 61, 6906.
37. Salbeck, J.; Bauer, J.; Weisso¨rtel, F. Macromol Symp 1997, 125, 121.
38. Chou, C.-H.; Reddy, D. S.; Shu, C.-F. J Polym Sci Part A: Polym Chem 2002, 40, 3615.
39. Wu, S.-C.; Shu, C.-F. J Polym Sci Part A: Polym Chem 2003, 41, 1160.
40. Korshak, V. V.; Vinogradova, S. V.; Vygodskii, Y. S. J Macromol Sci Rev Macromol Chem 1974, 11, 45.
41. Stevens, M. P. Polymer Chemistry: An Introduc-tion, 2nd ed.; Oxford University Press: New York, 1990; p 124.
42. Wu, F.-I.; Dodda, R.; Reddy, D. S.; Shu, C.-F. J Mater Chem 2002, 12, 2893.
43. Gilchrist, T. L. Heterocyclic Chemistry; Wiley: New York, 1985.
44. Haas, G.; Prolog, V. Helv Chim Acta 1969, 52, 1202.
45. Harris, R. M.; Padaki, S.; Sybert, P.; Stille, J. K. Polym Prepr 1978, 19, 7.
46. Milstein, D.; Stille, J. K. J Org Chem 1979, 44, 1613.
47. Eaton, D. Pure Appl Chem 1998, 60, 1107. 48. Jenekhe, S. A.; Osaheni, J. A. Science 1994, 265,
765.
49. Osaheni, J. A.; Jenekhe, S. A. Macromolecules 1994, 27, 739.
50. Pommerehne, J.; Vestweber, H.; Guss, W.; Mahrt, R. F.; Ba¨ssler, H.; Porsch, M.; Daub, J. Adv Mater 1995, 7, 551.