Ruthenium Vinylidene and σ-Acetylide Complexes
Containing 1,4,7-Trimethyl-1,4,7-triazacyclononane
(Me
3tacn): Synthesis and Alkyne-Coupling Reactivity
San-Ming Yang,
†Michael Chi-Wang Chan,
†Kung-Kai Cheung,
†Chi-Ming Che,*
,†and Shie-Ming Peng
‡Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, and Department of Chemistry, National Taiwan University, Taiwan
Received January 24, 1997X
The ruthenium(II) complexes [Ru(Me3tacn)(L)2X]PF6 (L)PMe3, X )O2CCF3 (1a); L ) PMe3, X)Cl (1b); L )
1/
2 dmpe, X )O2CCF3 (1c)) are prepared. Only 1a reacts with 1 equiv of RCtCH (R ) Ph and p-tolyl) to give the vinylidene complexes [Ru(Me3 tacn)-(PMe3)(O2CCF3){CdCH(R)}]PF6(R)Ph (2a) and p-tolyl (2b)) in refluxing 1,2-dichloroethane. Reaction of 2a and 2b with PMe3 in methanolic KOH solution give the corresponding σ-acetylide complexes [Ru(Me3tacn)(PMe3)2(CtCR)]PF6 (R ) Ph (3a) and p-tolyl (3b)). Similarly, treatment of 2a with P(OMe3)3 affords [Ru(Me3tacn)(PMe3)(P(OMe)3
)(CtCPh)]-PF6(3c). Oxidative cleavage of the vinylidene ligand in 2a by oxygen gives [Ru(Me3
tacn)-(PMe3)(O2CCF3)(CO)]PF6(4) and benzaldehyde. Complex 1b reacts with 2.5 equiv of RCtCH
(R)Ph, p-tolyl) and 1.5 equiv of KOH in methanol to yield the η
3-butenynyl species [Ru(Me 3
-tacn)(PMe3){η3-RC3dCH(R)}]PF6 (R ) Ph (5a) and p-tolyl (5b)). In addition, 2a and 2b react with RCtCH (R)Ph, p-tolyl) and KOH in methanol to give 5a and 5b, respectively. Treatment of 2a with p-tolylCtCH and KOH in methanol gives [(Me3tacn)Ru(PMe3){η3
-PhC3dCH(p-tolyl)}]PF6(5c) and [(Me3tacn)Ru(PMe3){η3-(p-tolyl)C3dCH(Ph)}]PF6(5c′) in a
1:1 ratio. Reacting 2b with PhCtCH similarly gives 5c and 5c′in equal amounts. Structures of 3c, 5a, and 5c/5c′are established by X-ray crystallography. Mechanistic insights from the isolated complexes suggest that hydrogen shift between vinylidene and acetylide moieties is an important process in the coupling of alkynes.
Reports on the reactivity of transition metal vinyl-idene and acetylide complexes demonstrate a close relationship between these organometallic interactions.1
Their interconversions are important in the dimeriza-tion of alkynes2and condensation of alkynes with allylic
alcohols3or carboxylic acids.4 Many d6metal vinylidene
complexes have been prepared by reaction of appropri-ate metal precursors with 1-alkynes.5 Theoretical
stud-ies suggest that initial side-on coordination of the 1-alkyne is followed by a 1,2-hydrogen shift to give the metal vinylidene complex.6,7 This usually spontaneous
rearrangement is driven by a repulsive interaction between the filled π⊥orbital of the alkyne and the filled dπ(t2g) metal orbital.8 However, Selegue and Bullock
reported that this rearrangement can require thermal initiation.9 Recently, 1,2-hydrogen shift initiated by an
electron transfer pathway was published.10
Ruthenium vinylidene complexes containing “soft” ligands11such as η-cyclopentadienyl,12η-benzene,13
bis-(diphenylphosphino)methane,14and CH
3(CH2)2N(CH2
-CH2PPh2)215have been synthesized. However, since the
unfavorable π-interaction between the dπ(Ru) and
†The University of Hong Kong.
‡National Taiwan University.
XAbstract published in Advance ACS Abstracts, May 15, 1997.
(1) (a) Bruce, M. I. Chem. Rev. 1991, 91, 197. (b) Antonova, A. B.; Ioganson, A. A. Russ. Chem. Rev. 1989, 58, 593. (c) Bruce, M. I.; Swincer, A. G. Adv. Organomet. Chem. 1983, 22, 59.
(2) (a) Yi, C. S.; Liu, N. H. Organometallics 1996, 15, 3968. (b) Bianchini, C.; Innocenti, P.; Peruzzini, M.; Romerosa, A.; Zanobini, F.; Frediani, P. Organometallics 1996, 15, 272. (c) Matsuzaka, H.; Takagi, Y.; Ishii, Y.; Nishio, M.; Hidai, M. Organometallics 1995, 14, 2153. (d) Barbaro, P.; Bianchini, C.; Peruzzini, M.; Polo, A.; Zanobini, F. Inorg. Chim. Acta 1994, 220, 5. (e) Bianchini, C.; Frediani, P.; Masi, D.; Peruzzini, M.; Zanobini, F. Organometallics 1994, 13, 4616. (f) Ducha-teau, R.; van Wee, C. T.; Meetsma, A.; Teuben, J. H. J. Am. Chem. Soc. 1993, 115, 4931. (g) Jun, C.-H.; Lu, Z.; Crabtree, R. H. Tetrahedron Lett. 1992, 33, 7119. (h) Bianchini, C.; Peruzzini, M.; Zanobini, F.; Frediani, P.; Albinati, A. J. Am. Chem. Soc. 1991, 113, 5453.
(3) (a) Dien, S.; Dixneuf, P. H. J. Chem. Soc., Chem. Commun. 1994, 2551. (b) Trost, B. M.; Martinez, J. A.; Kulawiec, R. J.; Indolese, A. F. J. Am. Chem. Soc. 1993, 115, 10402. (c) Trost, B. M.; Kulawiec, R. J. Am. Chem. Soc. 1992, 114, 5579. (d) Trost, B. M.; Dyker, G.; Kulawiec, R. J. Am. Chem. Soc. 1990, 112, 7809.
(4) (a) Doucet, H.; Martin-Vaca, B.; Bruneau, C.; Dixneuf, P. H. J. Org. Chem. 1995, 60, 7247. (b) Mitsudo, T. A.; Hori, Y.; Watanabe, Y. J. Organomet. Chem. 1987, 334, 157. (c) Ruppin, C.; Dixneuf, P. H. Tetrahedron Lett. 1986, 27, 6323.
(5) For Re(I): (a) Bianchini, C.; Marchi, A.; Marvelli, L.; Peruzzini, M.; Romerosa, A.; Rossi, R. Organometallics 1996, 15, 3804. (b) Kowalczyk, J. J.; Arif, A. M.; Gladysz, J. A. Organometallics 1991, 10, 1079. (c) Senn, D. R.; Wong, A.; Patton, A. L.; Marsi, M.; Strouse, C. E.; Gladysz, J. A. J. Am. Chem. Soc. 1988, 110, 6096. For Os(II): (d) Gamasa, M. P.; Gimeno, J.; Gonzalez-Cueva, M.; Lastra, E. J. Chem. Soc., Dalton Trans. 1996, 2547. (e) Bruce, M. I.; Koutsantonis, G. A.; Liddell, M. J.; Nicholson, B. K. J. Organomet. Chem. 1987, 320, 217. (6) Silvestre, J.; Hoffmann, R. Helv. Chim. Acta 1985, 68, 1461. (7) Wakatsuki, Y.; Koga, N.; Yamazaki, H.; Morokuma, K. J. Am. Chem. Soc. 1994, 116, 8105.
(8) (a) Templeton, J. L. Adv. Organomet. Chem. 1989, 29, 1. (b) Kostic, N. M.; Fenske, R. F. Organometallics 1982, 1, 974.
(9) (a) Lomprey, J. R.; Selegue, J. P. J. Am. Chem. Soc. 1992, 114, 5518. (b) Bullock, R. M. J. Chem. Soc., Chem. Commun. 1989, 165.
(10) Connelly, N. G.; Geiger, W. R.; Lagunas, M. C.; Metz, B.; Rieger, A. L.; Reiger, P.H.; Shaw, M. J. J. Am. Chem. Soc. 1995, 117, 12202. (11) Pearson, R. G. Hard and Soft Acids and Bases; Dowden, Hutchinson, and Ross: Stroudsburg, PA, 1973.
(12) (a) Bruce, M. I.; Wallis, R. C. Aust. J. Chem. 1979, 32, 1471. (b) Bruce, M. I.; Wallis, R. C. J. Organomet. Chem. 1978, 161, C1.
(13) (a) Le Bozec, H.; Ouzzine, K.; Dixneuf, P. H. Organometallics
1991, 10, 2768. (b) Dixneuf, P. H. Pure Appl. Chem. 1989, 61, 1763.
(14) (a) Touchard, D.; Haquette, P.; Pirio, N.; Toupet, L.; Dixneuf, P. H. Organometallics 1993, 12, 3132. (b) Haquette, P.; Pirio, N.; Touchard, D.; Toupet, L.; Dixneuf, P. H. J. Chem. Soc., Chem. Commun. 1993, 163.
S0276-7333(97)00046-0 CCC: $14.00 © 1997 American Chemical Society
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
π⊥(CtC) orbitals is the driving force for the alkyne -vinylidene rearrangement, we anticipated that synthe-sis of vinylidene complexes may be achieved by utilizing “hard” amine ligand systems. However, reaction be-tween [Ru(NH3)5(H2O)]2+and phenylacetylene yielded
the η2-alkyne complex [Ru(NH
3)5(η2-PhCtCH)]2+.16
We have been studying the chemistry of organo-ruthenium complexes containing the saturated tertiary amine 1,4,7-trimethyl-1,4,7-triazacyclononane.17,18 In
this work, ruthenium vinylidene and σ-acetylide deriva-tives are prepared, and reactions of the former with base and oxygen are studied. Coupling reactions with 1-alkynes resulting in carbon-carbon bond formation are observed.
Experimental Section
All reactions were carried out in a nitrogen atmosphere using standard Schlenk techniques unless otherwise stated. Ru(Me3tacn)Cl319 and [Ru(Me3tacn)(OH2)2(O2CCF3)](OTf)220
(OTf)trifluoromethanesulfonate) were prepared according to the published methods. Trimethylphosphine and 1,2-bis-(dimethylphosphino)ethane (dmpe) were purchased from Merck and used as received. PhCtCH and p-tolylCtCH were obtained from Aldrich and distilled before use. All solvents were reagent grade and used without further purification.
13C{1H}and1H NMR spectra were recorded on a JEOL 270
NMR, Bruker 300 DPX NMR, or Bruker 500 DRX FT-NMR spectrometer operating at 270, 300, or 500 MHz (1H)
and 67.5, 75, or 125 MHz (13C), respectively. Peak positions
were calibrated with Me4Si as internal standard. 31P{1H}
NMR spectra were recorded on the Bruker 500 DRX FT-NMR spectrometer operating at 202.4 MHz, and chemical shifts were measured relative to external 85% H3PO4 with downfield
values taken as positive. Fast atom bombardment (FAB) mass spectra were obtained on a Finnigan MAT 95 mass spectrom-eter with 3-nitrobenzyl alcohol matrix. Infrared spectra were recorded as Nujol mulls on a BIO RAD FT-IR spectrometer between KBr plates. Elemental analyses were performed by the Butterworth Laboratory Ltd, U.K.
[Ru(Me3tacn)(PMe3)2(O2CCF3)]PF6(1a). PMe3(0.16 g,
2.1 mmol) and zinc powder (0.50 g) were mixed in acetone (20 cm3). After 5 min, [Ru(Me
3tacn)(H2O)2(O2CCF3)](OTf)2(0.50
g, 0.69 mmol) was added to the stirred solution to give a yellow coloration instantaneously. The stirring was continued at room temperature for 1 h. After removal of zinc powder, the solvent was removed under reduced pressure and a saturated aqueous solution of NH4PF6 was added to give the titled
compound as a yellow solid (yield)0.22 g, 47%). Anal. Calcd for C17H39N3O2F9P3Ru: C, 30.02; H, 5.79; N, 6.11. Found: C,
29.86; H, 5.75; N, 6.15. 1H NMR (300 MHz, (CD
3)2CO): 1.43
(18H, virtual t, JPH)3.7 Hz, P(CH3)3), 2.76-3.53 (21H, m, Me3tacn). IR (cm-1): ν(CO) 1685. 31P{1H}NMR ((CD3)2CO):
-0.5. FAB mass spectrum: m/z 538 [M
+ -PF6], 462 [M + -PF6-PMe3], 386 [M + -PF6-2PMe3].
[Ru(Me3tacn)(PMe3)2Cl]PF6(1b). Ru(Me3tacn)Cl3(0.10
g, 0.26 mmol) and zinc powder (0.50 g) were added to a stirred ethanolic solution (20 cm3) of PMe
3(0.05 g, 0.66 mmol). The
mixture was refluxed for 18 h. The zinc powder was removed, and upon addition of NH4PF6, a yellow solid was formed. The
solid was filtered, washed with ethanol, water, and diethyl ether, and air-dried (yield) 0.10 g, 63%). Anal. Calcd for C15H39N3ClF6P3Ru: C, 29.78 ; H, 6.49; N, 6.95. Found: C,
29.95; H, 6.38; N, 6.85. 1H NMR (500 MHz, CD
3CN): 1.49
(18H, virtual t, JPH)3.7 Hz, P(CH3)3), 2.66-3.13 (21H, m, Me3tacn). 31P{1H} NMR (CD3CN): -1.0. FAB mass spec-trum: m/z 460 [M+
-PF6], 384 [M
+
-PF6-PMe3].
[Ru(Me3tacn)(dmpe)(O2CCF3)]PF6 (1c). Compared to 1a, the titled compound was prepared using dmpe instead of
PMe3(yield)0.19 g, 41%). Anal. Calcd for C17H37N3O2F9P3 -Ru: C, 30.00; H, 5.48; N, 6.18. Found: C, 30.05; H, 5.60; N, 6.15. 1H NMR (300 MHz, (CD 3)2CO): 1.42 (6H, d, J)9 Hz, P-CH3), 1.71 (6H, d, JPH ) 7.62 Hz, P-CH3), 2.67 (3H, s, N-CH3), 2.73-3.37 (22H, m, 2× N-CH3, N-CH2, P-CH2). IR (cm-1): ν(CO) 1685. 31P{1H}NMR (CD 3CN): 42.9. FAB mass spectrum: m/z 536 [M+ -PF6].
[Ru(Me3tacn)(PMe3)(O2CCF3){CdCH(Ph)}]PF6 (2a).
Complex 1a (0.20 g, 0.29 mmol) and PhCtCH (0.03 g, 0.29 mmol) were mixed in 1,2-dichloroethane (20 cm3). The solution
was refluxed for 2 h to give a red solution. The solvent was removed, and a methanolic solution of NH4PF6was added to
afford a red crystalline solid (yield)0.18 g, 87%). Anal. Calcd for C22H36N3O2F9P2Ru: C, 37.29; H, 5.12; N, 5.93. Found: C,
37.38; H, 5.12; N, 5.74. IR (cm-1): ν(CO) 1710 (m); ν(CdC) 1621 (m). 1H NMR (270 MHz, (CD 3)2CO): 1.59 (9H, d, JPH) 9.25 Hz, P(CH3)3), 3.25-3.65 (21H, m, Me3tacn), 5.47 (1H, d, JPH)4.39 Hz, dCH(Ph)), 7.14-7.46 (m, 5H, C6H5). 13C{1H} NMR (270 MHz, CD2Cl2): 363.7 (d, JPC)22.3 Hz, RudCdC), 129.4, 128.3, 127.7, 126.9 (C6H5), 117.3, 112.9 (CF3CO2), 111.6 (RudCdC), 63.6, 63.2, 61.4, 60.3, 59.5, 58.2, 56.5, 52.0, 51.7 (Me3tacn), 16.3 (d, JPC )30.9 Hz, P(CH3)3). 31P{1H}NMR
(CD3CN): -3.0. FAB mass spectrum: m/z 564 [M
+
-PF6], 462 [M+
-PF6-PhCtCH].
[Ru(Me3tacn)(PMe3)(O2CCF3){CdCH(p-tolyl)}]PF6(2b).
The procedure was similar to 2a except p-tolylCtCH was used instead of PhCtCH (yield)0.10 g, 50%). Anal. Calcd for C23H38N3O2F9P2Ru: C, 38.17; H, 5.26; N, 5.81. Found: C, 37.96, H, 5.52; N, 5.94. IR (cm-1): ν(CO) 1715 (m); ν(CdC) 1635 (m). 1H NMR (500 MHz, (CD 3)2CO): 1.63 (9H, d, JPH) 9.6 Hz, P(CH3)3), 2.30 (3H, s, C6H4CH3), 3.02-3.87 (21H, m, Me3tacn), 5.13 (1H, d, JPH )4.45 Hz, dCH(p-tolyl)), 7.04 -7.13 (dd, 4H, C6H4). 13C{1H} NMR (125 MHz, (CD3)2CO): 362.9 (d, JPC)23.3 Hz, RudCdC), 130.2, 130.1, 127.9, 126.7 (C6H4), 110.8 (RudCdC), 63.8, 57.7, 58.7, 60.1, 61.2, 61.3, 63.2, 52.6 (Me3tacn), 21.1 (C6H4CH3), 17.1 (d, JPC ) 32.6 Hz, P(CH3)3). 31P{1H}NMR ((CD3)2CO): -3.8. FAB mass spec-trum: m/z 578 [M+
-PF6], 462 [M
+
-PF6-p-tolylCtCH].
[Ru(Me3tacn)(PMe3)2(CtCPh)]PF6(3a). PMe3(0.10 g,
1.3 mmol) and potassium hydroxide (0.10 g, 1.8 mmol) were dissolved in anhydrous methanol (10 cm3). Complex 2a (0.10
g, 0.14 mmol) was then added, and the mixture was stirred for 2 h to give a yellow solution. The methanol was removed under reduced pressure. The residue was then dissolved in acetone, and a saturated aqueous solution of NH4PF6was
added. Slow evaporation of acetone gave a yellow crystalline solid (yield)0.05 g, 53%). Anal. Calcd for C23H44N3F6P3Ru: C, 41.19: H, 6.61; N, 6.27. Found: C, 40.91; H, 6.53; N, 6.10. IR (cm-1
): ν(CtC) 2065. 1H NMR (500 MHz, (CD
3)2CO): 1.64
(18H, virtual t, JPH)3.7 Hz, P(CH3)3), 2.75-3.34 (21H, m, Me3tacn), 6.95-7.00 (1H, m, para H), 7.11-7.19 (4H, m, ortho and meta H). 13C{1H} NMR (125 MHz, (CD
3)2CO): 131.3,
130.9, 128.7, 124.1 (C6H5), 131.0 (t, JPC)10 Hz, Ru-CtC), 108.9 (Ru-CtC), 62.5, 61.7, 60.1, 58.2, 55.4 (Me3tacn), 21.9
(virtual t, JPC)14.5 Hz, P(CH3)3).
31P{1H}NMR ((CD 3)2CO):
2.4. FAB mass spectrum: m/z 526 [M+
-PF6], 450 [M
+
-PF6-PMe3].
[Ru(Me3tacn)(PMe3)2{CtC(p-tolyl)}]PF6 (3b).
Com-pared to 3a, 2b was used instead of 2a as starting material (yield)0.04 g, 42%). Anal. Calcd for C24H46N3F6P3Ru: C, 42.10; H, 6.77; N, 6.14. Found: C, 41.95; H, 6.41; N, 5.98. IR (15) (a) Bianchini, C.; Peruzzini, M.; Romerosa, A.; Zanobini, F.
Organometallics 1995, 14, 3152. (b) Bianchini, C.; Innocenti, P.; Masi, D.; Peruzzini, M.; Romerosa, A.; Zanobini, F. J. Chem. Soc., Chem. Commun. 1994, 2219.
(16) Lehmann, H.; Schenk, K. J.; Chapuis, G.; Ludi, A. J. Am. Chem. Soc. 1979, 101, 6197.
(17) Yang, S. M.; Cheng, W. C.; Cheung, K. K.; Che, C. M.; Peng, S. M. J. Chem. Soc., Dalton Trans. 1995, 227.
(18) Yang, S. M.; Cheng, W. C.; Peng, S. M.; Cheung, K. K.; Che, C. M. J. Chem. Soc., Dalton Trans. 1995, 2955.
(19) Neubold, P.; Wieghardt, K.; Nuber, B.; Weiss, J. Inorg. Chem.
1989, 28, 459.
(20) Cheng, W. C.; Yu, W. Y.; Cheung, K. K.; Che, C. M. J. Chem. Soc., Chem. Commun. 1994, 1063.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
(cm-1 ): ν(CtC) 2065. 1H NMR (270 MHz, CD 3CN): 1.51 (18H, virtual t, JPH)3.8 Hz, P(CH3)3), 2.24 (3H, s, C6H4CH3), 2.66 -3.19 (21H, m, Me3tacn), 6.94-7.05 (4H, dd, C6H4). 13C{1H} NMR (67.5 MHz, CD3CN): 133.9, 130.9, 129.7, 128.4 (C6H4),
108.5 (Ru-CtC), 62.4, 61.7, 60.0, 58.3, 55.5 (Me3tacn), 21.9
(virtual t, JPC)14.5 Hz, P(CH3)3), 21.2 (C6H4CH3), Ru-CtC not resolved. 31P{1H}NMR (CD
3CN): 2.4. FAB mass
spec-trum: m/z 540 [M+
-PF6], 464 [M
+
-PF6-PMe3].
[Ru(Me3tacn)(PMe3)(P(OMe)3)(CtCPh)]PF6(3c).
Com-pared to 2a, P(OMe)3was used instead of PMe3(yield)0.06 g, 55%). Anal. Calcd for C23H44N3O3F6P3Ru: C, 38.43; H, 6.17;
N, 5.85. Found: C, 38.31; H, 6.16; N, 5.87. IR (cm-1 ): ν(CtC) 2056. 1H NMR (270 MHz, (CD 3)2CO): 1.57 (9H, d, JPH)8.6 Hz, P(CH3)3), 2.80-3.32 (21H, m, Me3tacn), 3.98 (9H, d, JPH )10.0 Hz, P(OCH3)3), 7.01-7.20 (5H, m, phenyl). 13C{1H} NMR (270 MHz, (CD3)2CO): 130.9, 130.8, 128.8, 124.5 (C6H5), 110.1 (Ru-CtC), 62.9, 62.2, 61.2, 60.6, 60.4, 59.5, 57.5, 55.6, 54.2 (Me3tacn), 54.3 (d, JPC)10.4 Hz, P(OCH3)3), 20.8 (d, JPC ) 31.2 Hz, P(CH3)3), Ru-CtC not resolved.
31P{1H} NMR
((CD3)2CO): 137.6 (d, JPP)70.5 Hz, P(OMe)3), 3.2 (d, JPP) 70.5 Hz, PMe3). FAB mass spectrum: m/z 574 [M+
-PF6], 498 [M+
-PF6-PMe3].
[Ru(Me3tacn)(PMe3)(O2CCF3)(CO)]PF6(4). Oxygen gas
was introduced into a 1,2-dichloroethane solution of 2a (0.08 g, 0.11 mmol) for 8 h. The color of the solution changed from red-orange to yellow. The solution was then concentrated to ca. 5 cm3under reduced pressure. The titled compound was
isolated as a yellow solid upon addition of diethyl ether and recrystallized from dichloromethane/diethyl ether (yield)0.03 g, 52%). Anal. Calcd for C15H30N3O3F9P2Ru: C, 28.39; H, 4.77;
N, 6.62. Found: C, 28.15; H, 4.62; N, 6.51. IR (cm-1): ν(CtO) 1964. 1H NMR (500 MHz, (CD 3)2CO): 1.60 (9H, d, JPH)9.2 Hz, P(CH3)3), 3.1-3.6 (21H, m, Me3tacn). 13C{1H}NMR (125 MHz, (CD3)2CO): 204.8 (d, J)18.4 Hz, CO), 63.8, 62.9, 61.4, 61.1, 59.9, 58.7, 58.1, 53.1, 52.7 (Me3tacn), 16.4 (d, J)10.9 Hz, P(CH3)3). 31P{1H}NMR ((CD3)2CO): -3.6. FAB mass spectrum: m/z 490 [M+
-PF6].
[Ru(Me3tacn)(PMe3){η3-PhC3)CH(Ph)}]PF6 (5a).
Method A. Complex 1b (0.15 g, 0.24 mmol), PhCtCH (0.06
g, 0.6 mmol), and KOH (0.02 g, 0.36 mmol) were refluxed in methanol (15 cm3) for 18 h to give a clear red solution. After
cooling, the solution was concentrated to ca. 5 cm3 under
reduced pressure. Upon addition of NH4PF6, a red
microcrys-talline solid was formed. The solid was filtered, washed with ice-cool ethanol and diethyl ether, and air-dried (yield)0.11 g, 67%).
Method B. Complex 2a (0.75 g, 1.0 mmol) was added
slowly to a hot methanolic KOH (0.06 g, 1.1 mmol) solution (10 cm3) over 15 min to give a clear yellow solution which was
refluxed for a further 5 min. PhCtCH (0.11 g, 1.1 mmol) in methanol (5 cm3) was then added dropwise to the yellow
solution to give a clear red solution which was refluxed for 5 h. The solution was then concentrated to ca. 5 cm3, and upon
addition of NH4PF6a red microcrystalline solid formed. The
solid was filtered, washed with ice-cool ethanol and diethyl ether, and then air-dried (yield)0.10 g, 62%). Anal. Calcd for C28H41N3F6P2Ru: C, 48.27; H, 5.93; N, 6.03. Found: C,
48.23; H, 5.96; N, 6.05. 1H NMR (500 MHz, CD
2Cl2) (the
num-bering scheme for the hydrogen and carbon resonances is given in ref 21): 0.94 (9H, d, JPH ) 7.9 Hz, P(CH3)3), 1.63 (3H, s, N-CH3), 2.40-3.61 (15H, m, Me3tacn), 3.81 (3H, s, N-CH3), 6.94 (1H, s, H4), 7.21 (1H, t, J)7.3 Hz, H8′), 7.33 (1H, t, J) 7.4 Hz, H8), 7.39 (2H, t, J)7.6 Hz, H7), 7.44 (2H, t, J)7.5 Hz, H7’), 7.77 (2H, d, J)7.4 Hz, H6′), 7.82 (2H, d, J)7.4 Hz, H6). 13C{1H}NMR (125 MHz, CD 2Cl2): 159.1 (d, J)7.6 Hz, C3), 138.2, 132.4, 130.8, 129.6, 129.3, 127.8, 126.2, 125.3 (2× C6H5), 124.6 (d, J)6.1 Hz, C1), 123.2 (C4), 62.4, 61.6, 61.4, 59.8, 59.4, 58.9, 58.7, 58.4 (Me3tacn), 57.2 (d, J)1.5 Hz, C2), 47.8 (N-CH3), 16.7 (d, J)28.6 Hz, P(CH3)3). 31P{1H}
NMR (CD2Cl2): 4.8. FAB mass spectrum: m/z 553 [M+
-PF6], 477 [M+
-PF6-PMe3].
[Ru(Me3tacn)(PMe3){η3-(p-tolyl)C3dCH(p-tolyl)}]PF6 (5b). Compared to 5a, this complex was synthesized by
method A using p-tolylCtCH instead of PhCtCH or by method B using complex 2b and p-tolylCtCH as the starting materials (yield)0.09 g, 52%). Anal. Calcd for C30H45N3F6P2 -Ru: C, 49.72; H, 6.22; N, 5.80. Found: C, 49.52; H, 6.46; N, 5.65. 1H NMR (500 MHz, CD 2Cl2): 0.93 (9H, d, JPH)7.9 Hz, P(CH3)3), 1.61 (3H, s, N-CH3), 2.33 (3H, s, H9), 2.39 (3H, s, H9′), 2.41-3.61 (15H, m, Me3tacn), 3.87 (3H, s, N-CH3), 6.88 (1H, s, H4), 7.20 (2H, d, J)7.9 Hz, H7), 7.26 (2H, d, J)7.9 Hz, H7′), 7.66 (2H, d, J)8.1 Hz, H6′), 7.72 (2H, d, J)8.1 Hz, H6). 13C{1H}NMR (125 MHz, CD 2Cl2): 157.5 (d, J)7.6 Hz, C3), 138.1 (C8′), 136.1 (C8), 135.7 (C5), 130.7 (C6′), 130.3 (C7), 129.9 (C7′), 129.5 (C5′), 125.2 (C6), 123.8 (d, J)5.4 Hz, C1), 122.7 (C4), 62.4, 61.6, 61.4, 59.8, 59.4, 58.9, 58.7, 58.4 (Me3tacn), 55.8 (d, J)1.6 Hz, C2), 47.7(N-CH3), 21.5(C9’), 21.3(C9), 16.7 (d, J ) 28.5 Hz, P(CH3)3). 31P{1H} NMR (CD2Cl2): 5.2. FAB mass spectrum: m/z 580 [M+
-PF6], 504 [M+
-PF6-PMe3].
[Ru(Me3tacn)(PMe3){η3-PhC3dCH(p-tolyl)}]PF6(5c) and [Ru(Me3tacn)(PMe3){η3-(p-tolyl)C3dCH(Ph)}]PF6 (5c′).
p-TolylCtCH was used in method B for 5a (yield)0.08 g, 48%). Anal. Calcd for C29H43N3F6P2Ru‚CH3OH: C, 48.51; H, 6.38; N, 5.66. Found: C, 48.22; H, 6.24; N, 5.57. 1H NMR (500 MHz, CD2Cl2): 0.94 (18H, d, JPH)7.8 Hz, P(CH3)3), 1.62 (6H, s, N-CH3), 2.39 (3H, s, CH3of p-tolyl), 2.45 (3H, s, CH3of p-tolyl), 2.50-3.59 (30H, m, Me3tacn), 3.87 (6H, s, N-CH3), 6.91 (1H, s, dCH), 6.93 (1H, s, dCH), 7.20-7.85 (18H, m, C6H5). 31P{1H}NMR (CD
2Cl2): 5.0. FAB mass spectrum: m/z 566
[M+
-PF6], 490 [M
+
-PF6-PMe3].
Structural Determination. X-ray quality crystals were
obtained by slow diffusion of diethyl ether into an acetone solution for 3c and a dichloromethane solution for 5a, respec-tively. Intensities and lattice parameters were measured on a Rigaku AFC7R or Enraf-Nonius CAD-4 diffractometer using the ω-2θ scan mode. Crystal parameters and details of data collection and refinement are given in Table 1. Intensity data were corrected for Lorentz and polarization effects. Empirical absorptions were based on the ψ-scan of five strong reflections. The structures were solved by the heavy-atom Patterson method and refined by full-matrix least squares and Fourier-difference syntheses using the MSC-Crystal Structure Package TEXSAN on a Silicon Graphic Indy computer.22 All non-H
atoms were refined anisotropically. The H atoms at calculated positions with thermal parameters equal to 1.3 times that of the attached C atoms were not refined. Selected bond dis-tances and angles of 3c and 5a are tabulated in Tables 2 and 3, respectively.
Results and Discussion
Zinc reduction of [Ru(Me3tacn)(OH2)2(O2CCF3)]2+in
acetone in the presence of PMe3 and dmpe gives
[Ru(Me3tacn)(PMe3)2(O2CCF3)]PF6 (1a) and [Ru(Me3
-tacn)(dmpe)(O2CCF3)]PF6 (1c), respectively. Similar (21) Numbering scheme for hydrogen and carbon atoms in 5a and
5b:
(22) PATTY & DIRDIF92: Beurskens, P. T.; Admiraal, G.; Beur-sken, G.; Bosman, W. P.; Garcia-Grand, S.; Gould, R. O.; Smits, J. M. M.; Smykalla C.(1992). The DIRDIF program system, Technical Report of the Crystallography Laboratory, University of Nijmegen, The Netherlands.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
ruthenium complexes of Me3tacn with π-acid ligands
have been reported.18 In addition, Ru(Me
3tacn)Cl3
reacts with PMe3in ethanol in the presence of zinc to
yield [Ru(Me3tacn)(PMe3)2Cl]PF6(1b) in moderate yield.
Attempts to prepare the bulkier PPh3analogues [Ru(Me3
-tacn)(PPh3)2X]PF6(X)Cl or O2CCF3) were unsuccess-ful.
Preparation of Vinylidene Complexes [Ru-(Me3tacn)(PMe3)(O2CCF3){CdCH(R)}]PF6(R)Ph
(2a); R)p-tolyl (2b)) and σ-Acetylide Complexes
[Ru(Me3tacn)(L)(PMe3)(CtCR)]PF6(R )Ph, L )
PMe3(3a); R)p-tolyl, L)PMe3(3b); R)Ph, L)
P(OMe)3(3c)). [Ru(Me3tacn)(PMe3)2(O2CCF3)]PF6(1a)
reacts with PhCtCH and p-tolylCtCH in refluxing 1,2-dichloroethane to give the vinylidene complexes [Ru-(Me3tacn)(PMe3)(O2CCF3){CdCH(R)}]PF6(R)Ph (2a) and p-tolyl (2b)) respectively. No desired products are obtained using alkylacetylenes such as 2-methyl-3-butyn-2-ol, tert-butylacetylene, (trimethylsilyl)acetylene, or 1-hexyne. Furthermore, no reaction was found between 1c and PhCtCH after refluxing in 1,2-dichlo-roethane or ethanol for 24 h. The chelating dmpe ligand in 1c is expected to resist dissociation and prevent subsequent reaction with the 1-alkyne. Hence dissocia-tion of PMe3 to generate a coordination vacancy is
presumably the first step in the formation of 2a,b. Reaction between 1b and PhCtCH in 1,2-dichloro-ethane gives impure [Ru(Me3tacn)(PMe3)(Cl){
CdCH-(Ph)}]PF6(identified by1H NMR) in very low yield (ca.
5%) after reflux for 10 h. The observation that the rate of formation for vinylidene complexes is faster for 1a than for 1b warrants further comment. Since 1a and
1b are coordinatively saturated 18-electron species,
dissociation of PMe3is likely to be the most endothermic
and hence the rate-determining step of vinylidene formation. In this system, ground state destabilization resulting in phosphine dissociation from [Ru(Me3
tacn)-(PMe3)2X]+ (X
) O2CCF3 (1a), Cl (1b)) is negligible because the steric requirement of X is small. In addi-tion, the rate of PMe3dissociation in CpRu(PMe3)2X (X
)halide, alkyl, hydride, amide, and hydroxy) have been studied by Bercaw23 and Caulton.24 They suggested
that π-donation from X can stabilize the 16-electron intermediate CpRu(PMe3)X, which results in a faster
dissociation rate. In both 1a and 1b, however, the ligand X has π-electrons which can stabilize the 16-electron species Ru(Me3tacn)(PMe3)X to a similar extent.
Hence this factor cannot account for the large difference in the phosphine dissociation rate between 1a and 1b. We attribute this to neighboring group participation by the trifluoroacetate anion. This phenomenon has been invoked previously in the oxidative addition and reduc-tive elimination of square-planar platinum25and
irid-ium26 complexes. Unlike in 1b, the lone pair of the
carboxylate group in 1a can interact with the metal which lowers the activation energy for the dissociation of PMe3to form an 18-electron intermediate I1 (Scheme
1). The η2-trifluoroacetate anion in I1 isomerizes to an η1-mode (I2) to provide a vacant site for the coordination
of RCtCH. The conversion between η1and η2bonding
modes for the carboxylate ligand is often observed in the generation of coordination vacancy.27 It is likely
that the RCtCH is initially bound to the ruthenium center in a side-on fashion (I3), and 1,2-hydrogen shift subsequently occurs to give the vinylidene complex.
(23) Bryndza, H. E.; Domaille, P. J.; Paciello, R. A.; Bercaw, J. E. Organometallics 1989, 8, 379.
(24) Caulton, K. G. New J. Chem. 1994, 18, 25.
(25) Constable, A. G.; Langrick, C. R.; Shabanzadeh, B.; Shaw, B. L. Inorg. Chim. Acta 1982, 65, L151.
(26) Miller, E. M.; Shaw, B. L. J. Chem. Soc., Chem. Commun. 1974, 480.
(27) (a) Braun, T.; Gevert, O.; Werner, H. J. Am. Chem. Soc. 1995, 117, 7291. (b) Scher, M.; Wolf, J.; Werner, H. J. Chem. Soc., Chem. Commun. 1991, 1341.
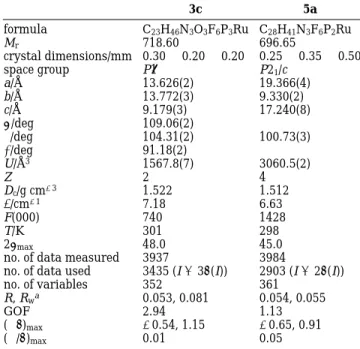
Table 1. Crystal Data for 3c and 5a
3c 5a formula C23H46N3O3F6P3Ru C28H41N3F6P2Ru Mr 718.60 696.65 crystal dimensions/mm 0.30× 0.20 × 0.20 0.25 × 0.35 × 0.50 space group P1h P21/c a/Å 13.626(2) 19.366(4) b/Å 13.772(3) 9.330(2) c/Å 9.179(3) 17.240(8) R/deg 109.06(2) β/deg 104.31(2) 100.73(3) γ/deg 91.18(2) U/Å3 1567.8(7) 3060.5(2) Z 2 4 Dc/g cm-3 1.522 1.512 µ/cm-1 7.18 6.63 F(000) 740 1428 T/K 301 298 2θmax 48.0 45.0
no. of data measured 3937 3984
no. of data used 3435 (I>3σ(I)) 2903 (I>2σ(I))
no. of variables 352 361 R, Rwa 0.053, 0.081 0.054, 0.055 GOF 2.94 1.13 (∆F)max -0.54, 1.15 -0.65, 0.91 (∆/σ)max 0.01 0.05 aR)∑(|Fo|-|Fc|)/∑|Fo|. Rw)(∑w(|Fo|-|Fc|)2/∑w|Fo|2)1/2.
Table 2. Selected Bond Distances (Å) and Angles (deg) for [Ru(Me3tacn)(PMe3)(P(OMe)3
)-(CtCPh)]PF6(3c) Ru-P(1) 2.225(2) Ru-P(2) 2.337(2) Ru-N(1) 2.256(6) Ru-N(2) 2.269(5) Ru-N(3) 2.226(5) Ru-C(1) 1.991(6) C(1)-C(2) 1.235(8) C(2)-C(3) 1.426(8) P(1)-Ru-P(2) 85.6(1) P(1)-Ru-N(1) 98.1(2) P(1)-Ru-N(2) 174.9(1) P(1)-Ru-N(3) 96.0(1) P(1)-Ru-C(1) 93.1(2) P(2)-Ru-N(1) 104.1(2) P(2)-Ru-N(2) 99.6(1) P(2)-Ru-N(3) 177.3(1) P(2)-Ru-C(1) 83.6(2) N(1)-Ru-N(2) 79.7(2) N(1)-Ru-N(3) 77.8(2) N(1)-Ru-C(1) 166.8(2) N(2)-Ru-N(3) 79.1(2) N(2)-Ru-C(1) 88.5(2) N(3)-Ru-C(1) 94.1(2) Ru-C(1)-C(2) 173.0(5) C(1)-C(2)-C(3) 176.6(6)
Table 3. Selected Bond Distances (Å) and Angles (deg) for [Ru(Me3tacn)(PMe3
)-{η3-PhC 3dCH(Ph)}]PF6(5a) Ru-P(1) 2.307(3) Ru-N(1) 2.181(6) Ru-N(2) 2.231(5) Ru-N(3) 2.203(7) Ru-C(16) 2.158(8) Ru-C(17) 2.114(8) Ru-C(18) 2.058(8) C(15)-C(16) 1.47(1) C(16)-C(17) 1.26(1) C(17)-C(18) 1.37(1) C(18)-C(19) 1.37(1) P(1)-Ru-N(1) 98.7(2) P(1)-Ru-N(2) 177.9(2) P(1)-Ru-N(3) 98.5(2) P(1)-Ru-C(16) 88.4(2) P(1)-Ru-C(17) 87.7(2) P(1)-Ru-C(18) 88.2(2) N(1)-Ru-N(2) 82.8(3) N(1)-Ru-N(3) 79.5(3) N(1)-Ru-C(16) 170.5(3) N(1)-Ru-C(17) 139.1(3) N(1)-Ru-C(18) 101.2(3) N(2)-Ru-N(3) 83.2(3) N(2)-Ru-C(16) 89.9(3) N(2)-Ru-C(17) 90.2(3) N(2)-Ru-C(18) 90.1(3) N(3)-Ru-C(16) 105.8(3) N(3)-Ru-C(17) 139.8(3) N(3)-Ru-C(18) 173.1(3) C(16)-Ru-C(17) 34.3(3) C(16)-Ru-C(18) 72.5(3) C(17)-Ru-C(18) 38.3(3) Ru-C(16)-C(15) 152.7(6) Ru-C(16)-C(17) 70.9(5) C(15)-C(16)-C(17) 136.1(8) Ru-C(17)-C(16) 74.8(5) Ru-C(17)-C(18) 68.7(5) C(16)-C(17)-C(18) 143.4(8) Ru-C(18)-C(17) 73.1(5) Ru-C(18)-C(19) 149.7(6) C(17)-C(18)-C(19) 137.2(8) C(18)-C(19)-C(20) 126.3(7)
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
The vinylidene complexes 2a and 2b are air-stable solids. Their IR spectra each contain bands at ca. 1710 and 1620 cm-1 which are typical for the stretching
frequencies of the CdO and CdC groups in η1-O 2CCF3
and vinylidene ligands, respectively. The31P{1H}NMR
spectra show a single resonance at-3.0 and-3.8 ppm for the coordinated PMe3 in 2a and 2b, respectively.
Their 13C{1H} NMR spectra each reveal a low-field
doublet at ca. 363 ppm (2J
PC≈ 23 Hz) for the
metal-bonded vinylidene carbon while the resonance for the
β-carbon is at ca. 110 ppm. The spectroscopic data
confirm that 2a is a vinylidene complex rather than a metallacyclic vinyl ester compound because the latter would display a lower13C resonance and ν(CdO) band
for CRand the CF3CO2group, respectively.
28 In the1H
NMR spectra, the chemical shifts of the vinylidene protons in 2a and 2b (5.47 and 5.13 ppm, respectively) are similar to the related complexes [Ru(η-C5H5)(PPh3)2 {CdCH(Ph)}]PF6 (5.43 ppm),11a [Ru(η-C6Me6)(PMe3
)-(Cl){CdCH(Ph)}]PF6(5.66 ppm),12aand [Ru(P(OMe)3)4
-(CtCPh){CdCH(Ph)}]PF6 (5.98 ppm).29
Nucleophilic attack at the R-carbon of vinylidene complexes to give heteroatom-stabilized carbene species is well established and can be affected by the steric and electronic properties of the spectator ligands.30 Complex 2a and 2b are stable in refluxing methanol, while only
deprotonation of the vinylidene ligand is observed upon
reaction with primary and secondary amines. Hence upon addition of tert-butylamine to an acetone-d6
solu-tion of 2a, the vinylidene proton signal in the1H NMR
spectrum vanishes and the σ-acetylide complex is formed (see below). This is in contrast to the report by Bianchini that primary and secondary amines react with ruthenium vinylidene derivatives to give amino-carbene and isocyanide complexes.14a We propose that
complexes 2a,b are resistant to nucleophilic addition as a result of electronic rather than steric factors: the auxiliary ligands in the present system do not appear to impart steric hindrance, while the high π-basicity of the [Ru(Me3tacn)] fragment is expected to lower the
electrophilicity of theR-carbon atom.
Reaction of 2a and 2b with methanolic KOH in the presence of phosphine L (L)PMe3or P(OMe)3) affords the σ-acetylide complexes [Ru(Me3tacn)(PMe3)L(CtCR)]+
(R)Ph, L)PMe3(3a), R)p-tolyl, L)PMe3(3b), R ) Ph, L ) P(OMe)3 (3c)). It is noteworthy that the expected formation of the σ-acetylide species via direct substitution of 1b with the appropriate Grignard re-agent does not give the desired products. The use of amines, e.g. triethylamine, tert-butylamine, as base results in lower yields. The vinylidene derivative 2a is first deprotonated by KOH to give the σ-acetylide intermediate; substitution of the CF3CO2 ligand by
PMe3then proceeds to give 3a. Complexes 3b and 3c
are presumably formed via similar reaction pathways. In the13C{1H}NMR spectra for complexes 3a
-c, a singlet is observed at 108-110 ppm for the β-acetylide carbon (hence no phosphorus coupling). 3a and 3b both show five resonances in the range 55-63 ppm which correspond to the Me3tacn ligand and suggest Cs
sym-metry. In complex 3c, nine carbon resonances are assigned to Me3tacn, and this implies the presence of Ci symmetry. Large coupling in the 31P{1H} NMR
spectrum (2J
PP)70.5 Hz) between PMe3and P(OMe)3 is evident. A triplet at ca. 131 ppm in the13C{1H}NMR
spectrum of 3a is assigned to the R-carbon, but an analogous signal for 3b and 3c is obscured by phenyl resonances. The IR spectra for 3a-c each show an intense absorption band at ca. 2060 cm-1for the CtC
moiety.
Introduction of dioxygen into a 1,2-dichloroethane solution of 2a affords [Ru(Me3tacn)(CO)(PMe3
)-(O2CCF3)]+(4) and benzaldehyde. Oxidative cleavage
of vinylidene ligands have been previously reported.31
We found that the incorporation of an electron-with-drawing group (e.g. NO2, Cl) into the para position of
the phenyl ring in 2a leads to longer reaction times.32
The stability of the vinylidene complexes toward oxida-tion therefore increases as the electron density at the CdC bond decreases. The FAB mass spectrum of 4 reveals a cluster at m/z 490 which corresponds to the parent cationic fragment [Ru(Me3tacn)(PMe3)(O2CCF3
)-(CO)]+. A low-field doublet at 204.8 ppm (2J PC ) 28.6Hz) in the13C NMR spectrum and a strong
absorp-tion at 1964 cm-1in the IR spectrum are characteristic
of a terminal carbonyl ligand.
Synthesis of η3-Butenynyl Complexes [Ru-(Me3tacn)(PMe3){η3-RC3dCH(R)}]PF6(R)Ph (5a),
(28) Daniel, T.; Mahr, N.; Braun, T.; Werner, H. Organometallics
1993, 12, 1475.
(29) Albertin, G.; Antoniutti, S.; Bordignon, E.; Cazzaro, F.; Ianelli, S.; Pelizzi, G. Organometallics 1995, 14, 4114.
(30) Bruce, M. I.; Swincer, A. G. Aust. J. Chem. 1980, 33, 1471.
(31) (a) Mezzetti, A.; Consiglio, G.; Morandini, F. J. Organomet. Chem. 1992, 430, C15. (b) Oro, L. A.; Ciriano, M. A.; Foces-Foces, C.; Cano, F. M. J. Organomet. Chem. 1985, 289, 117. (c) Bruce, M. I.; Swincer, A. G.; Wallis, R. C. J. Organomet. Chem. 1979, 171, C5.
(32) Yang S. M.; Che, C. M. Unpublished results. Scheme 1
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
p-tolyl (5b)). Reaction of excess PhCtCH and KOH with [Ru(Me3tacn)(PMe3)2Cl]PF6(1b) in refluxing
metha-nol gives a orange-red solution from which red crystals of [Ru(Me3tacn)(PMe3){η3-PhC3dCH(Ph)}]PF6(5a) are
obtained (method A). Similarly, [Ru(Me3tacn)(PMe3 )-{η3-(p-tolyl)C
3dCH(p-tolyl)}]PF6 (5b) is formed using p-tolylCtCH. Treatment of the vinylidene complexes
2a and 2b with KOH followed by RCtCH in refluxing
methanol also gives 5a and 5b, respectively (method B). Formation of η3-butenynyl complexes from well-defined
ruthenium2band tungsten33precursors have been
re-ported.
Using method A, we attempted to isolate the inter-mediate(s) of the reaction by adding diethyl ether to the mixture after reflux for 30 min to precipitate all ionic species present. A red solid and colorless solution were afforded, and the 1H NMR spectrum of the solid
consisted of three species: starting complex 1b (PMe3
protons at 1.49 ppm), a small amount of 5a (character-istic vinyl proton at 6.94 ppm), and small amounts of an unknown species with a doublet at ca. 5.5 ppm. Due to the similarities between these resonances and that of 2a, we suggest that this species is a vinylidene intermediate in the formation of 5a. This assertion is further supported by the successful synthesis of 5a from the vinylidene complex 2a via method B.
The 1H and 13C{1H} signals of the 1,4′
-di(p-tolyl)-butenynyl ligand in complex 5b have been assigned by
DEPT-135, HMBC, and HSQC 13C
-1H COSY NMR
experiments. In the 13C{1H} NMR spectrum, three
small doublets appearing at 55.8 ppm (2J
PC)1.5 Hz), 123.8 (2J
PC)5.4 Hz), and 157.5 ppm (
2J
PC) 7.6 Hz) correlate to the ruthenium-bonded C2, C1, and C3 atoms respectively. The1H,13C{1H}, and13C
-1H COSY
NMR spectra of 5a are similar to those of 5b, except the 1H resonances in 5b at 2.33 and 2.39 ppm are
attributed to the p-tolyl methyl groups. The1H NMR
spectra of 5a and 5b each contain a singlet at ca. 6.9 ppm which is assigned to the vinylic proton of the 1,4′ -disubstituted η3-butenynyl ligand. One Me
3tacn methyl
group appears at a higher field in both the 1H and 13C{1H}NMR spectra (ca. 1.6 and 48 ppm, respectively)
than other signals for the ligand (2.4-3.6 ppm for
1H
and 58-63 ppm for
13C). It is apparent from the X-ray
structure of 5a (vide infra) that this methyl substituent is located above one of the phenyl rings of the η3
-butenynyl moiety and is therefore shielded by the diamagnetic ring current.
[Ru(Me3tacn)(PMe3){η3-PhC3dCH(p-tolyl)}]PF6 (5c) and [Ru(Me3tacn)(PMe3){η3-(p-tolyl)C3
dCH-(Ph)}]PF6(5c′): Synthesis and Mechanism.
Reac-tion of 2a with p-tolylCtCH in methanolic KOH gives a red solid. The FAB mass spectrum shows a cluster around m/z 566 which can be assigned to the isomeric fragments [Ru(Me3tacn)(PMe3){η3-PhC3dCH(p-tolyl)}]+
(5c) or [Ru(Me3tacn)(PMe3){η3-(p-tolyl)C3dCH(Ph)}]+
(5c′). The 31P{1H} NMR spectrum shows a slightly
broad signal at 5.0 ppm, while the1H and13C{1H}NMR
spectra are uninformative due to overlapping signals. Nevertheless, the1H NMR spectrum shows two signals
of equal intensity at 6.91 and 6.93 ppm which are attributed to the vinylic protons of the η3-butenynyl
moieties in 5c and 5c′. In addition, two peaks of equal
intensity at 2.39 and 2.45 ppm are assigned to the methyl hydrogens of the p-tolyl groups.
In order to eliminate the possibility that the isolated red solid is an equimolar mixture of 5a and 5b, we have also recorded the1H and31P{1H}NMR spectra and the
FAB mass spectrum of such a mixture. In the1H NMR
spectrum, two signals at 6.94 and 6.88 ppm are visible for the vinylic protons of 5a and 5b, respectively, while the analogous resonances for 5c/5c′are absent. The methyl hydrogens for the p-tolyl substituents appear at 2.33 and 2.39 ppm. Moreover, the 31P{1H} NMR
spectrum shows two signals at 4.8 and 5.2 ppm which correspond to the PMe3 ligand in 5a and 5b,
respec-tively; again the corresponding peaks for 5c/5c′are not observed. The FAB mass spectrum does not show a cluster at m/z 566. Hence there is no signals corre-sponding to the red product from the reaction of 2a with
p-tolylCtCH, which is a 1:1 mixture of [Ru(Me3
tacn)-(PMe3){η3-PhC3dCH(p-tolyl)}]PF6 (5c) and [Ru(Me3
-tacn)(PMe3){η3-(p-tolyl)C3dCH(Ph)}]PF6(5c′). Finally,
the analogous reaction between complex 2b with phen-ylacetylene also gives 5c/5c′as a red solid with identical spectroscopic properties. The molecular structure of 5c/
5c′(see the Supporting Information) shows coordination of the η3-butenynyl fragment to the metal center as in
the structure of 5a.
Scheme 2 depicts our proposed mechanism for the formation of the η3-butenynylruthenium(II) complexes 5c and 5c′. The stepwise mechanism is related to others previously reported.34 However, the location of the p-tolyl substituent in the final products provide
inter-esting mechanistic information.
We have demonstrated that 1 molar equiv of KOH serves to deprotonate the vinylidene ligand in the preparation of 3a-c. We propose that the reaction of
(33) McMullen, A. K.; Selegue, J. P.; Wang, J. G. Organometallics
1991, 10, 3421.
Scheme 2. Proposed Mechanism for the Formation of 5c/5c′
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
2a with p-tolylacetylene in the presence of KOH yields
the (η2-p-tolylCtCH)(σ-CtCPh) intermediate IA.
Re-arrangement of p-tolylCtCH results in formation of the (vinylidene)(σ-acetylide) intermediate IB, and subse-quent 1,2-migratory insertion of the acetylide gives the observed complex 5c. The formation of the 5c′isomer gives greater insight into the reaction mechanism.35
Complex 5c′ is derived from the (σ-p-tolylacetylide)-(phenylvinylidene) intermediate IB′which is generated by the isomerization of IB through proton transfer. From a thermodynamic viewpoint, the strong basicity at the β-carbon of the acetylide moiety and the high acidity of the vinylidene proton will favor the proton migration, and this is further facilitated by the electron-donating nature of the [Ru(Me3tacn)] fragment. Such
proton transfer processes have not been observed by Bianchini.2b We believe that the isomerization is
ki-netically unfavored in aprotic solvents, while in our system the proton transfer/isomerization can be assisted by the methanol solvent (Scheme 3). Moreover, we assume that the C-C coupling is slower than the rate of proton transfer partly because of the weak trans effect of the Me3tacn ligand. Hence, the isomeric
intermedi-ates IB and IB′are generated in equilibrium, and this results in the formation of 5c and 5c′in equal propor-tions.
X-ray Crystal Structures of 3c and 5a. Figures 1
and 2 show perspective views of the cations in 3c and
5a respectively. Selected bond distances and angles are
presented in Tables 2 and 3, respectively.
The coordination geometry around the ruthenium center in 3c is a distorted octahedron with the metal atom surrounded by two phosphines, three nitrogen atoms of Me3tacn, and a σ-acetylide ligand. The three
Ru-N distances are comparable. The most evident distortion from idealized geometry is the bending of the acetylide moiety toward Me3tacn and PMe3(N(2)-Ru -C(1) 88.5(2)°, P(2)-Ru-C(1) 83.6(2)°). The Ru-P dis-tances are shorter for the more electron-accepting P(OMe)3(Ru-P(1) 2.225(2) Å) than for PMe3(Ru-P(2) 2.337(2) Å). Since the cone angles of P(OMe3)3 and
PMe3 are similar (107° and 118°, respectively),36 the
strong π-basicity of the [Ru(Me3tacn)] fragment
appar-ently results in a stronger bond with P(OMe3)3. The
ethynyl moiety is almost linear (Ru-C(1)-C(2) 173.0(5)°) and the Ru-C separation of 1.991(6) Å is within the range expected for ruthenium(II) σ-acetylide com-plexes.37 The high-energy IR stretch (2065 cm-1) of the
CtC bond is consistent with the C(1)-C(2) bond length of 1.235(8) Å, which is comparable to that in disubsti-tuted organic alkynes (ca. 1.20 Å)38and organometallic
alkynyl complexes (1.14-1.24 Å).
39
The molecular structure of 5a corresponds to that elucidated spectroscopically for the p-tolyl derivative 5b. The ruthenium center is in a distorted octahedral environment assuming the η3-butenynyl ligand is
oc-cupying two sites. The salient feature of 5a is the
(34) (a) Albertin, G.; Antoniutti, S.; Bordignon, E. J. Chem. Soc., Dalton Trans. 1995, 719. (b) Werner, H. J. Organomet. Chem. 1994, 475, 45. (c) Santos, A.; Lez, J.; Matas, L.; Ros, J.; Gal, A.; Echavarren, A. M. Organometallics 1993, 12, 4215. (d) Scher, M.; Mahr, N.; Wolf, J.; Werner, H. Angew. Chem., Int. Ed. Engl. 1993, 32, 1315. (e) Bianchini, C.; Bohanna, C.; Esteruelas, M. A.; Frediani, P.; Meli. A.; Oro, L. A.; Peruzzini, M. Organometallics 1992, 11, 3837. (f) Field, L.; George, A. V.; Purches, G. R.; Slip, I. H. M. Organometallics 1992, 11, 3019. (g) Wakatsuki, Y.; Yamazaki, H.; Kunegawa, N.; Satoh, J. Y.; Satoh, T. J. Am. Chem. Soc. 1991, 113, 9604. (h) Albertin, G.; Amendola, P.; Antoniutti, S.; Ianelli, S.; Pelizzi, G.; Bordignon, E. Organometallic 1991, 10, 2876. (i) Jia, G.; Meek, D. W. Organometallics
1991, 10, 1411.
(35) The possibility that 5c/5c′are interconvertible by acid- or
base-catalyzed isomerization was suggested by one reviewer. However, this
is ruled out since no changes are observed by1H NMR spectroscopy
for the treatment of 5b with CF3CO2D or CD3ONa in CD3OD.
(36) Tolman, C. A. Chem. Rev. 1977, 77, 313.
(37) Whittall, I. R.; Humphrey, M. G.; Hockless, D. C. R.; Skelton, B. W.; White, A. H. Organometallics 1995, 14, 3970 and reference therein.
Scheme 3
Figure 1. Perspective view of the cation in [Ru(Me3
tacn)-(PMe3)(P(OMe)3)(CtCPh)]PF6(3c).
Figure 2. Perspective view of the cation in [Ru(Me3
tacn)-(PMe3){η3-PhC3dCH(Ph)}]PF6(5a).
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
[RuC3] unit of the η3-butenynyl group. Structural
parameters associated with this fragment for several related ruthenium complexes are collected in Table 4. The small bend-back angle γ for 5a (136.1(8)°) falls in the range of metal-diphenylacetylene interactions (135 -140°)40and suggests strong interaction between C(16)/
C(17) and Ru. This is supported by the short corre-sponding bond distances e and f, while elongation of the C(16)-C(17) contact (distance c) to 1.260(1) Å is also observed. The greater interaction between the η3
-butenynyl unit and the ruthenium center in 5a com-pared to other examples in Table 4 is believed to be a consequence of the strong π-basicity of the [Ru(Me3
-tacn)] moiety.
Conclusion
The rate of PMe3 dissociation in [Ru(Me3
tacn)-(PMe3)2X]+(X
)O2CCF3(1a), Cl (1b)) is enhanced by
η1/η2 isomerization of the CF
3CO2 ligand in 1a. The
vinylidene complexes [Ru(Me3tacn)(PMe3)(O2CCF3 )-{CdCH(R)}]PF6 (R ) Ph (2a) and p-tolyl (2b)) are prepared by the reaction of 1a with the appropriate 1-alkyne. Due to the high π-basicity of the [Ru(Me3
-tacn)] moiety which lowers the electrophilicity of the vinylideneR-carbon, no nucleophilic addition across the CdC bond is observed. Alkyne coupling reactions to give η3-butenynyl complexes 5a, 5b, and 5c/5c′ are
studied. It is significant that, partly due to the weak
trans effect of the saturated triamine, coupling of the σ-acetylide and vinylidene groups is slower than proton
migration between these two ligands for IB and IB′ (Scheme 2) in methanol. An equilibrium between these isomeric intermediates is thus established and yields an unprecedented mixture of 5c and 5c′.
Acknowledgment. We thank The University of
Hong Kong and the Hong Kong Research Grants Council for support and S.-M.Y. is grateful for a Croucher Scholarship administrated by the Croucher Foundation of Hong Kong.
Supporting Information Available: Tables of final positional parameters, anisotropic displacement parameters and bond lengths and angles for 3c, 5a, and 5c/5c′(27 pages). Ordering information and Internet access instructions are given on any current masthead page.
OM970046+ (38) (a) Ittel, S. D.; Ibers, J. A. Adv. Organomet. Chem. 1976, 14,
33. (b) March, J. Advanced Organic Chemistry, 4th ed.; Wiley: New York, 1992.
(39) (a) Montoya, J.; Santos, A.; Lopez, J.; Echavarren, A. M.; Ros, J.; Romero, A. J. J. Organomet. Chem. 1992, 426, 383. (b) Bruce, M. I.; Humphrey, M. G.; Snow, M. R.; Tiekink, E. R. T. J. Organomet. Chem. 1986, 314, 213. (c) Consiglio, G.; Morandini, F.; Sironi, A. J. Organomet. Chem. 1986, 306, C45.
(40) (a) Decian, A.; Colin, A.; Schappacher, M.; Richard, L.; Weiss, R. J. Am. Chem. Soc. 1981, 103, 1850. (b) Sunkel, K.; Nagel, U.; Beck, W. J. Organomet. Chem. 1981, 222, 252.
(41) (a) Liles, D. C.; Verhoeven, P. F. M. J. Organomet. Chem. 1996, 522, 33. (b) Alcock, N. W.; Hill, A. F.; Melling, R. P.; Thompsett, A. R. Organometallics 1993, 12, 641. (c) Jia, G.; Gallucci, J. C.; Rheingold, A. L.; Haggerty, B. S.; Meek, D. W. Organometallics 1991, 10, 3459. (d) Jia, G.; Rheingold, A. L.; Meek, D. W. Organometallics 1989, 8, 1378.
Table 4. Comparison of Structural Data for the Complexes RuLn{η3-PhC3)CH(Ph)}
RuLn a (Å) b (Å) c (Å) d (Å) e (Å) f (Å) R(deg) β (deg) γ (deg) ref
[Ru(Me3tacn)(PMe3)]+(5a) 1.366 1.368 1.260 2.058 2.114 2.158 137.2 143.4 136.1 this work
[Ru(CO)2(PPh3)2]+ 1.319 1.371 1.244 2.170 2.233 2.320 138.1 147.4 148.7 41a [RuCl(Cyttp)] (syn-mer)a 1.335 1.416 1.220 2.040 2.229 2.558 129.2 154.3 156.7 41b [RuCl(Cyttp)] (anti-mer)a 1.343 1.396 1.248 2.084 2.169 2.319 130.4 148.2 147.0 41b [Ru(CtCPh)(Cyttp)]a 1.339 1.379 1.249 2.200 2.191 2.258 133.1 148.7 144.6 41c [Ru(PNP)(CtCPh)] (anti-mer)b 1.34 1.41 1.23 2.06 2.19 2.39 130 150 154 2b [Ru(PMe2Ph)4]+ 1.341 1.401 1.229 2.119 2.226 2.510 131.1 155.3 155.8 41d aCyttp)PhP{(CH2)3P(C6H11)2}2.bPNP)CH3(CH2)3P(CH2CH2PPh2)2.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
![Figure 1. Perspective view of the cation in [Ru(Me 3 tacn)- tacn)-(PMe 3 )(P(OMe) 3 )(CtCPh)]PF 6 (3c).](https://thumb-ap.123doks.com/thumbv2/9libinfo/8843839.239800/7.918.525.786.54.339/figure-perspective-view-cation-tacn-tacn-pme-ctcph.webp)
