國立高雄大學應用化學系(碩士班)
口試論文
高吸附性三氧化鉬的製備與其廢水處理之應用
Synthesis and Characterization of Highly Adsorptive
Molybdenum Trioxide and Their Application to Water
Treatment
研究生:李松樺 撰
指導教授:莊琇惠 博士
I
謝誌
在碩士的生涯當中,最感謝老師 莊琇惠細心與耐心的指導,從無到 有,一路慢慢的將我提拔上來,讓我在研究學習上有明確的方向。然而, 在日常生活也給予極大的關心與幫助。 接著也要感謝實驗室的夥伴們,黃信智學長各種實驗技巧與觀念的 傳承,平常實驗有任何的瓶頸,學長都會熱心的給予幫助,並一同解決 問題。也感謝其他的夥伴,陳家卉學姊、蔡浩瑋同學、林聖翔學弟、許 賢澤學弟和吳尚霖學弟,感謝大家在我實驗繁忙的時候,能夠從一旁協 助我,減輕我的負擔。 另外也要感謝其他實驗的夥伴,大家互相協助幫忙,一同為了目標 而努力。更要感謝有機光電材料實驗室,平時有事沒事就往那裡跑,是 個串門子與放鬆的好地方,大家聚在一起互相聊天,一起討論各自的實 驗。感謝應化系六樓的夥伴,彼此之間互相的協助,讓我在碩士生涯不 孤單。 非常感謝我能夠在國立高雄大學應用化學系,完成我的碩士學業, 這幾年來其實得到許多人的幫助以及勉勵,讓我在碩士生涯能夠更加的 順利。也祝福各位,在未來的人生當中,能夠為了自己的目標更加努力。 李松樺 中華民國 一百零五年 三月二八日II
目錄
第一章 緒論 ... 1 1.1 前言 ... 1 1.2 材料介紹 ... 2 第二章 理論說明與文獻回顧 ... 4 2.1 文獻探討 ... 4 2.1.1 氧化鉬之合成 ... 4 2.1.2 吸附材料的發展 ... 12 2.2 理論說明 ... 16 2.2.1 微波法 ... 16 2.2.2 光致螢光 (Photoluminescence, PL) ... 17 2.2.3 Langmuir 等溫吸附模式 ... 18 2.2.4 表面電位 (Zeta Potential) ... 20 第三章 材料與方法 ... 22 3.1 實驗藥品及儀器設備 ... 22 3.1.1 實驗藥品 ... 22 3.1.2 實驗儀器設備 ... 23 3.2 MoO3合成步驟 ... 24 3.3 染料吸附實驗步驟 ... 26 3.3.1 亞甲藍檢量線測定之溶液配製 ... 26 3.3.2 亞甲藍吸附速率測定之溶液配製 ... 27III 3.3.3 亞甲藍吸附量測定之溶液配製 ... 27 3.3.4 Langmuir adsorption 之溶液配製 ... 28 3.4 儀器分析條件 ... 29 第四章 結果與討論 ... 31 4.1 改變反應時間對於氧化鉬的影響 (0.04 M AHM) ... 31 4.1.1 X-ray 粉末繞射法分析 ... 31 4.1.2 掃描式電子顯微鏡圖像分析 ... 33 4.1.3 顯微拉曼光譜分析 ... 35 4.1.4 光致螢光光譜分析 ... 36 4.2 改變反應時間對於氧化鉬的影響 (0.01 M AHM) ... 37 4.2.1 X-ray 粉末繞射法分析 ... 37 4.2.2 掃描式電子顯微鏡圖像分析 ... 39 4.2.3 顯微拉曼光譜分析 ... 41 4.2.4 光致螢光光譜分析 ... 43 4.3 氧化鉬對亞甲籃吸附之研究 ... 45 4.3.1 吸附速率之探討 ... 45 4.3.2 最大吸附容量之探討 ... 48 4.3.3 表面電位之探討 ... 51 第五章 結論 ... 57 第六章 參考文獻 ... 58
IV
圖目錄
圖 1-1 八面體示意圖。...2 圖 2-1 利用傳統加熱的方式,在 85°C 下反應合成氧化鉬之 SEM 圖。…5 圖 2-2 利用傳統加熱的方式,在 120°C 下反應合成氧化鉬之 SEM 圖。..5 圖 2-3 利用超音波法合成氧化鉬之 SEM 圖。………...….6 圖 2-4 利用超音波法合成氧化鉬之 SEM 圖。………...….6 圖 2-5 使用水及乙醇當作溶劑,以溶劑熱法合成氧化鉬之 SEM 圖。….7 圖 2-6 利用水熱法於鹽酸水溶液中合成氧化鉬之 SEM 圖。……….8 圖 2-7 利用水熱法於醋酸水溶液中合成氧化鉬之 SEM 圖。……….8 圖 2-8 利用水熱法合成氧化鉬之 SEM 圖: (a) 添加氯化鉻,(b) 添加 PVP………...9 圖 2-9 利用微波法合成氧化鉬之 SEM 圖: (a,b) 無添加 Zr+離子,(c,d) Zr+ 離子的輔助。…..………...………..………….10 圖 2-10 利用微波法合成氧化鉬之 SEM 圖。………...10 圖 2-11 亞甲藍之結構圖。………...………..…..14 圖 2-12 加熱方式示意圖: (A,左圖) 傳統加熱,(B,右圖) 微波加熱。....16 圖 2-13 光致螢光電子-電洞對複合之示意圖。………...17 圖 2-14 Langmuir 等溫吸附模式圖。………...…...….19 圖 2-15 電雙層之示意圖。………...…...…..…...…....21 圖 3-1 MoO3合成流程圖。………...………..……..24 圖 3-2 亞甲藍水溶液之檢量線。………...….…..26 圖 4-1 0.04 M 鉬酸銨溶液,反應溫度 120o C,反應時間為 10、30 和 90V 分鐘所得產物之 X-ray 粉末繞射圖。………...…….32 圖 4-2 0.04 M 鉬酸銨溶液,反應溫度 120o C,反應時間為 10、30 和 90 分鐘所得產物之 SEM 圖像 (5K)。………...…....……34 圖 4-3 六角柱示意圖。………...………....34 圖 4-4 0.04 M 鉬酸銨溶液,反應溫度 120o C,反應時間為 10、30 和 90 分鐘所得產物之 Raman 圖譜。…………...……….…….35 圖 4-5 0.04 M 鉬酸銨溶液,反應溫度 120o C,反應時間為 10、30 和 90 分鐘所得產物之 PL 圖譜。………...………...36 圖 4-6 0.01 M 鉬酸銨溶液,反應溫度 120o C,反應時間為 10、30 和 90 分鐘所得產物之 X-ray 粉末繞射圖譜。………...38 圖 4-7 0.01 M 鉬酸銨溶液,反應溫度 120o C,反應時間為 10、30 和 90 分鐘所得產物之 SEM 圖像 (10K)。………...….…40 圖 4-8 0.01 M 鉬酸銨溶液,反應溫度 120o C,反應時間為 10、30 和 90 分鐘所得產物之 Raman 圖譜。………...….………42 圖 4-9 0.01 M 鉬酸銨溶液,反應溫度 120o C,反應時間為 10、30 和 90 分鐘所得產物之 PL 圖譜。………...………44 圖 4-10 不同產物對亞甲藍吸附速率在 UV-Vis 圖譜上的表現。……....46 圖 4-11 不同產物對亞甲藍的吸附圖。………...47 圖 4-12 不同產物對亞甲藍之最大吸附量的量測曲線圖。……….……50 圖 4-13 不同產物對亞甲藍之 Langmuir 等溫吸附模式圖。………...…50 圖 4-14 不同產物於不同 pH 值下的表面電位。………..51 圖 4-15 最大吸附容量與表面電位之關係圖。……….52
VI
圖 4-16 陽離子型染料之結構圖。……….….55 圖 4-17 陰離子型染料之結構圖。……….….56 圖 4-18 MoO-90L 對於不同染料之吸附量關係圖。……….….56
VII
表目錄
表 1-1 氧化鉬多形態同素異形體。...3 表 1-2 各晶相氧化鉬的晶格常數。...3 表 2-1 氧化鉬之合成方式及其形貌與晶型整理表。………..11 表 2-2 各文獻對亞甲藍的吸附效果整理表。……….………...15 表 3-1 實驗條件整理。...25 表 3-2 亞甲藍水溶液之各濃度訊號。………...26 表 4-1 亞甲藍溶液進行吸附之吸收度變化。………...48 表 4-2 比表面積、晶型與最大吸附容量之整理表。………...49 表 4-3 比表面積、表面電位與最大吸附容量之整理表。………..52 表 4-4 各文獻之比表面積與吸附量整理表。………..54VIII
高吸附性三氧化鉬的製備與其廢水處理之應用
指導教授:莊琇惠 博士 國立高雄大學應用化學系 學生:李松樺 國立高雄大學應用化學系碩士班 摘要 在廢水處理中染料的去除是一大課題。近年來學者們紛紛將金屬材料運用於染料 吸附,本研究即是探討氧化鉬在這方面的潛力。首先利用微波法,以鉬酸銨溶液為起 始劑,與硝酸進行反應,合成出一系列之氧化鉬。所得產物利用粉末 X-ray 繞射儀、 掃描式電子顯微鏡、顯微拉曼光譜儀及光致螢光光譜儀鑑定之。由結果得知,在 0.04 M 鉬酸銨溶液的環境下,隨著反應時間的增加,其形狀由錐狀轉變為柱狀,但產物 結構皆為 h-MoO3。然而,在較低濃度 0.01 M 鉬酸銨溶液的環境下,隨著反應時間的 增加,其形狀由柱狀轉變為帶狀,而產物結構則由 h-MoO3轉變為α-MoO3。最後, 將氧化鉬產物應用於亞甲藍染料吸附,其最大吸附容量為 613.5 mg/g,單位面積最大 吸附容量為 469.5 mg/m2。而影響氧化鉬對染料吸附的主要因素則為產物的表面電 位。 關鍵字: 氧化鉬、微波法、染料吸附、亞甲藍、表面電位。IX
Synthesis and Characterization of Highly Adsorptive
Molybdenum Trioxide and Their Application of Water
Treatment
Advisor: Dr. Shiow-Huey Chuang Department of Applied Chemistry National University of Kaohsiung
Student: Sung-Hua Li Department of Applied Chemistry National University of Kaohsiung
Abstract
Dye removal in wastewater treatment has been a challenging problem and researchers who have applied transition metal materials in dye absorption have identified oxidized molybdenum as a potential solution. In the present study, ammonium molybdate
tetrahydrate under microwave-assisted heat treatment was used as an initiator to react with nitric acid to synthesize a series of molybdenum oxide products. These products were tested using an X-ray diffractometer, scanning electron microscopy, micro-Raman spectroscopy, and photoluminescence spectroscopy. The results show that as the reaction time increased in an ammonium molybdate tetrahydrate solution with a concentration of 0.04 M, the pyramidal products became columnar in shape and had h-MoO3 structures.
However, as the reaction time increased in an ammonium molybdate tetrahydrate solution with a concentration of 0.01 M, the columnar products became ribbon-shaped and their
X
h-MoO3 structures transformed into α-MoO3. Finally, all the molybdenum oxide products
were used to adsorb methylene blue dye, with maximum adsorption capacities and maximum unit adsorption capacities of 613.5 mg/g and 469.5 mg/m2, respectively. The main factor that influences dye adsorption is the surface charges on these products.
Keyword: molybdenum oxide, microwave-assisted heat treatment, dye adsorption, methylene blue, surface charge.
1
第一章
緒論
1.1 前言
隨著時代的進步、科技的日新月異,使得人類的生活更加的舒適與 便利,卻也相繼帶來人口暴增、能源消耗與環境汙染的問題。近年來環 保意識的抬頭,人們更加重視環保議題,許多的研究學者與企業也開始 朝著節能、綠色能源與降低汙染為目標。 染整工業結合了織物染色與化學工業,在紡織工業中扮演關鍵的角 色。然而,在製程當中常會使用到大量的水、染料與化學助劑,造成大 量的廢水汙染。而且廢水的水質複雜,具有高色度及生物難分解的特性, 造成廢水處理不易。因此,如何改善製程或提升廢水處理效率等方式, 以符現代社會的綠色及環保概念,是急需解決的問題之一。 現行廢水處理技術,按原理可以分為: 物理處理法、化學處理法及生 物處理法。其中,物理處理法乃是透過分離和去除水中不溶解的懸浮汙 染物。此方法的優點在於操作方便、分離效果良好及設備簡易,故使用 上極為廣泛。因此,在許多的文獻中,常會利用物理吸附的方式對染料 進行汙水處理之研究。 吸附材料是直接影響染料吸附量的關鍵,因此吸附材料的種類為一 重大因素,以下列舉幾項吸附材料做介紹。活性碳1-3因其具有高度的孔 洞性質,因此廣泛的作為吸附材料。然而,活性碳對於非水溶性染料的 吸附較差,且價格昂貴及再生成本高等問題,因此逐漸有其他的取代材 料出現,如紅泥4,5、黏土6、粉煤灰7及沸石 8。取代材料的出現得以解 決成本的問題,但是吸附效果並不如預期的好。隨著科技的進步,半導2 體產業開始蓬勃發展,過渡金屬材料作為吸附材料也受到更多研究者的 關切,如 TiO2 9、Fe 2O3 10、MnO 2 11、CeO 2 12、NiO13與 WO 3 14,15。近年來也 有研究使用氧化鉬作為水處理的應用16-20,可得到顯著的效果,表示氧化 鉬在水處理的應用上是相當具有潛力的材料。本研究將對氧化鉬進行探 討與其在染料吸附上的應用。
1.2 材料介紹
氧化鉬: 常見的型態為三氧化鉬,其分子量為 143.94 g/mol、密度為 4.7 g/cm3、熔點 795°C 和沸點 1155°C。不溶於水,可溶於氫氟酸、濃硫 酸及氨水和強鹼溶液中。 在單位晶格中,氧化鉬的鉬原子連接六個氧原子形成八面體 (圖 1-1)。 然而,氧化鉬的結構會因製備溫度不同,或是原子比例不同造成結構扭 曲。氧化鉬具有相當多的晶相,可藉由改變製備的溫度或其他反應條件, 得到不同晶相的氧化鉬,例如:熱穩定型態的斜方晶相 (orthorhombic, α-MoO3) 及兩種次穩定型態的結構,六方晶相 (hexagonal, h-MoO3) 和單斜晶相 (monoclinic, β-MoO3) 21-23
(表 1-1 及表 1-224)。其中以斜方晶相最 為常見。
3
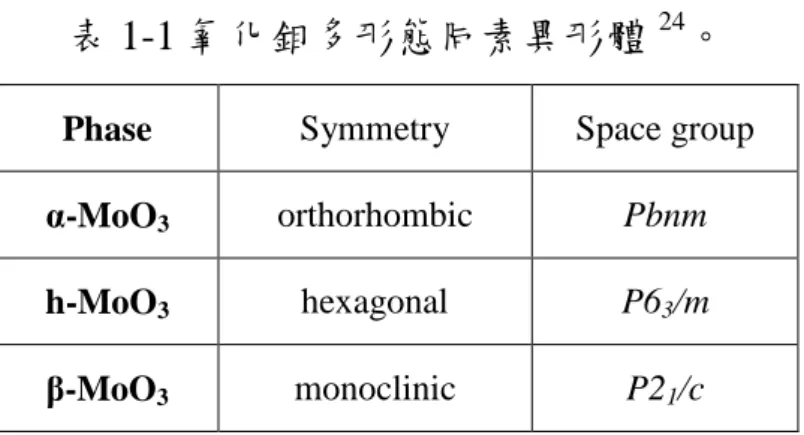
表 1-1 氧化鉬多形態同素異形體24。
表 1-2 各晶相氧化鉬的晶格常數24。
Phase Symmetry Space group
α-MoO3 orthorhombic Pbnm
h-MoO3 hexagonal P63/m
β-MoO3 monoclinic P21/c
Lattice constant (Å ) α-MoO3 h-MoO3 β-MoO3
a 3.963 10.531 7.118
b 13.856 10.531 5.366
4
第二章
理論說明與文獻回顧
2.1 文獻探討
關於文獻回顧,將分兩部份進行探討,材料製備及吸附材料的發展 過程。2.1.1 氧化鉬之合成
關於氧化鉬的製備,列舉如下: (1) 火焰合成法 (flame synthesis method)2011 年 Zheng25研究團隊利用火焰合成法氧化鉬。以 Mo 金屬做為起
始物,將 Mo 金屬置於燃燒器當中,利用火焰燃燒起始物,可得 MoOx
蒸氣。將其冷卻後可得片狀氧化鉬 (α-MoO3)。
(2) 噴霧熱分解法 (spray pyrolysis method)
2012 年 Carreño26研究團隊利用噴霧熱分解法合成氧化鉬。使用鉬酸
銨作為起始物,將鉬酸銨溶液噴灑在不同溫度 (420-670 K) 的基板上,
進行熱分解。將其冷卻後可得到氧化鉬薄膜 (α-MoO3)。
(3) 微波電漿法 (microwave plasma method)
2012 年 Thongtem27研究團隊利用微波電漿法合成氧化鉬。以鉬酸銨
作為起始物,在 900 瓦的微波電漿下照射 5 分鐘,再透過後續的熱處理
5 (4) 傳統加熱法 2009 年 Ramana28研究團隊利用傳統加熱的方式合成氧化鉬。將鉬酸 銨溶於去離子水中,並加入硝酸混合,在 85°C 下反應 1 小時,可合成出 柱狀氧化鉬 (h-MoO3) (圖 2-1)。 圖 2-1 利用傳統加熱的方式,在 85°C 下反應合成氧化鉬之 SEM 圖。28 2013 年 Bose22研究團隊利用傳統加熱的方式合成氧化鉬。將鉬酸銨 溶於去離子水中,並加入硝酸混合,在 120°C 下反應 3 小時,可合成出 柱狀氧化鉬 (h-MoO3) (圖 2-2)。 圖 2-2 利用傳統加熱的方式,在 120°C 下反應合成氧化鉬之 SEM 圖。22
6 (5) 超音波法 (ultrasonic approach) 2011 年 Wang29研究團隊利用超音波法合成氧化鉬。將(NH4)2Mo2O7 溶於去離子水中,並加入鹽酸混合,藉由超音波振盪 80 分鐘,可合成出 奈米柱狀氧化鉬 (h-MoO3) (圖 2-3)。 圖 2-3 利用超音波法合成氧化鉬之 SEM 圖。29 2012 年 Luo 與 Li30研究團隊利用超音波法的方式合成氧化鉬。將鉬
酸銨溶於去離子水中,並加入硝酸、SDS (sodium dodecyl sulfate) 及尿素 混合,藉由超音波法在 75°C 下進行反應。反應 50 分鐘可得到奈米柱狀 氧化鉬 (h-MoO3) (圖 2-4)。
7
(6) 溶劑熱法 (solvothermal method)
2010 年 Hong31研究團隊利用溶劑熱法合成氧化鉬。將 MoCl5溶於不
同比例的水與乙醇溶液中,在 180°C 下反應 10 小時,可得到片狀氧化鉬 (α-MoO3) 及似球狀氧化鉬 (monoclinic MoO2) (圖 2-5)。
圖 2-5 使用水及乙醇當作溶劑,以溶劑熱法合成氧化鉬之 SEM 圖: 水:乙醇 (mL);(a) 50:0, (b) 40:10, (c) 25:25, (d) 0:50。31
8 (7) 水熱法 (hydrothermal method) 2007 年 Zheng32研究團隊利用水熱法合成氧化鉬。將鉬酸溶於氨水 溶液中,並加入鹽酸混合,在 100°C 下反應 8 小時,可得到柱狀氧化鉬 (h-MoO3) (圖 2-6)。 圖 2-6 利用水熱法於鹽酸水溶液中合成氧化鉬之 SEM 圖。32 2012 年 Zaghib33研究團隊利用水熱法合成氧化鉬。將鉬酸銨與醋酸 水溶液混合,在 170 及 185°C 下反應 6 天,可得到纖維狀氧化鉬 (α-MoO3)。 在 170°C 下所得到的纖維狀較不完整,而在 185°C 下可以得到較完整的 纖維狀形貌,直徑為 50-80 nm,長度可達到 μm 的等級 (圖 2-7)。 圖 2-7 利用水熱法於醋酸水溶液中合成氧化鉬之 SEM 圖: (a) 170°C,(b) 185°C。33
9 2013 年 Gou34研究團隊利用水熱法合成氧化鉬。將鉬酸銨溶於去離 子水,再加入硝酸混合。並加入氯化鉻或 PVP 作為添加劑,在 180°C 下 反應 24 小時,分別可得到網狀 (圖 2-8a) 或顆粒狀 (圖 2-8b) 氧化鉬 (α-MoO3)。 圖 2-8 利用水熱法合成氧化鉬之 SEM 圖: (a) 添加氯化鉻,(b) 添加 PVP。34
(8) 微波法 (microwave-assistant heat treatment)
2012 年 Osaka35研究團隊利用微波法合成氧化鉬。將金屬鉬與硝酸混 合反應,並且添加 ZrO(NO3)2·2H2O 作為 Zr +離子的來源,在 80°C 下反應 15-30 分鐘。在沒有 Zr+離子的輔助下,得到片狀氧化鉬,其晶型為 MoO3·1/2H2O。若加入 Zr +離子,則會得到片狀及海膽狀的 h-MoO 3與 MoO3·1/2H2O 混合。研究 Zr +離子的輔助下改變氧化鉬的結構與形貌 (圖 2-9)。
10 圖 2-9 利用微波法合成氧化鉬之 SEM 圖: (a,b) 無添加 Zr+離子,(c,d) Zr+離子的輔助。35 2014 年 Bose23研究團隊利用微波法合成氧化鉬。將鉬酸銨溶於去離 子水中,並加入硝酸混合,在微波輻射下反應 2 分鐘,可得到柱狀氧化 鉬 (h-MoO3) (圖 2-10)。
圖 2-10 利用微波法合成氧化鉬之 SEM 圖。23
11 由上述的文獻可以得知,目前已有許多的合成方法可以得到氧化鉬。 然而不同的合成方式或加熱環境,所得到的氧化鉬其形貌、晶型與特性 都不盡相同,對於後續應用都有可能得到不同的效果。 而在最初實驗設計的時候,考慮到加熱的效率,選用微波法作為本 實驗的合成方法。利用微波的特殊加熱方式,能夠有效地提升加熱效率, 大幅地縮短反應的時間。 表 2-1 氧化鉬之合成方式及其形貌與晶型整理表。 合成方法 形貌與晶型 文獻 火焰合成法 片狀α-MoO3 25 噴霧熱分解法 α-MoO3 26 微波電漿法 板狀α-MoO3 27 傳統加熱法 柱狀 h-MoO3 28 傳統加熱法 柱狀 h-MoO3 22 超音波法 柱狀 h-MoO3 29 超音波法 柱狀 h-MoO3 30 溶劑熱法 片狀α-MoO3/ 球狀 monoclinic MoO2 31 水熱法 柱狀 h-MoO3 32 水熱法 纖維狀α-MoO3 33 水熱法 網狀或顆粒狀α-MoO3 34 微波法 片狀 h-MoO3/ 海膽狀 MoO3·1/2H2O 35 微波法 柱狀 h-MoO3 23
12
2.1.2 吸附材料的發展
亞甲藍為染整工業中常用的試劑之一,也是環境污染的一大元凶。 在此介紹亞甲藍去除的相關文獻,並討探吸附材料的發展。 2001 年 Shioya1研究團隊利用活性碳進行亞甲藍吸附。活性碳的比表 面積高達 761 m2 /g,其對於亞甲藍的吸附也可達到 486 mg/g,是一個吸 附效果相當良好的材料。然而,活性碳的價格昂貴及再生成本高等問題, 使得學者們往其他成本較低廉的材料進行研究。2005 年 Wang5研究團隊利用紅泥 (red mud) 進行亞甲藍吸附。但因
為紅泥本身的比表面積僅有 21 m2 /g,造成其吸附量也只有 2.1 mg/g。也 有其他研究團隊7利用粉煤灰 (fly ash) 進行亞甲藍吸附,然而吸附量也 只達到 16.6 mg/g。雖然這一系列的材料,成本較低廉,但是吸附效果並 不理想。 2006 年 Wang8研究團隊利用沸石 (zeolite MCM-22) 進行亞甲藍吸 附。雖然此材料具有較高的比表面積,可達到 490 m2 /g,但是對於亞甲 藍的吸附量,並沒有有效地提升,僅有 67.3 mg/g。 隨著半導體產業蓬勃發展之後,過渡金屬材料也逐漸列為染料吸附 的材料之一。2010 年 Ni36研究團隊利用鈦酸鹽奈米管 (titanate nanotubes) 進行亞甲藍吸附。鈦酸鹽奈米管的比表面積為 158 m2 /g,其吸附量可達 到 133 mg/g。
13 2010 年 Yong14研究團隊利用氧化鎢 (WO2/W18O49) 進行亞甲藍吸附。 氧化鎢的比表面積為 114 m2 /g,其吸附量可達到 139 mg/g。同時研究顯 示當氧化鎢之比表面積為 78、88 及 93 m2 /g 時,其吸附量分別為 67、112 及 118 mg/g。由此可明顯看出,比表面積越高的樣品,對亞甲藍的吸附 量也越高。 2013 年 Su37研究團隊利用網狀奈米線的氧化鎢 (W18O49) 進行亞甲 藍吸附。其產物的比表面積可達到 223 m2 /g,吸附量可至 201 mg/g。顯 示比表面積較大的材料,有助於提升染料的吸附量。 2013 年 Liu38研究團隊利用中空管狀的氧化鎢 (h-WO3) 進行亞甲藍 吸附。其產物的比表面積僅有 27.83 m2 /g,然而對染料的吸附量卻有 75 mg/g。相較先前的文獻,此產物的比表面積雖然不高,但吸附量卻意外 地有良好的表現。 2013 年 Zhao39研究團隊利用 N-doped H2W1.5O5.5·H2O 進行亞甲藍吸 附。其產物的比表面積僅有 7.4 m2 /g,然而對亞甲藍的吸附量卻高達 376.9 mg/g。其研究顯示,並非只有比表面積會影響亞甲藍的吸附量,表面電 位也是其中一項重要的因素。 2014 年 Wang40研究團隊利用海膽狀的氧化鎢 (WO3·0.33H2O) 進行 亞甲藍吸附。其產物的比表面積為 78.4 m2 /g,對亞甲藍的吸附量卻有 247.3 mg/g。 由上述的文獻可得知,對於染料吸附,比表面積是個相當重要的因 素。比表面積越高的樣品,其吸附量越高。然而,近年來也發現,在相 對具低比表面積的材料對染料仍有不錯的吸附效果,經研究發現,其影
14 響因素是材料本身的表面電位39,40。當材料的表面電位偏負值的情況下, 對於亞甲藍離子 (MB+ , 圖 2-11) 或是其他陽離子染料,可產生庫倫引力, 有利於吸附效果的增加。因此比表面積與表面電位,兩者都是影響該材 料吸附染料的重要因素。各文獻對亞甲藍的吸附效果整理表如表 2-2。 近年來,氧化鉬逐漸受到研究者的青睞,也有文獻使用氧化鉬應用 於水處理16-20。2015 年 Lei19研究團隊利用奈米帶的氧化鉬 (α-MoO 3) 進 行亞甲藍吸附與催化。雖然氧化鉬的吸附效率僅有 6.98%,但是,添加 過氧化氫與氧化鉬混合後,吸附效率可提升至 30.02%。 2015 年 Bi 與 Qi20研究團隊利用奈米帶的氧化鉬 (α-MoO3) 進行亞甲 藍吸附。利用 30 mg 的產物,可用 60 分鐘的時間,將亞甲藍溶液 (20 mg L-1, 30 mL) 吸附至將近 100%。 雖然目前氧化鉬對於吸附亞甲藍的文獻極為有限19-20,但是如何藉由 增加比表面積或降低表面電位的方式,來提升氧化鉬對於亞甲藍的吸附 效果,則會是一項值得挑戰的研究。因此本實驗將對氧化鉬進行探討與 其在亞甲藍吸附上的應用。 圖 2-11 亞甲藍之結構圖。
15 表 2-2 各文獻對亞甲藍的吸附效果整理表。 材料 比表面積 (m2 /g) 吸附量 (mg/g) 文獻 活性碳 761 486 1 紅泥 21 2.1 5 沸石 490 67.3 8 鈦酸鹽(titanate) 158 133 36 WO2/W18O49 114 139 14 W18O49 223 201 37 h-WO3 27.83 75 38 N-doped H2W1.5O5.5·H2O 7.4 376.9 39 WO3·0.33H2O 78.4 247.3 40
16
2.2 理論說明
2.2.1 微波法
微波法是以電磁場的方式將能量供給物質,此方式能夠將能量迅速 穿透物質減少熱梯度的影響,並可達到加熱迅速、減少製程時間與節省 能源的目的。一般傳統加熱 (圖 2-12,A),能量是以傳導、對流或是輻射 的方式,由物質的表面將能量傳遞到物質內部。而微波加熱 (圖 2-12,B) 則是利用電磁波穿透物質至物質的內部,使分子產生極化與電子震盪現 象,這些運動造成分子高速旋轉進而摩擦生熱,使溫度上升。是一種將 電磁場轉換為熱能的能量轉換程序。由於電磁波能深達物質內部,因此 能給予物質均勻且迅速的加熱,而且相較於在高溫爐中進行水熱法或是 溶劑熱法的反應,微波法更具有加熱迅速,溫度點準確的優勢。 圖 2-12 加熱方式示意圖: (A,左圖) 傳統加熱,(B,右圖) 微波加熱。4117
2.2.2 光致螢光 (Photoluminescence, PL)
以一外部的激發光源照射待測樣品,使樣品中的價電子從價帶 (valence band) 躍遷到更高能階的導帶 (conduction band),而原本的價帶 留下一個電洞,形成電子-電洞對。而在非常短的時間內,大部分位於高 能階電子會藉由釋放光子或其他方式釋放能量,遷移至較低能態。當電 子-電洞以輻射複合 (recombination) 的方式結合而放出光子,此稱之為螢 光。又因其電子躍遷的能量來自外加光源,因此稱之為光致螢光。但在 材料中可能存在著雜質能階或缺陷能階,此時電子-電洞對可能不以輻射 複合方式放出光子,而是以非輻射複合的方式放出熱能或其他能量。 光致螢光為一分析迅速,且非破壞性的檢測技術。常用於檢測材料 的組成、雜質、摻雜分布與缺陷程度。一般半導體材料的量測常利用高 於材料能隙的光源,使電子進行躍遷。若材料中無缺陷或摻雜的影響, 則電子由導帶遷移至價帶的能量差即為此材料的能隙。但若材料中存在 缺陷或是有其他的摻雜,則會在價帶與導帶間形成次能階 (sub-band), 而改變電子遷移的機制。 圖 2-13 光致螢光電子-電洞對複合之示意圖。42
18
2.2.3 Langmuir 等溫吸附模式
吸附 (adsorption) 此現象是利用固體其表面之作用力,將流體中某 些物質吸附並附著於固體表面上。具有此表面吸附力之固體稱為吸附劑 (adsorbent),而被吸附於此固體表面上的物質則稱為吸附質 (adsorbate)。 而吸附劑的吸附能力稱為吸附容量 (adsorptive capacity, qm),其定義為單 位吸附劑所能夠吸附的溶質量,通常以 x/m 表示 (x 為被吸附物質的質量, 而 m 為吸附劑的重量,x/m 單位通常以 mg/g 來表示)。 Langmuir 等溫吸附模式則是吸附實驗上常用來檢測吸附容量的方式。 在定溫的條件下,將吸附劑與被吸附溶質混合,於平衡狀態之吸附量 (qe) 與溶液濃度關係,繪製成圖。Langmuir 等溫吸附模式的公式如下: (1)Ce: 溶液中亞甲藍平衡濃度 (equilibrium MB concentration, mg L-1); qe: 氧化鉬對亞甲藍之平衡吸附量
(the amount of MB adsorbed at equilibrium, mg g-1)
qm: 氧化鉬對亞甲藍之最大吸附容量 (maximum adsorption capacity mg g-1)
K: 平衡常數 (equilibrium constant, L mg-1)
將公式經過轉換之後,可得式(2):
19 此時以 Ce 為 x 軸,Ce/qe為 y 軸繪圖,可得一線性關係的圖形。而 由此圖形之斜率 (1/qm) 即可得到最大吸附容量 (qm)。因此可利用實驗的 方法來求得 Langmuir 等溫吸附模式中的最大吸附容量 (maximum adsorptive capacity, qm)。 圖 2-14 Langmuir 等溫吸附模式圖。
20
2.2.4 表面電位 (Zeta Potential)
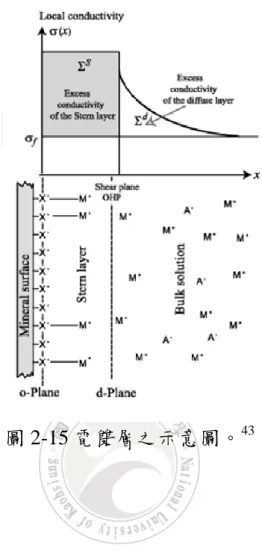
Zeta Potential 的量測方式是在毛細管兩側施加一電場,帶電粒子會 往反方向的電極移動,並測量其電泳的移動率,再藉由 Henry equation 計算表面電位,如式(3): (3) Ue: 電泳遷移速率 ε: 介電常數 : 表面電位 (Zeta Potential) η: 溶液的黏度 f(κα): Henry’s function 在膠體化學中,膠體粒子之表面常會具有晶格缺陷和吸附特殊離子 等因素,往往造成膠體粒子表面帶有靜電荷。而表面的靜電荷經由溶液 中的電荷平衡後,形成電雙層 (electrical double layer),依其電子分布可 分為固定層 (stern layer) 和擴散層 (diffuion layer),如圖 2-15。而流動在 固定層與擴散層之間的剪面 (shear plane),其電位稱之為 Zeta Potential。 Zeta Potential 為固定層與擴散層之間剪面的電位,且此滑動的剪面位於 液體內某處,並非為固體與夜體的相界面上。21
圖 2-15 電雙層之示意圖。43
22
第三章
材料與方法
3.1 實驗藥品及儀器設備
3.1.1 實驗藥品
本研究使用的藥品如下: 藥品名稱 化學式 製造廠商 純度 Ammonium molybdate tetrahydrate 鉬酸銨 (AHM) (NH4)6Mo7O24·4H2O STREM 99.98% Ethanol 乙醇 C2H5OH Sigma-Aldrich 95% Nitric acid 硝酸 HNO3 J. T. Baker 15.7 M Molybdenum(Ⅵ) oxide三氧化鉬 (標準品) α-MoO3 Alfa Aesar 99.95%
Methylene Blue 亞甲藍 (MB) C16H18N3SCl·3H2O ACROS 95% Rhodamine B 羅丹明 B (RhB) C28H31ClN2O3 Sigma 95% Sunset Yellow 日落黃 (SY) C16H10N2Na2O7S2 Aldrich 90% Methyl Blue 甲基藍 C37H27N3Na2O9S3 Sigma
23
3.1.2 實驗儀器設備
儀器名稱 儀器廠商及型號
微波合成系統
Microwave synthesis system Milestone STARTs X- ray 粉末繞射儀
(X-ray diffractometer, XRD) Rigaku Multiflex 掃描式電子顯微鏡
(Scanning electron microscopy, SEM) JEOL JSM-6330TF 顯微拉曼光譜儀
(Micro-Raman spectroscopy, Raman) PTT BWII RAMaker 比表面積分析儀
(Surface Area and Porosity Analyzer, BET) Micromeritics ASAP 2020 近紫外光可見光光譜儀
(Ultraviolet/Visible Spectroscopy) Hitachi U-3010 微光致螢光光譜儀
(Micro-PL spectroscopy, PL) UniRam
動態光散射粒徑分析儀
24
3.2 MoO
3合成步驟
圖 3-1 MoO3合成流程圖。 實驗步驟: 取鉬酸銨 (0.500 或 0.156 g) 與 10 mL 水混合,攪拌至鉬酸銨完全溶 解,得到鉬酸銨溶液 (0.04 或 0.01 M)。再加入 10 mL 的 2.2 M 硝酸,混 合均勻,得透明澄清溶液。將溶液轉移至微波消化管內,鎖上蓋子,置 入微波系統中,在 120°C 下反應 10、30 或 90 分鐘,得到深淺不一的灰 白色沉澱物。產物過濾後再以水及乙醇清洗,再放入烘箱於 60°C 下乾燥 一天。實驗流程如圖 3-1,產物編號及其實驗條件整理如表 3-1。25 表 3-1 實驗條件整理。 產物編號 AHM (M) 硝酸 (2.2 M, mL) 反應時間 (min) 反應溫度 (°C) MoO-10 0.04 10 10 120 MoO-30 0.04 10 30 120 MoO-90 0.04 10 90 120 MoO-10L 0.01 10 10 120 MoO-30L 0.01 10 30 120 MoO-90L 0.01 10 90 120
26
3.3 染料吸附實驗步驟
3.3.1 亞甲藍檢量線測定之溶液配製
取亞甲藍 0.187 g 加去離子水溶解,配置 500 mL 溶液,可得 1 mM 亞甲藍水溶液。取 1mM 亞甲藍水溶液 10 mL,加至 100 mL 容量瓶中, 配置 100 mL 溶液,可得 0.1 mM 亞甲藍水溶液。重複上述步驟,分別配 製 0.03 mM、0.02 mM、0.01 mM、0.005 mM 及 0.001 mM 亞甲藍水溶液。 將上述濃度之亞甲藍水溶液分別以 UV-Vis 光譜儀測量。並以 664 nm 之 訊號數值繪製檢量線,可得其濃度對應吸收度得直線方程式, y=0.74026x+0.039,R2=0.9943。如表 3-2 及圖 3-2。 表 3-2 亞甲藍水溶液之各濃度訊號 MB 濃度 (mM) 0.03 0.02 0.01 0.005 0.001 UV 訊號 (a.u.) 2.189 1.581 0.861 0.430 0.085 圖 3-2 亞甲藍水溶液之檢量線。27
3.3.2 亞甲藍吸附速率測定之溶液配製
取亞甲藍 0.187 g 加去離子水溶解,配置 500 mL 溶液,可得 1 mM 亞甲藍水溶液。取 1 mM 亞甲藍水溶液 10 mL,加至 100 mL 容量瓶中, 配置 100 mL 溶液,可得 0.1 mM 亞甲藍水溶液。重複上述步驟,配製 0.025 mM 亞甲藍水溶液。 取產物 20 mg,並加入 1 mL 乙醇與產物混合。再緩慢加入 0.025 mM 亞甲藍水溶液 50 mL,持續攪拌並開始計時。每隔特定時間取出 2 至 3 mL 混合溶液進行離心。離心 30 分鐘後,取上層澄清溶液以 UV 量測。3.3.3 亞甲藍吸附量測定之溶液配製
取產物 10 mg,並加入 1 mL 乙醇與產物混合。再緩慢加入 0.050 mM 亞甲藍水溶液 500 mL,持續攪拌並開始計時。MoO-10、MoO-30 和 MoO-90 隔七天後取出 2 至 3 mL 混合溶液離心。離心 30 分鐘後,取上 層澄清溶液以 UV 量測。MoO-10L、MoO-30L 和 MoO-90L 隔一天後取 出 2 至 3 mL 混合溶液離心。離心 30 分鐘後,取上層澄清溶液以 UV 量 測。利用吸附前及吸附後的吸收度差值,估算出吸附量 (qe)。28
3.3.4 Langmuir adsorption 之溶液配製
取產物 10 mg,並加入 1 mL 乙醇與產物混合。再緩慢加入亞甲藍水 溶液 500 mL,持續攪拌並開始計時。分別與不同的濃度之亞甲藍水溶液 進行吸附,濃度分別為 0.005、0.010、0.020、0.030、0.040、0.050 和 0.060 mM。 MoO-10、MoO-30 和 MoO-90 隔七天後取出 2 至 3 mL 混合溶液離心。 離心 30 分鐘後,取上層澄清溶液以 UV 量測。MoO-10L、MoO-30L 和 MoO-90L 隔一天後取出 2 至 3 mL 混合溶液離心。離心 30 分鐘後,取上 層澄清溶液以 UV 量測。利用吸附前及吸附後的吸收度差值,分別計算 出於不同濃度下的吸附量 (qe)。並作出平衡濃度與平衡吸附量之關係圖 求得最大吸附容量 (qm)。29
3.4 儀器分析條件
3.4.1 粉末 X-ray 繞射儀分析法 (X-ray diffractometer, XRD)
使用 X-ray 繞射儀 (Rigaku Multiflex 2Kw) 分析,實驗使用燈源為 Cu Kα1 (λ=1.541838Å ),Ni 濾波器,D.S.=0.5o、S.S.=1o、R.S.=0.15 mm,
掃描範圍為 2θ=5o
~60o,掃描速率為 1o/min。將所得之繞射光譜使用
JCPDS (Joint Committee on Power Diffraction Standards) 資料庫做比對, 以分析及鑑定在不同實驗條件製成的氧化鉬粉末。利用 XRD 得知產物是 否具結晶相及純相。
3.4.2 掃描式電子顯微鏡分析法 (Scanning electron microscopy, SEM)
用掃描式電子顯微鏡 (JOEL JSM-6330TF) 進行分析。將待測粉末均 勻灑在碳膠上並固定於載台上,再將樣品表面上鍍金,增加樣品的導電 性,以加速電壓 10 KeV,進行樣品表面形態的觀測。 3.4.3 顯微拉曼光譜儀分析法 (Micro-Raman spectroscopy,Micro-Raman) 使用顯微拉曼光譜儀 (PTT BWII RAMaker) 進行分析。取適量的待 測樣品,放在玻璃載台上,即可進行顯微拉曼光譜分析,選用 633 nm (He-Ne Laser) 光源,能量為 2 mW,掃描範圍為 70~1100 cm-1,掃描時間 為 30 秒。使用顯微拉曼光譜,主要是為了偵測待測樣品中,原子和原子 之間的振動模式。
30 3.4.4 紫外-可見光光譜儀分析法 (Ultraviolet/Visble spectroscopy, UV-Vis) 使用紫外-可見光光譜儀 (Hitachi U-3010) 進行分析。取約2 mL待測 溶液置於石英槽內,將石英槽置於紫外-可見光光譜儀內的插槽,進行 UV-Vis光譜分析。掃描範圍400 nm至800 nm,掃描速率300 nm/min。利 用所得的UV-Vis圖譜,得知待測樣品之吸收度變化。
3.4.5 粉末比表面積分析法 (Surface Area and Porosity Analyser, BET)
使用粉末比表面積分析儀 (Micromeritics ASAP 2020) 進行分析。秤 取 200 mg 待測樣品置於試料瓶中,安裝於除氣槽進行除氣步驟。在持續 抽氣的狀態下,以升溫速率 10o C/min 至 90oC,並持溫 3 小時,將待測樣 品中所含的水分或其他吸附物去除。除氣完成後,將試料瓶移至分析處 並浸入液態氮阱內,以定量的氬氣充入試料瓶中,測量在不同相對壓力 下的氬氣吸附量,再由電腦 BET 公式計算出樣品的比表面積。 3.4.6 微光致螢光分析 (Micro-PL spectroscopy) 取適量的待測樣品粉末,進行微光致螢光光譜儀的檢測。儀器使用 的光源為 325 nm 的雷射光,掃描範圍 350 nm 至 800 nm。衰減片選用 100% (雷射光可通過強度為 100%,無衰減),grating 使用 300,曝光時間為 1 秒,掃描次數為 60 次。
31
第四章
結果與討論
4.1 改變反應時間對於氧化鉬的影響 (0.04 M AHM)
在 0.04 M 鉬酸銨溶液下,利用微波法合成氧化鉬。取鉬酸銨 (0.500 g) 作為起始物與水 (10 mL)、硝酸 (2.2 M, 10 mL) 混合反應,利用微波 法在 120°C 下反應 10、30 或 90 分鐘。得到一系列的產物 MoO-10、MoO-30 和 MoO-90,並利用 XRD、SEM、Micro-Raman 及 PL 鑑定產物。4.1.1 X-ray 粉末繞射法分析
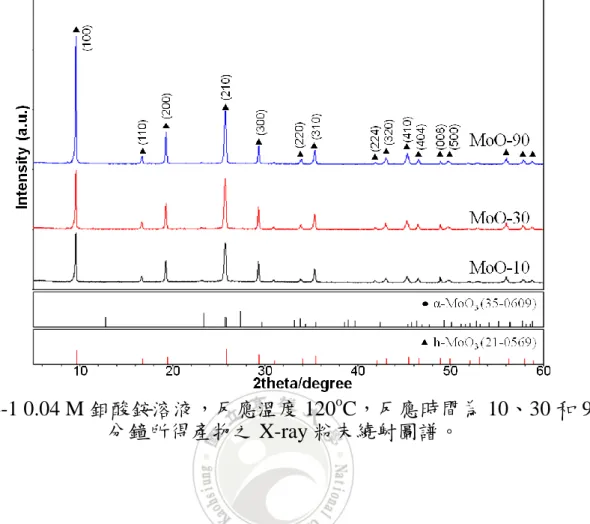
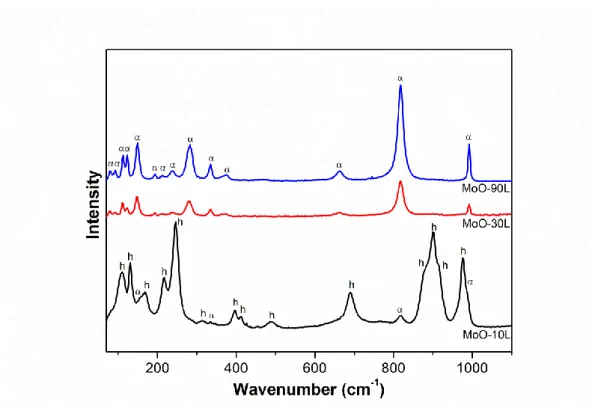
觀察圖 4-1 可以得知,反應 10 分鐘至 90 分鐘所得的產物訊號,皆 得到相似的繞射峰。根據資料庫的對照之後,可以發現這些繞射峰是由 次穩定態的六方晶相氧化鉬 (h-MoO3, JCPDS No.21-0569) 所組成。由此 可得知,在 0.04 M 鉬酸銨溶液下,不論反應時間的長短,皆可得到六方 晶相的氧化鉬。然而,隨著反應時間的增加,可以觀察到位於 9.6o的繞 射峰相對強度,逐漸增大,而其對應的晶相面為 (100)。因此推測可能是 產物有著優選方向的成長,才導致 9.6o的繞射峰相對強度有逐漸增大的 趨勢。32
圖 4-1 0.04 M 鉬酸銨溶液,反應溫度 120o
C,反應時間為 10、30 和 90 分鐘所得產物之 X-ray 粉末繞射圖譜。
33
4.1.2 掃描式電子顯微鏡圖像分析
由圖 4-2 可以觀察到,在 0.04 M 鉬酸銨溶液下合成氧化鉬,其形貌 為柱狀與錐狀的聚集體。當反應時間為 10 分鐘,產物的形貌主要為錐狀, 且其表面容易有缺陷,形狀較不完整。反應時間為 30 分鐘時,可得較完 整錐狀的產物。而當反應時間增長至 90 分鐘時,錐狀產物僅剩少量,主 要以柱狀為主。由上述的現象可以得知,隨著反應時間增長,產物的形 貌逐漸從錐狀轉變為柱狀,其表面也更加完整。 參考 XRD 數據可以發現,反應時間增長,促使柱狀形貌增加,(100) 也隨之增加。從六角柱示意圖 (圖 4-3) 可以發現,{100} 面族屬於其中 之一的面相。當形貌由錐狀逐漸成長為柱狀,使得產物往 {100} 面族方 向排列,形成完整的六角柱狀,因此在 XRD 的數據上可以觀察到優選方 向的成長。根據文獻44,可以藉由增加硫脲溶液的濃度,促使形貌由錐 狀轉變為柱狀。在本研究當中,則是可以運用反應時間的不同,改變錐 狀與柱狀的形貌變化。34
圖 4-2 0.04 M 鉬酸銨溶液,反應溫度 120o
C,反應時間為 10、30 和 90 分鐘所得產物之 SEM 圖像 (5K)。
35
4.1.3 顯微拉曼光譜分析
觀察圖 4-4 可以得知,反應時間 10、30 和 90 分鐘的產物,其拉曼 圖譜的訊號皆可對應至 h-MoO3的訊號 45,46,其中 114、133、174、218 及 248 cm-1訊號峰為 MoO 4的振動訊號。而 319、398、413、489 及 691 cm -1 訊號峰為 O-Mo-O 的振動訊號。而 882、901、912 及 977 cm-1訊號峰為 Mo=O 的振動訊號。 與前面 XRD 偵測所得到的結果互相對照,推測在 10 分鐘的短時間 反應下,已可得到結構完整的 h-MoO3結構,導致在 XRD 及 Raman 的偵 測結果上都可以得到相當明顯的訊號。 圖 4-4 0.04 M 鉬酸銨溶液,反應溫度 120o C,反應時間為 10、30 和 90 分鐘所得產物之 Raman 圖譜。36
4.1.4 光致螢光光譜分析
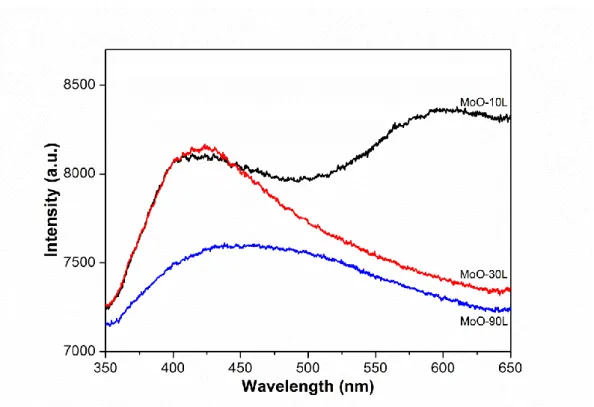
觀察圖 4-5 可以得知,利用光源為 325 nm 的雷射光照射 MoO-10、 MoO-30 及 MoO-90 這三個產物,在 PL 圖譜上可以得到兩個較寬廣的訊 號,分別位於 415-437 nm 及 575-603 nm。並且根據文獻47可以得知, 415-437 nm 為氧化鉬本身的能隙 (band gap) 所產生的訊號,而能隙的大 小為 2.92 eV。575-603 nm 則為氧化鉬的氧缺陷所產生的訊號。且隨著反 應時間的增加,575-603 nm 的訊號逐漸增強,代表隨著反應時間的增加, 氧化鉬中的氧缺陷逐漸增加。 由上述各數據顯示,在 0.04 M 鉬酸銨溶液的環境下,我們可得知反 應時間的增加,促使產物形貌從錐狀轉變為六角柱狀。其結構不受影響, 仍舊為六方晶相的氧化鉬。而在反應時間增加的同時,也使氧化鉬中的 氧缺陷也逐漸增加。 圖 4-5 0.04 M 鉬酸銨溶液,反應溫度 120o C,反應時間為 10、30 和 90 分鐘所得產物之 PL 圖譜。37
4.2 改變反應時間對於氧化鉬的影響 (0.01 M AHM)
在較低濃度 0.01 M 鉬酸銨溶液下,利用微波法合成氧化鉬。取鉬酸 銨 (0.156 g) 作為起始物與水 (10 mL)、硝酸 (2.2 M, 10 mL) 混合反應, 利用微波法在 120°C 下反應 10、30 或 90 分鐘。得到一系列的產物 MoO-10L、MoO-30L 和 MoO-90L,並利用 XRD、SEM、Micro-Raman 及 PL 鑑定產物。
4.2.1 X-ray 粉末繞射法分析
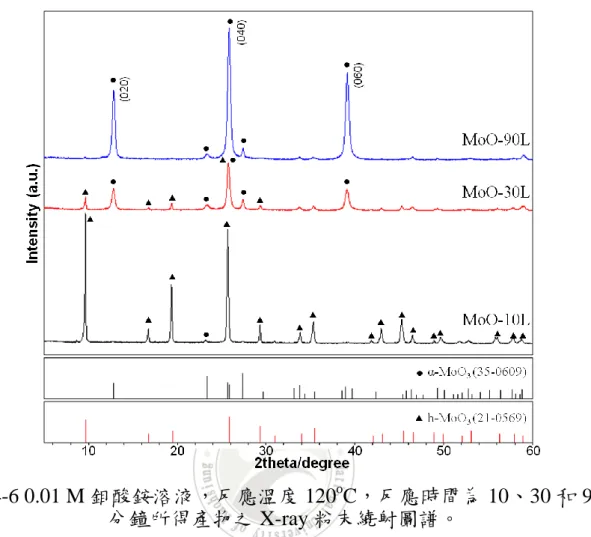
觀察圖 4-6 可以得知,在較低鉬酸銨濃度的環境下,反應 10 分鐘,
所得的產物為 h-MoO3。然而,反應時間增長至 30 分鐘時,產物則以正
交晶相的氧化鉬 (α-MoO3, JCPDS No.35-0609) 為主,同時存在 h-MoO3。
當反應再增長至 90 分鐘,產物僅存 α-MoO3在 MoO-90L 中。其中三個繞 射峰較為明顯,分別為 (020)、(040) 及 (060) 的面相,此現象表示產物 具有優選方向的成長。由 XRD 圖譜顯示,在 0.01 M 鉬酸銨溶液下,增 長反應時間,會促使氧化鉬的結構從 h-MoO3逐漸轉為α-MoO3。 與 0.04 M 鉬酸銨溶液的環境下互相比較,以較高濃度的鉬酸銨溶液 反應可以得到 h-MoO3。然而,降低鉬酸銨溶液的濃度,起始物的量相對 變少,但是反應條件仍舊是 120O C,這代表每單位的起始物,在加熱的 過程可以獲得更多的能量。隨著反應時間的增加,一旦獲得足夠的能量, 就能使結構從次穩定態的 h-MoO3轉變為熱穩定態的α-MoO3。
38
圖 4-6 0.01 M 鉬酸銨溶液,反應溫度 120o
C,反應時間為 10、30 和 90 分鐘所得產物之 X-ray 粉末繞射圖譜。
39
4.2.2 掃描式電子顯微鏡圖像分析
由圖 4-7 可以觀察到,在 0.01 M 鉬酸銨溶液下反應 10 分鐘,其形貌 為柱狀,並且有少許的奈米帶附著於柱狀上。隨著反應時間增長至 30 分 鐘,產物的形貌逐漸轉為奈米帶為主,柱狀形貌僅剩少量。最後將反應 增長至 90 分鐘,形貌全部轉變為奈米帶。由上述可知,隨著反應時間增 長,產物的形貌由柱狀轉為奈米帶。 參考 XRD 數據並與先前 MoO-10、MoO-30 及 MoO-90 的數據一同 分析。當產物的結構為 h-MoO3時,產物的形貌容易呈柱狀或錐狀。而且 在較低濃度鉬酸銨溶液下,隨著反應時間的增長,氧化鉬的結構從 h-MoO3逐漸轉為α-MoO3,同時其形貌也由柱狀轉變為奈米帶,故可認 定的柱狀或錐狀產物為 h-MoO3,奈米帶產物為α-MoO3。40
圖 4-7 0.01 M 鉬酸銨溶液,反應溫度 120o
C,反應時間為 10、30 和 90 分鐘所得產物之 SEM 圖像 (10K)。
41
4.2.3 顯微拉曼光譜分析
觀察圖 4-8 可以得知,反應時間為 10 分鐘的產物,其拉曼光譜主要 可對應至 h-MoO3的訊號,同時存在微弱的α-MoO3的訊號 46,48,49。其中 110、132、168、217 及 246 cm-1訊號峰為 MoO4的振動訊號。而 314、335、 397、415、490 及 690 cm-1訊號峰為 O-Mo-O 的振動訊號。而 816、881、 900、912、976 及 992 cm-1訊號峰為 Mo=O 的振動訊號。MoO-10L 雖然 在 XRD 中觀測到 α-MoO3的訊號相當微弱,但從 SEM 圖中可以看到奈 米帶的α-MoO3,造成此現象的原因可能是在α-MoO3形成的初期,其結 晶性不佳且量少導致其繞射峰不夠明顯所致,因此在拉曼光譜上α-MoO3 的訊號也較微弱。 隨著反應時間的增加,氧化鉬逐漸由六方晶相轉變為正交晶相,因 此在拉曼圖譜上可以發現,MoO-30L 及 MoO-90L 兩個產物的訊號皆為 α-MoO3,且因時間的增加其結構也更加完整。其中 80、93、113、124、 149、194 及 213 cm-1訊號峰為 MoO4的振動訊號。而 240 及 282 cm -1訊 號峰為 O=Mo=O 的振動訊號。而 335、376、463 及 662 cm-1訊號峰為 O-Mo-O 的振動訊號。而 818 及 992 cm-1訊號峰為 Mo=O 的振動訊號。 雖然在 MoO-30L XRD 的繞射峰有少量的 h-MoO3訊號,然而在拉曼光譜 中,因 h-MoO3的振動訊號過於微弱,而不易被觀察到。42
圖 4-8 0.01 M 鉬酸銨溶液,反應溫度 120o
C,反應時間為 10、30 和 90 分鐘所得產物之 Raman 圖譜。
43
4.2.4 光致螢光光譜分析
觀察圖 4-9 可以得知,利用光源為 325 nm 的雷射光照射 MoO-10L、 MoO-30L 及 MoO-90L 這三個產物,在 PL 圖譜上可以得到兩個較寬廣的
訊號,分別位於 400-440 nm 及 575-603 nm。並且根據文獻47可以得知,
400-440 nm 為產物本身的能隙 (band gap) 所產生的訊號,MoO-10L、 MoO-30L 和 MoO-90L 的能隙大小分別為 2.95、2.94 和 2.75 eV。575-603 nm 為產物中的氧缺陷所產生的訊號。然而在 0.01 M 鉬酸銨溶液下,隨 著反應時間的增加,575-603 nm 的訊號卻急遽下降,代表產物中的氧缺 陷大幅減少。配合 XRD 的結果可以推測,當反應時間增加,產物由 h-MoO3 轉變為α-MoO3,在結構上會進行重排。而在此過程中因起始物濃度較低, 使其在反應過程中,隨著反應時間的增加,可與充份的氧原子反應,因 而大幅改善產物中氧缺陷的現象。 由上述的各數據可以得知,反應時間與鉬酸銨的濃度都會對氧化鉬 的合成產生影響。在 0.04 M 鉬酸銨溶液的環境下,反應時間的增加,產 物結構皆為 h-MoO3,但其形狀由錐狀轉變為柱狀。 然而,在較低濃度 0.01 M 鉬酸銨溶液的環境下,會有利於每單位的 起始物,在加熱的過程中獲得更多的能量,因此使產物從柱狀的 h-MoO3 轉變為帶狀的α-MoO3。同時也可與較多的氧原子反應,在充足的反應時 間後,可得到幾乎不具氧缺陷的α-MoO3。
44
圖 4-9 0.01 M 鉬酸銨溶液,反應溫度 120o
C,反應時間為 10、30 和 90 分鐘所得產物之 PL 圖譜。
45
4.3 氧化鉬對亞甲籃吸附之研究
4.3.1 吸附速率之探討
取 20 mg 之氧化鉬置入亞甲藍溶液中 (2.5*10-5 M,50 mL) 進行吸附 速率實驗。間隔一定時間對亞甲藍溶液進行 UV-Vis 之量測,藉此探討氧 化鉬對亞甲藍之吸附效果,並將所得數據繪製成圖 4-10 與圖 4-11。 觀察 UV-Vis 數據的變化,可發現在 0.04 M 鉬酸銨溶液 (MoO-10、 MoO-30 和 MoO-90) 這一系列所得的產物,其吸附速率較差,需要經過 長達 7 小時的吸附時間,才有辦法將亞甲藍完全吸附。但是在 0.01 M 鉬 酸銨溶液 (MoO-10L、MoO-30L 和 MoO-90L) 這一系列所得的產物,就 明顯看出其吸附速率較為快速。吸附 5 分鐘後,吸附率分別為 95.9%、 97.3%和 98.2%,吸附效果相當良好。 吸附染料屬於一物理現象,因此比表面積為一主要的因素。為了探 討兩系列產物在吸附速率上面的差異,在此也進行了比表面積的分析。 MoO-10、MoO-30 和 MoO-90 其比表面積分別為 0.87、1.19 和 1.92 m2/g。 在 0.04 M 鉬酸銨溶液下合成氧化鉬,其產物皆屬於柱狀與錐狀的聚集體, 尺寸較大,因此可以發現其表面積較低。而 MoO-90L 的產物為奈米帶, 其比表面積為 12.25 m2 /g。由此結果可以得知,比表面積較大的產物,對 於亞甲藍的吸附速率較快。46
47
48
4.3.2 最大吸附容量之探討
針對氧化鉬產物,探討其對亞甲藍的吸附量 (qe) 測試。亞甲藍起始
濃度為 5.0*10-5
M,體積為 500 mL 進行吸附。MoO-10、MoO-30 和 MoO-90 間隔 7 天後進行 UV-Vis 量測。MoO-10L、MoO-30L 和 MoO-90L 間隔 1 天後進行 UV-Vis 量測,觀察亞甲藍溶液吸收度的變化後,再計算出其吸 附量。各產物吸附前後之吸收度變化如表 4-1。
表 4-1 亞甲藍溶液進行吸附之吸收度變化。
MoO-10 MoO-30 MoO-90 MoO-10L MoO-30L MoO-90L 吸附前 (a.u.) 3.062 3.062 3.062 3.062 3.062 3.062 吸附後 (a.u.) 2.245 1.072 1.005 0.831 0.900 0.953 吸附量 (qe, mg/g) 213.4 519.7 537.2 582.6 564.6 550.8 利用吸附量 (qe) 進行更精確地測量,可計算出其最大吸附容量 (qm)。 在不同濃度的亞甲藍溶液中,加入固定 10 mg 的氧化鉬進行附吸,可以 得到吸附飽和後的吸附量 (qe),與溶液中亞甲藍的平衡濃度,將其繪製
成圖 4-12。並且利用 Langmuir adsorption model,如式(2),再利用 Ce/qe
對 Ce 作圖可繪製成圖 4-13。用此方式作圖,可得一線性關係的圖形,再
由圖形的斜率 (1/qm),可得知該樣品的最大吸附容量 (qm)
9,38,50-52。
49
Ce: 溶液中亞甲藍平衡濃度 (equilibrium MB concentration, mg L-1); qe: 氧化鉬對亞甲藍之平衡吸附量
(the amount of MB adsorbed at equilibrium, mg g-1)
qm: 氧化鉬對亞甲藍之最大吸附容量 (maximum adsorption capacity mg g-1)
K: 平衡常數 (equilibrium constant, L mg-1)
利用 Langmuir adsorption model 可以得知各產物對亞甲藍的吸附容 量。MoO-10、MoO-30 和 MoO-90 對亞甲藍的最大吸附容量分別為 226.2、 558.7 和 565.0 mg/g。而另一系列 MoO-10L、MoO-30L 和 MoO-90L 對亞 甲藍的最大吸附容量分別為 613.5、602.4 和 581.4 mg/g。其中以 MoO-10L 對亞甲藍的吸附效果最佳,其最大吸附容量可達 613.5 mg/g。藉由以往 的文獻我們可以了解到,比表面積的大小常常是影響染料吸附的一個關 鍵因素。在比表面積越高的情況下,越有機會得到高吸附量的效果。然 而,在本實驗中所得的氧化鉬其比表面積遠低於其他材料,但是對於亞 甲藍的吸附卻有更加卓越的效果。因此,在本實驗中影響吸附效果的主 因並非為比表面積。而且從表 4-2 也發現,不同的氧化鉬晶型對最大吸 附容量並沒有明顯的變化,因此晶型也不是主要的因素,可能有其他因 素造就了氧化鉬具有低比表面積卻有高吸附量的特性。 表 4-2 比表面積、晶型與最大吸附容量之整理表。
產物編號 MoO-10 MoO-30 MoO-90 MoO-10L MoO-30L MoO-90L
比表面積
(m2/g) 0.87 1.19 1.92 15.44 14.07 12.25
晶型 h-MoO3 h-MoO3 h-MoO3
h-MoO3(主) +α-MoO3 α-MoO3(主) + h-MoO3 α-MoO3 最大吸附容量 (mg/g) 226.2 558.7 565.0 613.5 602.4 581.4
50
圖 4-12 不同產物對亞甲藍之最大吸附量的量測曲線圖。
51
4.3.3 表面電位之探討
亞甲藍在水溶液中通常以離子型態 (MB+) 呈現,而氧化鉬屬於 n-type 的材料,所以在兩者之間可能存在正負電的吸引力,因此氧化鉬 的表面電位可能是影響染料吸附的一大主因。為了證實最大吸附容量與 表面電位的關係,故在此也進行了表面電位的分析,如圖 4-14 與表 4-3。 可發現,MoO-10、MoO-30 和 MoO-90 其與實驗 pH 值條件相仿的表面 電位分別為 -10.3、 -13.8 和 -19.9 mV,對亞甲藍的最大吸附容量分別為 226.2、558.7 和 565.0 mg/g。而 MoO-10L、MoO-30L 和 MoO-90L 其與實 驗 pH 值條件相仿的表面電位分別為 -27.5、 -25.6 和 -21.3 mV,對亞甲 藍的最大吸附容量分別為 613.5、602.4 和 581.4 mg/g。可發現當產物之 表面電位越低,其最大吸附容量也越大。其中 MoO-10L 為所有產物中具 有最低的表面電位 (-27.5 mV),而其對亞甲藍的吸附量也最大 (613.5 mg/g)。 圖 4-14 不同產物於不同 pH 值下的表面電位。52
表 4-3 比表面積、表面電位與最大吸附容量之整理表。
產物編號 MoO-10 MoO-30 MoO-90 MoO-10L MoO-30L MoO-90L 比表面積 (m2 /g) 0.87 1.19 1.92 15.44 14.07 12.25 表面電位 (mv) -10.3 -13.8 -19.9 -27.5 -25.6 -21.3 最大吸附容量 (mg/g) 226.2 558.7 565.0 613.5 602.4 581.4 圖 4-15 最大吸附容量與表面電位之關係圖。 進一步比較不同材料對亞甲藍之吸附效果,從表 4-4 可看出,一般 文獻值的單位面積最大吸附容量介在 0.1-3.2 mg/m2,僅在 Zhao39的研究 中有 50.9 mg/m2的卓越表現。而在本研究中的產物 MoO-10、MoO-30 和 MoO-90 其單位面積最大吸附容量分別為 260.0、469.5 和 294.3 mg/m2。 MoO-10L、MoO-30L 和 MoO-90L 其單位面積最大吸附容量分別為 39.7、 42.8 和 47.5 mg/m2。其中以 MoO-30 具最大值 469.5 mg/m2,此數值遠高
53 過於目前我們所知的文獻報導值。而且市售的α-MoO3,其最大吸附容量 與單位面積最大吸附容量,分別僅有 21.4 mg/g 及 17.6 mg/m2,顯示本研 究中合成出的氧化鉬其吸附效果遠遠高於市售的α-MoO3。當材料的表面 電位為負值時,對於亞甲藍離子或是其他陽離子染料,容易產生庫倫引 力,有利於吸附。若表面電位越低,則庫倫引力越大,吸附量也就越大, 但會趨近平穩,如圖 4-15。本研究中所有產物的比表面積雖小,但其表 面電位低,且再加上氧化鉬本身的特性,因此有利於提升吸附亞甲藍離 子之效果。
54 表 4-4 各文獻之比表面積與吸附量整理表。 材料 比表面積 (m2/g) 最大吸附容量 (mg/g) 單位面積最大 吸附容量 (mg/m2) 文獻 活性碳 761 486 0.6 1 粉煤灰 440 70.4 0.2 5 沸石 490 67.3 0.1 8 MnO2 70.7 63.4 0.9 11 Mn3O4 53.8 68.4 1.3 11 TiO2 242 196.8 0.8 9 WO2/W18O49 114 138.9 1.2 14 鈦酸鹽 158 133.3 0.9 36 W18O49 223 201 0.9 37 h-WO3 27.83 75 2.7 38 N-doped H2W1.5O5.5·H2O 7.4 376.9 50.9 39 WO3·0.33H2O 78.4 247.3 3.2 40 α-MoO3標準品 1.22 21.4 17.6 本研究 MoO-10 0.87 226.2 260.0 本研究 MoO-30 1.19 558.7 469.5 本研究 MoO-90 1.92 565.0 294.3 本研究 MoO-10L 15.44 613.5 39.7 本研究 MoO-30L 14.07 602.4 42.8 本研究 MoO-90L 12.25 581.4 47.5 本研究 註: 單位面積最大吸附容量皆為本研究經過計算所得之數據。
55
4.3.4 表面電位對於不同染料之影響
為了進一步探討表面電位對於染料吸附的影響,故在此也進行了不 同染料的吸附。利用 MoO-90L 分別與亞甲藍 (陽離子型)、羅丹明 B (陽 離子型)、日落黃 (陰離子型) 和甲基藍 (陰離子型) 進行吸附,染料之結 構圖如圖 4-16 及圖 4-17。MoO-90L 對於亞甲藍、羅丹明 B、日落黃和甲 基藍之吸附量分別為 550.8、71.8、16.1 和 18.3 mg/g,如圖 4-18。由實驗 結果可知,當氧化鉬與陰離子型的染料進行吸附時,由於同性電相斥的 性質,其吸附效果相當差。反觀,當氧化鉬與陽離子型的染料進行吸附 時,由於異性電相吸的性質,其吸附效果遠遠高於陰離子型的染料。羅 丹明 B 的吸附量為 71.8 mg/g,與陰離子型的染料比較,可高達 4 倍以上 的吸附量,而亞甲藍的吸附量為 550.8 mg/g,更是高達 30 倍以上的吸附 量,由此顯示氧化鉬對於陽離子型的染料有良好的吸附效果,對於亞甲 藍更有專一性的吸附表現。在此也證實,氧化鉬的表面電位對於陰離子 型及陽離子型染料的吸附效果,有明顯的差異。 圖 4-16 陽離子型染料之結構圖。56
圖 4-17 陰離子型染料之結構圖。
圖 4-18 MoO-90L 對於不同染料之吸附量關係圖。 (MoO-90: 10 mg,染料: 0.050 mM, 500mL)
57
第五章
結論
本實驗成功利用微波法,合成出兩個不同晶相之氧化鉬。並探討在 起始物濃度不同與反應時間不同時,對於產物的影響。最後,將所得產 物應用於亞甲藍染料之吸附,可得到高吸附的效果。 將鉬酸銨溶液與硝酸以微波法合成氧化鉬。在 0.04 M 鉬酸銨溶液的 環境下,反應時間為 10 至 90 分鐘,產物結構皆為 h-MoO3,但隨著反應 時間的增加,其形狀則由錐狀轉變為柱狀。然而,在較低濃度 0.01 M 鉬 酸銨溶液的環境下,會有利於每單位的起始物,在加熱的過程中獲得更 多的能量。反應時間為 10 至 90 分鐘,隨著反應時間的增加,產物結構 從 h-MoO3轉變α-MoO3,其形狀也由柱狀轉變為帶狀。 最後,將所得產物應用於亞甲藍染料之吸附。發現氧化鉬之比表面 積遠低於其他材料,但對於亞甲藍的吸附卻有更佳卓越的效果,因此認 為比表面積並非影響吸附效果的主因。進一步研究發現,表面電位為影 響最大吸附容量的一大因素。當表面電位越低時,則庫倫引力越大,其 最大吸附容量也就越大。其中 MoO-10L 可得到 613.5 mg/g 的最大吸附容 量,而 MoO-30 則具最大的單位面積最大吸附容量 469.5 mg/m2,與其他 材料的文獻比較,本實驗所得的氧化鉬具有高效能的表現。預期此產物 未來將可應用於染整工業的廢水處理,以符現代社會的綠色及環保概 念。58
第六章
參考文獻
1. Yamashita, J.; Shioya, M.; Kikutani, T.; Hashimoto, T., Carbon 2001, 39, 207. 2. Hernandez-Ramirez, O.; Holmes, S. M., J. Mater. Chem 2008, 18, 2751.
3. Sabio, E.; González, E.; González, J. F.; González-García, C. M.; Ramiro, A.; Gañan, J., Carbon 2004, 42, 2285.
4. Bhatnagar, A.; Vílar, V. J. P.; Botelho, C. M. S.; Boaventura, R. A. R., Environ.
Technol. 2011, 32, 231.
5. Wang, S.; Boyjoo, Y.; Choueib, A.; Zhu, Z. H., Water Res. 2005, 39, 129.
6. Bagane, M.; Guiza, S.; Ann. Chim., Annales de Chimie Science des Matériaux 2000,
25, 615.
7. Wang, S.; Ma, Q.; Zhu, Z. H., Fuel 2008, 87, 3469.
8. Wang, S.; Li, H.; Xu, L., J. Colloid Interface Sci. 2006, 295, 71. 9. Wang, R.; Cai, X.; Shen, F., Appl. Surf. Sci. 2014, 305, 352. 10. Cao, S.-W.; Zhu, Y.-J., J. Phys. Chem. C 2008, 112, 6253. 11. Chen, H.; He, J., J. Phys. Chem. C 2008, 112, 17540.
12. Yu, X.-F.; Liu, J.-W.; Cong, H.-P.; Xue, L.; Arshad, M. N.; Albar, H. A.; Sobahi, T. R.; Gao, Q.; Yu, S.-H., Chem. Sci. 2015, 6, 2511.
13. Zhao, J.; Tana, Y.; Su, K.; Zhao, J.; Yang, C.; Sang, L.; Lu, H.; Chen, J., Appl. Surf.
Sci. 2015, 337, 111.
14. Jeon, S.; Yong, K., J. Mater. Chem. 2010, 20, 10146.
15. Luo, J. Y.; Cao, Z.; Chen, F.; Li, L.; Lin, Y. R.; Liang, B. W.; Zeng, Q. G.; Zhang, M.; He, X.; Li, C., Appl. Surf. Sci. 2013, 287, 270.
59
16. Chiang, T. H.; Chen, M. Y.; Li, M. H.; Yen, M. Y., J Mater Sci 2013, 48, 6994. 17. Liu, T.; Li, B.; Hao, Y.; Yao, Z., Chem. Eng. J. 2014, 244, 382.
18. Manivel, A.; Lee, G.-J.; Chen, C.-Y.; Chen, J.-H.; Ma, S.-H.; Horng, T.-L.; Wu, J. J., Mater. Res. Bull. 2015, 62, 184.
19. Y.F.Zhou; K.Bi; L.Wan; X.Ji; C.Wen; K.Huang; C.Liang; Z.B.Sun; D.Y.Fan; Yang, H. J.; Y.G.Wang; M.Lei, Mater. Lett. 2015, 154, 132.
20. Ma, Y.; Jia, Y.; Jiao, Z.; Wang, L.; Yang, M.; Bi, Y.; Qi, Y., Mater. Lett. 2015, 157, 53.
21. Chithambararaj, A.; Bose, A. C., Beilstein J. Nanotechnol. 2011, 2, 585.
22. Chithambararaj, A.; Sanjini, N. S.; Bose, A. C.; Velmathi, S., Catal. Sci. Technol.
2013, 3, 1405.
23. Chithambararaj, A.; Rameshbabu, N.; Bose, A. C., Sci. Adv. Mater. 2014, 6, 1. 24. Pan, W.; Tian, R.; Jin, H.; Guo, Y.; Zhang, L.; Wu, X.; Zhang, L.; Han, Z.; Liu, G.;
Li, J.; Rao, G.; Wang, H.; Chu, W., Chem. Mater. 2010, 22, 6202. 25. Cai, L.; Rao, P. M.; Zheng, X., Nano Lett. 2011, 11, 872.
26. Martínez, H. M.; J.Torres; M.E.Rodríguez-García; Carreño, L. D. L., Physica B
2012, 407, 3199.
27. Klinbumrung, A.; Thongtem, T.; Thongtem, S., Journal of Nanomaterials 2012, 2012, 1.
28. Ramana, C. V.; Atuchin, V. V.; Troitskaia, I. B.; Gromilov, S. A.; Kostrovsky, V. G.; Saupe, G. B., Solid State Commun. 2009, 149, 6.
60
30. Bai, S.; Chen, S.; Chen, L.; Zhang, K.; Luo, R.; Li, D.; Liu, C. C., Sens. Actuators, B
2012, 174, 51.
31. Kim, W.-S.; Kim, H.-C.; Hong, S.-H., J. Nanopart. Res. 2010, 12, 1889. 32. Song, J.; Ni, X.; Gao, L.; Zheng, H., Mater. Chem. Phys. 2007, 102, 245.
33. Hashem, A. M.; Groult, H.; Mauger, A.; Zaghib, K.; Julien, C. M., J. Power Sources
2012, 219, 126.
34. Gou, Z.; Liu, T.; Zeng, W.; Yu, W.; Chen, Y., J. Mater. Sci. - Mater. Electron. 2013,
24, 1018.
35. Osaka, M.; Tanaka, K.; Sekine, S.; Akutsu, Y.; Suzuki, T.; Mimura, H., J. Nucl.
Mater. 2012, 427, 384.
36. Xiong, L.; Yang, Y.; Mai, J.; Sun, W.; Zhang, C.; Wei, D.; Chen, Q.; Ni, J., Biochem.
Eng. J. 2010, 156, 313.
37. Gao, X.; Xiao, F.; Yang, C.; Wang, J.; Su, X., J. Mater. Chem. A 2013, 1, 5831. 38. Li, J.; Liu, X.; Han, Q.; Yao, X.; Wang, X., J. Mater. Chem. A 2013, 1, 1246. 39. Zhu, R.; Cong, S.; Tian, Y.; Li, H.; Chen, M.; Huang, Y.; Zhao, Z.; Li, Q., Chem.
Commun. 2013, 49, 5787.
40. Liu, B.; Wang, J.; Wu, J.; Li, H.; Li, Z.; Zhou, M.; Zuo, T., J. Mater. Chem. A 2014, 2, 1947.
41. Komarneni, S.; Rajha, R. K.; Katsuki, H., Mater. Chem. Phys. 1999, 61, 50. 42. Liqiang, J.; Yichun, Q.; Baiqi, W.; Shudan, L.; Baojiang, J.; Libin, Y.; Wei, F.;
Honggang, F.; Jiazhong, S., Sol. Energy Mater. Sol. Cells 2006, 90, 1773. 43. Revil, A. Water Resour. Res. 2012, 48, 1.
61
44. Kumar, V.; Wang, X.; Lee, P. S., CrystEngComm 2013, 15, 7663.
45. Atuchin, V. V.; Gavrilova, T. A.; Kostrovsky, V. G.; Pokrovsky, L. D.; Troitskaia, I. B., Neorg. Mater. 2008, 44, 714.
46. Dieterle, M.; Weinberg, G.; Mestl, G., Phys. Chem. Chem. Phys. 2002, 4, 812. 47. Song, J.; Ni, X.; Zhang, D.; Zheng, H., Solid State Sciences 2006, 8, 1164. 48. Wei, G.; Qin, W.; Zhang, D.; Wang, G.; Kim, R.; Zheng, K.; Wang, L., J. Alloys
Compd. 2009, 481, 417.
49. Chen, Y.; Lu, C.; Xu, L.; Ma, Y.; Hou, W.; Zhu, J.-J., CrystEngComm 2010, 12, 3740.
50. Vadivelan, V.; Kumar, K. V., J. Colloid Interface Sci. 2005, 286, 90.
51. Hameed, B. H.; Ahmad, A. L.; Latiff, K. N. A., Dyes and Pigments 2007, 75, 143. 52. Basnet, P.; Zhao, Y., J. Mater. Chem. A 2014, 2, 911.




![TraditionalMLCalgorithmsmainlytacklethebatchMLCproblem,wheretheinputdataarepresentedinabatch[24,28].Nevertheless,inmanyMLCapplicationssuchase-mailcategorization[22],multi-labelexamplesarriveasastream.Onlineanalysisistherefore dimensionreducermotivatedbyma](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)