I
非鉑系觸媒應用在鹼性燃料電池中氧氣還原
反應之研究
學生:張瀠方
指導教授:林鵬 博士
吳樸偉 博士
國立交通大學 材料科學與工程研究所碩士班中文摘要
發展非鉑系觸媒催化氧氣還原反應之研究對於燃料電池普及化 與便利化具有極重大的影響。本研究分為三個主軸進行,首先以不同 Ru 比例之鈣鈦礦氧化物(La0.6Ca0.4CoxRu1-xO3)做為氧的氧化與還原反 應之雙效催化劑。另一方面,合成不同深寬比的棒狀奈米銀結構與不 同碳厚度的立方體奈米銀(Ag@C)結構,分別探討其形狀對於催化氧 氣還原反應之影響。 在鈣鈦礦氧化物觸媒部分,利用固態燒結法合成不同 Ru 比例之 鈣 鈦 礦 氧 化 物,經 由 XRD 證 實 成 功 的 將 不 同 Ru 比 例 參 雜 至 La0.4Ca0.6CoO3鈣鈦礦氧化物中,其元素組成 EDX 分析結果亦與合成 時比例相符。接著,利用充、放電極化曲線分析不同 Ru 比例的鈣鈦II 礦氧化物之催化活性,最後,量測長時間定電流充放電測試確認所合 成的雙效觸媒有其實際應用的可行性。研究結果 La0.6Ca0.4Co0.4Ru0.6O3 觸媒在空氣燃料電池電極中,具有最佳雙效性氧化還原催化特性。 接下來,棒狀奈米銀觸媒部分,利用晶種合成法合成。由 TEM 結果確認藉由控制晶種體積成功的合成不同深寬比的奈米銀結構。經 由電化學分析,深寬比越大之奈米銀結構對於催化氧氣還原反應具有 較佳之效果,進一步利用旋轉電極分析氧氣還原反應的路徑,確認棒 狀奈米銀結構催化氧氣還原之途徑為 4 電子反應路徑。 最後,立方體奈米銀觸媒部分則利用水熱法合成,藉由改變溫度 與反應時間來合成高產率立方體奈米銀結構,經由 TEM 與 SEM 圖 譜確認合成 95%以上顆粒大小 70 nm 的立方體奈米銀,由 XRD 與 HRTEM 分析探討立方體奈米銀為單晶的 FCC 結構,其表面皆為 {200}。此外,進一步利用兩段式升溫,調控合成出核殼狀與碳顆粒 點綴於立方體奈米銀的結構。最後電化學分析中,以碳顆粒點綴於立 方體奈米銀具有最佳催化效果。
III
Non-platinum Based Electrocatalysts for
Oxygen Reduction Reaction for Alkaline Fuel
Cell
Student:Yin-Fang Chang Advisor:Dr. Pang Lin Dr. Pu-Wei Wu Department of Materials Science and Engineering
National Chiao Tung University
Abstract
Non-platinum based electrochemical active materials were developed to catalyze oxygen reduction reaction in an alkaline electrolyte. First, a variety of
perovskite oxide (La0.6Ca0.4CoxRu1-xO3) was fabricated as the bifunctional catalysts
for both oxygen reduction and evolution reaction. Second, silver nanorods in various
aspect ratio, as well as silver nanocubes (Ag@C) with different carbon overcoats were
synthesized and studied for theis oxygen reduction abilities.
In La0.6Ca0.4CoxRu1-xO3 syntheis, we employed a solid state reaction method to
form oxide powder with distinct ruthenium ratios. X-ray diffraction pattern (XRD) of
the as-synthesized powders exhibited a majority phase identical to that of
La0.6Ca0.4CoO3, indicating successful incorporation of Ru4+ at the Co cation sites, and
Energy Dispersive X-ray Spectrometer (EDX) also exhibited consistent results.
Images from Scanning Electron Microscopy (SEM) on the resulting powders
displayed particles in irregular shape at 100-250 nm in size with substantial
IV
were supported on Black Pearl 2000 (BP2000) and deposited on commercially
available gas diffusion electrodes with a loading of 1.2 mgcm-2. In galvanodynamic
curves, the La0.6Ca0.4CoxRu1-xO3/BP2000 revealed significant enhanced bifunctional
catalytic abilities over those of La0.6Ca0.4CoO3/BP2000. Similar behaviors were
obtained in galvanostatic charging and discharging measurements at current densities
of 10 and 200 mAcm-2 for 10 min. We determined La0.6Ca0.4Co0.4Ru0.6O3 to possess
superior bifunctional catalytic activity for the oxygen reduction and evolution. In
addition, the La0.6Ca0.4Co0.4Ru0.6O3/BP200 delivered stable and sustainable lifetime
behaviors.
In the second part, a seed-mediated growth method in aqueous solution was used
to prepare silver nanorod. Transmission Electron Microscopy (TEM) confirmed that
various aspect ratios of silver nanorod structures were successfully synthesized by
controlling the volume of seeds. The galvanodynamic curves demonstrated that the
oxygen reduction was improved with increasing the aspect ratio of silver nanorods.
Similar behaviors were obtained in galvanostatic discharging measurements at current
densities of 10 and 200 mAcm-2 for 10 min. The mechanism for the ORR catalyzed by
silver nanorods was identified through a four-electron reaction path by rotating disk
electrode.
Hydrothermal synthesis was used to produce silver nanocubes. Varied by
processing temperature and time, the production yield for the silver nanocubes were
rather high. From TEM and SEM images, 95% of silver nanocubes were particles in
70 nanometers. In addition, they exhibited a face-centered cubic (FCC) lattice on
{200} facet at the surface. Furthermore, carbon decorated silver nanocube in a
core-shell structure was also formed by a two-stage in hydrothermal synthesis. We
observed that the silver nanocube with core-shell structure displayed an improved
V
致謝
兩年的研究生生活就在轉眼間過去了,回顧這 700 多天的日子, 受到了許多人的幫助與指導,使自己可以帄安順遂的碩士班畢業,首 先要感謝兩位指導教授:林鵬教授、吳樸偉教授,在這兩年的研究生 活中給予無私的指導與幫助,讓我體會到正確的學習態度與培養邏輯 思考能力,讓我受益良多。感謝我的父母、家人給予我經濟與心靈上 的支持,讓我無後顧之憂的進行研究。感謝高嘉駿學長、張雲閔學長、 謝育淇學長、葉耕余學長在實驗上給予的建議與討論,謝謝你們這兩 年的幫助與陪伴,讓這兩年的碩士班研究可以帄安順利的完成。也要 感謝陪伴我度過這兩年研究生涯的同伴:梁茹夢、李佳勳、陳佑慈、 陳境妤、賴俊翰同學們,在這兩年與你們一同努力、一同生活,要跟 你們說一聲:「認識你們真好」。另外還有陳致源學弟、張詠策學弟謝 謝你們忍受我們壓力大下無理取鬧的要求,也謝謝你們的努力與幫忙。 此外,還要特別感謝前來口試的口試委員們,周長彬教授、朝春光教 授、王禎翰教授們給予的指導與建議,讓論文內容更為周全完整。 受到太多人的幫助,要感謝的人也太多了,誠摯的向大家說聲謝 謝,因為有大家的存在與幫助讓我可以順利度過碩士班生涯,讓我有 足夠的勇氣面臨人生下一個階段的考驗,只能再次說聲謝謝,有你們 真好。VI
目錄
中文摘要
Ⅰ英文摘要
Ⅲ致謝
Ⅴ主目錄
Ⅵ圖目錄
Ⅹ表目錄
XV第一章
前言
1 1-1 研究背景 1 1-2 研究動機 2第二章
文獻回顧
4 2-1.燃料電池 4 2-1.1 燃料電池簡介 4 2-1.2 燃料電池分類 5 2-1.3 鹼性燃料電池簡介 8 2-1.4 鹼性燃料電池工作原理 9 2-2.電極動力學 12 2-2.1 電池極化反應現象 12VII 2-2.2 氫氣氧化反應 13 2-2.3 氧氣還原反應 13 2-3 氧化物之催化劑 19 2-3.1 金屬氧化物 20 2-3.2 鈣鈦礦氧化物 21 2-4 奈米結構銀之合成 24 2-4.1 polyol process 合成 24 2-4.2 光還原合成法 26 2-4.3 晶種成長還原法 27 2-4.4 水熱法 29
第三章
實驗方法與流程
33 3-1 實驗藥品與設備 33 3-1.1 實驗藥品與氣體 33 3-1.2 實驗設備 34 3-2 實驗流程 35 3-2.1 總實驗流程 35 3-2.2 鈣鈦礦氧化物 36 3-2.3 棒狀奈米銀合成 37 3-2.4 線狀奈米銀合成 38VIII 3-2.5 立方體奈米銀合成 39 3-3 催化劑合成 40 3-3.1 製備 La0.6Ca0.4CoxRu1-xO3 催化劑-固態燒結法 40 3-3.2 棒狀奈米銀結構合成-晶種成長法 41 3-3.3 立方體奈米結構銀合成-水熱法 43 3-4 分析儀器 44 3-4.1 定性分析 44 3-4.2 電化學分析 46
第四章
結果與討論
50 4-1 La0.6Ca0.4CoxRu1-xO3 催化劑對氧氣還原反應催化活性探討 50 4-1.1 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物 XRD 分析 50 4-1.2 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物 SEM 形貌與 EDX 分析 52 4-1.3 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物比表面積測試 55 4-1.4 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物催化劑之對氧氣還原反 應催化活性比較 56 4-1.5 La0.6Ca0.4CoxRu1-xO3催化劑之對氧氣生成反應催化活性比 較 62 4-2 不同深寬比棒狀奈米銀結構對氧氣還原反應之電催化活性 碳討 68IX 4-2.1 不同深寬比棒狀奈米銀結構之電子顯微鏡觀測結果 69 4-2.2 不同深寬比棒狀奈米銀顏色變化 75 4-2.3 不同深寬比棒狀奈米銀之紫外光-可見光吸收光譜分析 77 4-2.4 不同深寬比棒狀奈米銀之電化學測試與催化活性比較 79 4-3 立方體奈米銀對氧氣還原反應之電催化活性碳討 87 4-3.1 立方體奈米銀之合成 87 4-3.2 立方體奈米銀性質分析 96 4-3.3 電化學分析 101
第五章
結論
109第六章
參考文獻
112X
圖目錄
圖 2.1 各種燃料電池簡易示意圖 6 圖 2.2 鹼性燃料電池之示意圖 9 圖 2.3 模電極組 (MEA)示意圖 10 圖 2.4 電池放電極化曲線之示意圖 13 圖 2.5 氧氣還原反應機制示意圖 15 圖 2.6 模擬氧氣吸附在催化劑表面產生還原反應可能進行的反 應路徑 16 圖 2.7 Pauling model 氧氣還原反應機制 17 圖 2.8 Bridge model 氧氣還原反應機制 18 圖 2.9 RuO2結構示意圖 21 圖 2.10 理想鈣鈦礦結構(Perovskite)之單位晶胞示意圖 23 圖 2.11 polyol process 方法合成不同形狀的奈米銀結構 25 圖 2.12 光還原金屬機制示意圖 26 圖 2.13 40W 螢光下照射不同時間之 TEM 圖 27 圖 2.14 不同硝酸銀濃度於 30W 汞燈還原之 TEM 圖譜 27 圖 2.15 晶種成長法之示意圖 28 圖 2.16 晶種成長法棒狀結構成長示意圖 28XI 圖 2.17 不同合成條件下利用晶種成長法合成銀奈米棒之 TEM 圖譜 29 圖 2.18 不同合成條件下利用水熱法合成不同形狀結構奈米銀 31 圖 3.1 全部實驗流程圖 35 圖 3.2 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物合成流程圖 36 圖 3.3 棒狀奈米銀合成流程圖 37 圖 3.4 線狀奈米銀合成流程圖 38 圖 3.5 立方體奈米銀合成流程圖 39 圖 3.6 電化學裝置 47 圖 3.7 電化學裝置 48 圖 3.8 電化學裝置 49 圖 4.1 不同比例 La0.6Ca0.4CoxRu1-xO3之 XRD 比較圖 51 圖 4.2 不同 Ru 比例鈣鈦礦氧化物與晶格常數趨勢圖 52 圖 4.3 La0.6Ca0.4CoxRu1-xO3 之 SEM 影像 54 圖 4.4 La0.6Ca0.4Co0.2Ru0.8O3 SEM 影像與 EDX 結果 54 圖 4.5 La0.6Ca0.4Co0.2Ru0.8O3 EDX 之 mapping 結果 55 圖 4.6 La0.6Ca0.4CoxRu1-xO3氧氣還原 IV-極化曲線圖 57 圖 4.7 La0.6Ca0.4Co0.4Ru0.6O3催化劑與市售的催化劑極化曲線圖 59 圖 4.8 La0.6Ca0.4Co1-xRuxO3定電流放電測試 61
XII 圖 4.9 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物充電反應極化曲線圖 63 圖 4.10 La0.6Ca0.4Co1-xRuxO3鈣鈦礦觸媒之定電流測試圖 65 圖 4.11 La0.6Ca0.4Co0.4Ru0.6O3長時間充放電測試圖 67 圖 4.12 La0.6Ca0.4Co0.4Ru0.6O3長時間充放電電化學測試前後之 SEM 圖 68 圖 4.13 銀晶種溶液之 HRTEM 圖譜 70 圖 4.14 不同深寬比奈米棒狀銀之 TEM 圖譜 71 圖 4.15 線狀奈米銀結構之 SEM 圖譜 72
圖 4.16 棒狀奈米銀 HRTEM 和 SAED pattern 分析圖譜 74 圖 4.17 棒狀奈米銀 EDX 分析結果 75 圖 4.18 晶種溶液與不同深寬比奈米銀成長溶液顏色照片 76 圖 4.19 不同深寬比奈米棒狀銀之 UV-VIS 吸收光譜圖 78 圖 4.20 不同深寬比奈米棒狀銀氧氣還原反應極化曲線圖 80 圖 4.21 晶種體積為 0.5 mL 定電流放電測試 81 圖 4.22 晶種體積為 0.13mL 定電流放電測試 82 圖 4.23 晶種體積為 0.06 mL 定電流放電測試 82 圖 4.24 線狀奈米銀之定電流放電測試 83 圖 4.25 奈米棒狀銀催化劑於不同轉速下 RDE 測試 ORR 反應之 陰極極化曲線圖 85
XIII 圖 4.26 於不同電壓下,奈米棒狀銀催化劑之 Koutecky-Levich plot 86 圖 4.27 120℃下反應時間 8 小時之 SEM 與 TEM 圖譜 88 圖 4.28 120℃下反應時間為 10 小時之 TEM 圖譜 89 圖 4.29 120℃下反應時間 18 小時之 HRTEM 與 SEM 圖譜 89 圖 4.30 合成溫度 120℃下,不同反應時間之 TEM 與 HRTEM 圖 譜 92 圖 4.31 合成溫度 140℃下,不同反應時間之 TEM 與 HRTEM 圖 譜 93 圖 4.32 合成溫度 160℃下,不同反應時間之 TEM 與 HRTEM 圖 譜 93 圖 4.33 合成條件 120℃加熱 18 小時後,再升溫至 160℃加熱 2 小時後之 TEM 與 HRTEM 圖譜 95 圖 4.34 合成條件 120℃加熱 18 小時後,再升溫至 160℃加熱 4 小時後之 TEM 與 HRTEM 圖譜 95 圖 4.35 立方體奈米銀結構之 XRD 分析 97 圖 4.36 立方體奈米銀之 UV-VIS 吸收光譜圖 98
圖 4.37 立方體奈米銀之 HRTEM 圖譜、SAED 分析與 EDX 成分
分析 100
XIV 圖 4.38 立方體奈米銀之 HRTEM 分析圖譜與程式模擬轉換後 之圖譜 101 圖 4.39 立方體奈米銀於不同催化劑量下陰極極化曲線圖 103 圖 4.40 立方體奈米銀電化學測試前後之 TEM 與 HRTEM 圖譜 104 圖 4.41 不同碳化時間的立方體奈米銀觸媒 HRTEM 圖譜 105 圖 4.42 不同碳化時間的立方體奈米銀觸媒陰極極化曲線圖 106 圖 4.43 線狀結構與不同碳膜形貌立方體結構陰極極化曲線比較 圖 107 圖 4.44 不同結構質量活性極化曲線圖比較圖 108
XV
表目錄
表 4.1 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物之晶格常數 52 表 4.2 La0.6Ca0.4Co0.2Ru0.8O3元素組成 55 表 4.3 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物比表面積 56 表 4.4 La0.6Ca0.4CoxRu1-xO3放電反應電壓差異值 57 表 4.5 La0.6Ca0.4Co0.4Ru0.6O3定電流放電測試輸出電壓 60 表 4.6 La0.6Ca0.4Co0.4Ru0.6O3充電反應電壓差異值 63 表 4.7 不同深寬比奈米棒狀銀 i-V 極化曲線輸出電壓差異表 80 表 4.8 不同深寬比奈米棒狀銀定電流放電輸出電壓差異表 83 表 4.9 Koutecky-Levich line 之數據 86 表 4.10 反應溫度 120℃下,反應時間對立方體結構之影響 90 表 4.11 不同反應時間與溫度下立方體奈米銀顆粒大小 921
第一章 前言
1-1 研究背景
人類發展歷史中,隨著文明的演進,科技日新月新,能源使用的 量也大幅提升。於二十世紀初開始,石油的應用趨於廣泛,很快成為 主要的能源供給,石化燃料成為現今支配全球能源使用中最重要的來 源。但近幾年來,國際原油價格持續攀升,地球所蘊藏的石油有朝一 日終究會消耗殆盡,石油短缺已逐漸成為不可忽視的議題,而尋找替 代能源取代石化燃料已成為目前科學家研究之主流。而這些石化燃料 的大量使用也影響著地球的自然環境,由於二氧化碳的大量排放所產 生的溫室效應,使得地球氣候異常改變,造成全球暖化、兩極冰川融 化使得海帄面的上升、許多物種瀕臨滅絕等重大環境影響。因此,尋 找潔淨的替代能源與再生能源的發展,以及二氧化碳的減量方法似乎 成為各國政府與科學家研究之主軸。 潔淨的替代能源以低汙染、低二氧化碳排放、低成本以及能源轉 換效率高為發展主軸。其中以燃料電池、太陽能電池、風力發電等研 究最受科學家們高度重視。燃料電池的來源為氫氣,氫氣為零污染環 境的燃料,其陽極產生氫氣氧化反應;陰極產生氧氣還原化學反應提 供電能,其最終產物為水,為目前替代能源中極具發展潛力的一環。 然而追朔燃料電池發展是由十九世紀 William Grove 發明,至今已一2 個世紀的歷史,但是由於價格昂貴、能源轉換率低等因素限制其發展, 使得降低成本成為目前燃料電池研究領域中極為重要的一環。而在鹼 性燃料電池系統中,其催化劑受腐蝕的程度較低,因此可使用非鉑系 觸媒來替代,使得成本有效的降低。 因此,本篇研究的目的為利用非鉑系觸媒應用於鹼性燃料電池 系統中探討陰極氧氣還原反應,並更進一步研究非鉑系觸媒之催化活 性。
1-2 研究動機
鹼性燃料電池為目前燃料電池系統中能源轉換效率最高的 (55 -60%),且氧氣還原反應速率於鹼性條件下較酸性環境迅速,也由 於電解液為鹼性環境,金屬的腐蝕速度相較於酸性溶液下大幅降低許 多。因此,可以使用非鉑系催化劑來取代高價的貴重金屬催化劑,例 如以銀、鎳、鈣鈦礦氧化物、氧化鈷錳等催化劑取代白金觸媒,已達 降低成本的目的。 本篇研究為發展非鉑系觸媒應用於鹼性燃料電池系統中氧氣還 原反應之研究,分為三個主軸進行。第一部分為探討不同 Ru 比例的 鈣鈦礦氧化物 (La0.6Ca0.4Co1-xRuxO3)做為雙效性觸媒之研究,接下來 探討不同深寬比之棒狀奈米銀觸媒其氧氣還原反應之催化活性,最後 研究合成正方體奈米銀結構,進一步探討奈米結構型狀對於氧氣還原3 反應之影響。 首先先以固態燒結法合成不同 Ru 比例之鈣鈦礦氧化物 (La0.6Ca0.4Co1-xRuxO3),研究在不同 Ru 比例之觸媒對於氧氣還原與氧 氣生成反應之影響,探討其做為雙效性觸媒應用於空氣燃料電池之催 化活性。 此外,銀系觸媒在鹼性燃料系統中對氧氣還原反應之影響已於文 獻中大幅被報導,文獻中指出銀系觸媒對於氧氣還原反應有不錯的催 化活性,且相較於高價的貴重金屬(例:鉑)其成本較低。此研究的目 的在於合成不同形狀的奈米銀顆粒探討其在鹼性電解液條件下對於 氧氣還原反應的影響。由於不同的奈米結構有其獨特的特性,例如在 電學、光學、生物標記、催化特性方面等,而目前文獻較少研究探討 關於特殊的銀奈米結構觸媒應用於氧氣還原反應中之影響,因此本篇 研究發展合成不同深寬比的棒狀結構與立方體形狀的奈米銀顆粒為 催化劑,應用於鹼性燃料電池氧氣還原反應中,進一步探討奈米結構 對於催化活性之影響。
4
第二章 文獻回顧
2-1 燃料電池
2-1.1 燃料電池簡介 近年來由於人類大量消耗石化燃料,因其生產及消耗所排放的汙 染物,造成環境的破壞,使得自然環境的復原能力無法負荷,危及人 類世代的永續發展,為了減緩或防止環境被破壞,限制石化燃料使用 量和積極開發再生能源變成人類發展史上不可避免的途徑,但目前在 實用化方面還有許多問題有待克服。而燃料電池因原理簡單且不易受 外在環境影響,逐漸受到大家的重視,近年來燃料電池研究非常熱門。 燃料電池具有以下優點: (1) 能源使用效率高:因燃料電池利用氫與氧的化學反應產生電流, 由化學能直接轉換為電能,由文獻得知其轉換效率約35%左右,為 非常具有潛力的發電系統。 (2) 低汙染發電裝置:燃料電池運作理論上以氫氣為燃料、氧氣為氧 化劑,產物為水,對環境造成的汙染降至最低。且為低噪音的發 電系統,其工作環境極為安靜,燃料電池對環境友好性是其具有 長遠發展性的主要原因。 (3) 穩定、安全的能源供應裝置:燃料電池只要有穩定的燃料供給便 能提供電力。且與內燃機等機械循環系統相比,其轉動構件少,5 系統上更加安全可靠。 燃料電池為一種直接將化學能轉為電能的裝置,電化學反應是伴 隨著電荷轉移的化學作用,電子及離子以不同的反應路徑完成電荷的 轉移。由於此能源直接轉換的特性促使燃料電池具有極高的轉換效率, 燃料通常是氫氣、甲醇、乙醇、天然氣或其他的碳氫化合物,氧化劑 則可以用空氣中的氧氣,而副產物是熱、純水或較少量的二氧化碳。 若要將燃料電池普及化與便利化,其目標在於如何降低售價成本 與使其供電效率足以取代現有電力來源。目前燃料電池瓶頸在於電池 性能的提升、提高電池壽命、替代貴金屬催化劑開發、低成本量產技 術及周邊系統的建立。而燃料電池其低污染的特性,對於目前各國重 視的環保與替代能源開發問題都具有極大的開發潛力,有絕對必要發 展之價值。 2-1.2 燃料電池分類 一般依據其操作溫度範圍不同分為高溫型燃料電池、中溫型燃料 電池以及低溫型燃料電池。結合操作溫度、反應物選用及電解液的不 同,又可區分為固態氧化物燃料電池 (SOFC)、熔融碳酸鹽碳酸鹽燃 料電池 (MCFC)、磷酸燃料電池 (PAFC)、鹼性燃料電池 (AFC)、質 子交換薄膜燃料電池 (PEMFC)等五種如圖2.1 [1]。
6 圖 2.1 各種燃料電池簡易示意圖。 a. 固態氧化物燃料電池 (SOFC): 為高溫型燃料電池,以氧化鈣、氧化鋯固體氧化物為電解質,因 氧化物須於高溫下才有足夠的氧離子傳導性,所以操作溫度約為 500℃~1200℃。陽極部分為鎳與陶瓷的混和材料(Ni-YSZ);陰極 部分目前最廣為使用之觸媒為 LaMnO4。 anode:2O2-2 +2 H2→2H2O +4e- cathode:O2 + 4e- → 2O-2 b. 熔融碳酸鹽碳酸鹽燃料電池 (MCFC): 利用碳酸鉀、碳酸鋰等兩種以上碳酸鹽類混合物作為電解質,當 溫度約為 650℃時,會熔融成液體狀態,產生碳酸根離子,從陰極 流向陽極,與氫結合生成水、二氧化碳和電子。
7 anode:H2+ 2 CO3-2+ CO → 3 CO2+ H2O + 4e -cathode:4e-+ 2 CO2 + O2 → 2 CO3-2 c. 磷酸燃料電池 (PAFC): 利用多孔碳化矽和聚四氟乙烯製備微孔結構隔膜,以液態H3PO4 電解液,故長期運轉下會有腐蝕與電解液漏液的問題,其操作溫 度為150℃~200℃,屬於中溫型燃料電池。其電極是使用白金作為 電催化反應觸媒,不但價格昂貴,而且容易與出現在燃料中的一 氧化碳反應而發生中毒現象,導致發電性能衰退。 d. 鹼性燃料電池 (AFC) 鹼性燃料電池陰、陽極分別為氫氣、氧氣氣體擴散電極,其電解 質為氫氧化鉀鹼性溶液。其電極反應式: anode:2 H2+ 4 OH- → 4 H2O+ 4e -cathode:O2+ 4e-+ H2O → 4 OH -操作溫度約為70℃,屬於低溫型燃料電池。 e. 質子交換薄膜燃料電池 (PEMFC) 又稱為固態聚合物電解質燃料電池,其電解質為質子交換膜。以 質子交換膜研究來說,目前主要突破為杜邦 (Dupont)公司發展出 的Nafion薄膜,主要是以四氟乙烯為基礎構造。此燃料電池為低溫 型燃料電池,其操作溫度約85~105℃。此燃料電池兩電極皆為多
8 孔性的氣體擴散電極,確保氣體可以充分的供應到觸媒層已產生 反應 [2]。 2-1.3 鹼性燃料電池簡介 鹼性燃料電池早期被運用在太空計畫中,是燃料電池在發展初期 第一波引起注意的研究。相較於其他燃料電池,鹼性燃料電池的能量 轉化效率較高,高達55-60%,主要是因為在鹼性電解質中氧氣還原 反應速度較酸性電解質高,因此鹼性燃料電池可以使用非鉑系觸媒, 例如:雷尼鎳、銀、環狀非貴金屬錯合物等。而鹼性燃料電池若是以 氫氧化鉀為電解質,可稱之為鹼液燃料電解質,電解質若以傳導氫氧 離子的高分子模,則稱鹼性膜燃料電池 (AMFC)。圖2.2為鹼性燃料 電池之示意圖。 電極反應式: anode:2 H2+ 4 OH- → 4 H2O+ 4e -cathode:O2+ 4e-+ H2O → 4 OH -總反應:O2 + 2 H2 → 2H2O 鹼性燃料電池優點為操作時所需溫度並不高,大約70℃左右,為 低溫型燃料電池,其轉換效率好,可使用之觸媒種類多價格又便宜, 但由於空氣中的二氧化碳會使電解質產生碳酸化的影響,影響電解質 的品質,導致發電性能衰退。因此若使用空氣為氧化劑,必須先去除
9 空氣中的二氧化碳,造成操作複雜度與成本的上升,且在操作過程中 還必須將電化學反應所生成的水及時排出以維持水的帄衡,這是造成 最近各國燃料電池開發競賽中,無法成為主要開發對象的原因,因此 鹼性燃料電池還須繼續投入研究與開發改善上述缺點,才能由特殊的 太空任務導向使用轉為商業化應用 [2]。 圖 2.2 鹼性燃料電池之示意圖 [3]。 2-1.4 鹼性燃料電池工作原理 燃料電池基本上是由三個基本組建構而成:陽極 (anode)、陰極 (cathode)、以及兩電極之間電解質 (electrolyte),通常由氫氣為燃料、 氧氣為氧化劑。燃料與氧化劑不可混合,而是分開進入燃料電池系統 中,個別在電池的陽極及陰極產生反應。燃料經電化學反應後產生電
10
子,電子經由電極及外線路流向另一電極與氧化劑反應,氧化劑接受 電子後產生反應,並由電池內的電解質傳導離子,形成電池運轉的迴 路。而燃料電池是由這樣的基本組建串連組成較大功率的電極組,燃 料電池基本構造模電極組 (Membrane Electrode Assembly;MEA)如圖 2.3所示。 圖 2.3 模電極組 (MEA)示意圖 [4]。 模電極組主要有五層結構,最外層分別為陽極燃料氣體擴散層、 陰極氧氣擴散層,中間為電解質隔膜,位於電解質隔膜兩側的分別為 陰、陽極觸媒層。 a) 觸媒層:電化學反應皆在觸媒的表面上產生,觸媒表面必須 與電解質接觸,因電化學反應是在兩者的界面產生,故不能 添加太多的疏水層材料。而反應所需的氫離子皆由電解質傳
11 遞,所以如果觸媒與電解質接觸不良,將不利反應的進行。 觸媒層夾在高分子膜與氣體擴散層之間,其製造方法是先將 適量的觸媒與高分子膜的溶液均勻混合,可塗在氣體擴散層 上。 b) 氣體擴散層:使氣體均勻分布擴散進入催化層,並添加疏水 性材料以防止電解液漏液。氣體擴散層的疏水化處理,通常 添加四氟乙烯 (PTFE)於其中,由於四氟乙烯不親水,所以電 極模組的水氣不會凝聚於此,可以避免水氣阻礙氣體擴散, 四氟乙烯亦具有接合劑的功能,其化學性質穩定,不會因電 位操作改變其結構與性質。一般氣體擴散層材料為碳布或碳 紙,兩者須經高溫石墨化,使其具有導電性並增加抗蝕性。 由於電化學反應所需的電子需由氣體擴散層傳遞,故其必須 能傳導電子且導電性好,由於四氟乙烯導電性不佳,所以其 所添加比例須謹慎控制,一般多添加高導電度的碳粉來增加 導電度,來提高電池效能。 c) 隔離膜:其作用為避免陰陽極直接接觸而短路,並同時傳導 離子,所以厚度必須越小越好。 在燃料電池電化學反應發生時,陰極相對於陽極有較大的電 流及電壓損失,因此,陰極電極的優劣扮演極重要的角色,所以
12 一般選擇具有高的比表面積、高孔隙度且能提供穩定的汽液固三 相反應介面作為陰極電極。電極反應步驟為:氣體的溶解→擴散 →吸附→電化學反應→反應物脫離進入溶液。
2-2 電極動力學
2-2.1 電池極化反應現象介紹 當有電流通過電極時,其電極電動勢偏離帄衡值的現象稱極化現 象。在理想狀態下,燃料電池應具備很高的發電效率,但實際上卻只 有 30~60%之間,造成效率降低的主要原因是來自於電極反應所產生 的過電位 (overpotential)或極化 (polarization)現象。理想電極 反應為一帄衡狀態,但過電位存在時,會產生不可逆的電位,導致能 量損失 [5]。 燃料電池發電過程中常伴隨著下列幾種極化現象如圖:(一) 活性極化 (Activation polarization;ηactivation):電極反應過 程中所產生的阻力。由於牽涉到反應物電荷轉移過程的限 制,造成電荷轉移困難,又稱電化學極化。
(二) 歐姆極化 (Ohmic polarization;ηohmic):電解質或是電極的
內電阻使得電解液與電極界面所產生的電位降 (Potential drop)。
13 所產生的濃度梯度導致的電位降,或稱擴散極化。 由上述可知,總電極反應所產生的損失,因此實際所產生的能量 必須減去所損失的能量 [6]。 ion concentrat ohmic activation loss E loss ideal actual E E E 圖 2.4 電池放電極化曲線之示意圖 [7]。
2-2.2 氫氣氧化反應 (Hydrogen oxidation reaction;HOR)
在燃料電池的陽極部分為氫氣氧化反應 (HOR),其反應式為: H2 + 2H2O → 2H3O + + 2e -氫氣為燃料,由氣體擴散層擴散到觸媒與電解質交會的表面,產生氧 化反應,生成電子與質子 (proton)。電子由電極引導到外電路,產生 電流;質子則藉由電解液傳導到陰極。
2-2.3 氧氣還原反應 (Oxygen reduction reaction;ORR)
14
(direct 4-electron pathway)與過氧化物路徑 (peroxide pathway) [8]。 direct 4-electron pathway (4-電子路徑):
a) 酸性條件下: O2 + 4H + + 4e-→ 2H2O E0=1.229 V vs. NHE b) 鹼性條件下: O2 + 2H2O + 4e →4OH- E0=0.401 V vs. NHE peroxide pathway (2-電子路徑): a) 酸性條件下: O2 + 2H + + 2e- → H2O2 E0=0.670 V vs. NHE 過氧化氫再發生還原或是分解 H2O2 + 2H + + 2e- → 2H2O E0=1.770 V vs. NHE 2H2O2 → 2H2O + O2 b) 鹼性條件下: O2 + H2O +2e →HO2 + OH- E0= -0.065 V vs. NHE 過氧化氫再發生還原或是分解 HO2 + 2H2O + 2e → 3OH- E0=0.867 V vs. NHE 2HO2 - 2OH- + O2
氧氣還原反應中直接4-電子路徑 (direct 4-electron pathway)通常 伴隨著許多步驟,其還原步驟中可能伴隨著吸附在催化劑上的過氧化 氫中間產物 (peroxide intermediate),但是這些中間產物不會在水溶液 中發現;而相反的二電子 (peroxide pathway)路徑則會脫附過氧化氫 分子在水溶液中,可以利用旋轉電極 (Rotating Ring Disk Eelectrode) 法來辨別反應中是否有雙氧水等中間產物產生的方法,環電極(ring
15 electrode)用來監測過氧化氫離子是否於圓盤電極(disk electrode)上產 生,此方法常用來判定氧氣還原反應反應的路徑 [9-11]。 而實際上電極表面氧氣還原的反應機構極為複雜,隨著觸媒種類 不同、電池材料、電解質的pH環境相異而有不同的機制。氧氣還原 反應所牽涉到的反應機制如圖2-5所示大致上可以分為氧氣由溶液中 擴散至電極表面,吸附至活性觸媒表面與電解質交會的界面上,接著 吸附之反應物進行電荷轉移步驟 (charge transfer),氧氣接受電子發生 還原反應成生成物,生成物進一步還原或是分解後擴散至電解液中。 圖 2.5 氧氣還原反應機制示意圖 [12]。 Yeager總結文獻提出三種模式,模擬氧氣吸附在催化劑表面產生 還原反應可能進行的反應路徑,如圖2.6所示:
16 圖2.6 模擬氧氣吸附在催化劑表面產生還原反應可能進行的反應路 徑 [12]。 a) Griffiths model:氧氣分子的π軌域和催化劑的空dz2軌域互 相產生影響。若催化劑和氧氣分子間吸附鍵結越強,則造 成O-O鍵長變長,O-O鍵結變弱,可能導致O-O斷鍵。為圖 2.6中pathwayⅠ。 b) Pauling model:氧氣分子吸附在催化劑表面上,以end-on 的形式出現,部分的電子轉移產生過氧化物的中間產物。 在此模式下產物為水的為4-電子反應;產物為過氧化氫的 為2-電子反應。為圖2.6中pathwayⅡ。Evans、Tseung和Bevan
17 提出氧分子有可能會吸收熱能而分解為氧原子,氧分子會 先偽分離 (pseudo-splitting)形成氧原子,並吸附在電極表 面上:而後在kink site的氧原子和水分子進行還原反應。如 圖2.7所示 圖 2.7 Pauling model氧氣還原反應機制 [12]。 c) Bridge model:為圖2.6中pathwayⅢ。Goldstein和Tseung表 示兩個氧原子都化學吸附在催化劑表面上,以side-on的形 式出現。由於在鹼性電解液中,催化劑表面會吸附很多OH -離子,氧分子通過擴散層吸附在電極表面上且從外界電路 進行部分電子轉移,以單鍵鍵結成O-O (Bridge model),
18 上有晶格缺陷或不純物產生易形成kink site (高催化位),而 在kink site的OH-則容易和鄰近的水分子形成氫鍵,電子傳 遞而將O-O鍵和水分子打斷,最後則會打斷OH-鍵並在電 極表面上重新排列。如圖2.8所示。 圖 2.8 Bridge model氧氣還原反應機制 [12]。 在電化學反應發生時,陰極部分的氧氣還原反應相對於陽極有較 大的電流與電壓的損失,因此陰極電極的優劣扮演極重要的角色,而 氧氣還原反應中電子轉移步驟為速率決定步驟,如何找出加速其反應 的催化劑一直是目前大家致力研究的目標,目前氧氣還原反應的催化 劑大致可分為金屬 (例如:Pt [13],Ag [14-18]等)、混和金屬合金 (例
19
如:Pt-Ni,Pt-Pd等)、金屬氧化物 (metal oxides),例如RuO2 [19-20]、
MnO2、CoO,等等),鈣鈦礦化合物perovskites (LaCoO3、La0.6Ca0.4CoO3 [21-24]) ,尖晶石化合物spinels(NixAl1-xMn2O4、Ni2Co2O4、 Mn3xCo3-3xO4),以及金屬螯合物型催化劑pyrolyzed N-4 chelate compound (CoTMPP)。而其中貴重金屬催化劑多半屬於四個電子轉移 的直接還原反應,如Pt、Pd、Ag等等。而過渡金屬氧化物與一般的碳 材多屬於二電子的雙氧水間接反應,如石墨、碳黑、二氧化錳、氧化 鈷,另外包括金屬螯合物催化劑。由於氧氣還原反應相較於陽極有較 大的電流電壓、損失,目前許多科學家投入大量時間、新血研究,尋 找高效能的催化劑已達改善燃料電池效能,更進一步將燃料電池實用 化。
2-3 氧化物之催化劑
目前有許多文獻報導可以用來加速氧氣還原反應的催化劑有許 多種,分為金屬、混和金屬合金、金屬氧化物、鈣鈦礦化合物、尖晶 石化合物,以及金屬螯合物型催化劑,其中還原效率最高的為鉑或鉑 的合金,但由於白金的價格過於昂貴,使得研究趨向於尋找能替代白 金且具有高催化活性的催化劑。鈣鈦礦化合物 (perovskites)對許多反 應都有其特殊的催化活性,特別在氧氣活化部分,因此常應用於燃料 電池催化劑。La0.6Ca0.4CoO3-x鈣鈦礦結構的氧化物在氧氣還原反應中,20 已被了解具有良好的催化性質,且可以減少氧氣還原與生成反應的過 電壓,作為雙功能性電極已是文獻報導中氧氣還原與生成反應良好的 催化劑 [21-24]。近年來,S. Trasatti等人研究發現RuO2金屬氧化物在 電化學反應中,有卓越的造氧能力 [25]。因此本篇研究嘗試以鈣鈦 礦結構之鑭鈣鈷氧化物摻雜釕金屬,期待發展雙功能性空氣陰極電極, 對於氧的氧化與還原皆有良好電化學效果。
2-3.1
金屬氧化物
鈣鈦礦 (perovskite)、尖晶石 (spinel)、綠燒石 (pyrochlores)、金 屬氧化物催化劑等皆為目前文獻報導中與白金觸媒相比較為便宜且 對於氧氣還原反應也具有高催化活性的觸媒 [26]。
a) 尖晶石 (spinel):為AB2O4 結構,目前常見有Co3O4、NiCo2O4。
b) 綠燒石 (pyrochlores):為A2B2O7 結構,可分為有參雜 (A2B2-xBxO7) 和無參雜兩種 (A2B2O7)。
c) 金屬氧化物:主要為過渡金屬氧化物。NiO+Li2O、Co2O3、RhO3、
Ni-MnO2、Co3O4、RuO2等。S. Trasatti等人研究發現RuO2金屬氧化
物在電化學反應中,有卓越的造氧能力,將RuO2和TiO2混合為催
化劑在造氧方面有其獨特的催化性 [25]。圖2.9為RuO2結構示意圖
21 圖 2.9 RuO2結構示意圖 [27]。 2-3.2 鈣鈦礦氧化物 在各類的催化劑中,鈣鈦礦氧化物的研究相當受到重視,鈣鈦礦 結構的氧化物在氧氣還原中,已逐漸被了解具有良好的催化性質,其 結構為ABO3,並可能摻雜不等比例不同金屬於A或B中,來增加整體 導電度。圖2.10為理想鈣鈦礦結構 (Pervoskite)之單位晶胞示意圖 [28],A 是離子半徑較大之陽離子,通常為鹼土金屬類,例如:La、 Ca、Sr等,A 離子在單位晶胞中心;B 是離子半徑較小之陽離子, 通常為過渡金屬,例如:Co、Mn、Fe、Cu等;B 離子在八個角落, X 則是陰離子,在六個面心上;a 是晶格長度。可以分為有參雜 (doping)其他金屬元素與無參雜其他元素兩類。無參雜其他元素之鈣
鈦礦常見有ANiO3 (A=La、Nd、Pr、Sm、Eu)、LaBO3 (B=Mn、Co、
22
a) AA’BO3:常見有 La1-xCaxCoO3、La1-xLnxO3。
b) ABB’O3:例如:LaNi1-xMO3 (M=Fe、Co、V)。
c) AA’BB’O3:例:La1-xSrxCo1-yFeyO3。
常見的合成方法可分為檸檬酸鹽法 (Amorphous Citrate Precursor; ACP) 與固態燒結法 (Solid State Reaction Method,;SSRM)。
1.檸檬酸鹽法 (ACP) [29]:1967 年,Pechini學者發表提出,利用含 多官能基的醇類,例:乙二醇 (Ethylene glycol),以水溶液中檸檬酸 (Citric acid)分子的官能基來螫合鍵結金屬離子, 藉由加熱進行聚酯 化反應使其形成各成份陽離子皆均勻且為原子級,類似凝膠,然後再 經過加熱分解去除有機成份後,形成單相之金屬氧化物。只藉由檸檬 酸而不加入乙二醇,仍然可以獲得透明澄清的凝膠,許多金屬離子可 與檸檬酸生成配位,其先配製含金屬離子的檸檬酸鹽前驅化合物,再 經乾燥、加熱可製得金屬氣化物粉體。此種製程粉末的顆粒大小較均 一、純度高且製程簡單,可大量生產具開發潛力。
2.固態燒結法 (Solid State Reaction Method;SSRM):
固態燒結法為主體材料和含有摻雜雜質之前驅物,依所需比例混 合研磨均勻後,將其在低於樣品熔點的溫度條件下,進行燒結過程之 熱處理,此製成方法極為簡單且被廣為應用。而反應物的化學組成與 晶體結構是決定燒結過程中反應速率的重要因素之一。
23 目前LaCoO3參雜Ca的鈣鈦礦氧化物在氧氣還原反應中,已逐漸 被了解具有良好的催化性質,亦是在造氧方面也被研究學著廣為探討 的催化劑,目前La0.6Ca0.4CoO3鈣鈦礦氧化物已被認可在鹼性環境下可 作為雙效性空氣燃料電池的催化劑 [21-24]。 本實驗利用固態燒結法依莫爾比例分別秤取硝酸鑭、硝酸鈣、硝 酸鈷、氧化釕粉末均勻混合利用研缽研磨均勻製成,嘗試改變硝酸鈷 與氧化釕之比例製成不同比例之鈣鈦礦氧化物,以合成鈣鈦礦結構之 鑭鈣鈷氧化物摻雜釕金屬。嘗試以鈣鈦礦結構之鑭鈣鈷氧化物摻雜釕 金屬,主要目的為開發一種具有對於氧的氧化與還原皆有良好電化學 效果之新型雙效型鈣鈦礦結構觸媒,並製作成陰極空氣電極,以分析 其雙效性能。 圖 2.10 理想鈣鈦礦結構(Perovskite)之單位晶胞示意圖 [28]。
24
2-4 奈米結構銀合成
目前有許多文獻報導用來加速氧氣還原反應的催化劑有許多種, 其中還原效率最高的為鉑或鉑的合金,但由於白金的價格過於昂貴且 對反應的選擇性低等缺點,使得研究趨向於尋找能替代白金且具有高 催化活性的催化劑。而銀系觸媒相較於白金成本較為低廉,且有不錯 的催化效果。奈米銀顆粒對於催化氧氣還原反應已有許多文獻報導指 出有不錯的催化效果,而目前文獻較少研究探討關於特殊的銀奈米結 構應用於催化氧氣還原反應的影響,由於特殊的奈米結構有其獨特的 特性,例如在電學、光學、生物標記、催化特性方面等,因此本篇研 究合成不同形狀奈米銀顆粒做為催化劑,應用於鹼性燃料電池氧氣還 原反應中,進一步探討其結構對於催化活性之影響。而常見的合成不 同結構奈米銀顆粒方法有Polyol process合成、光還原法、晶種成長法、 水熱法等。 2-4.1 Polyol process 合成利用PVP (poly vinyl pyrrolidone)為包覆劑,乙二醇 (ethylene glycol)為溶劑加熱還原金屬離子的一種化學還原方法。實驗架構主要
將AgNO3和PVP之 ethylene glycol溶液分別以緩慢、固定的速率加入
至溫度為140-160℃ ethylene glycol 溶液中,利用ethylene glycol 為還 原劑,其隨著溫度升高還原力增強。可以藉由控制反應時間、硝酸銀
25
及PVP濃度、比例改變、反應溫度不同,還原成不同形狀的奈米銀顆 粒。由文獻指出利用此方法可以獲得高產率的不同形狀奈米銀結構 [30-33]。圖2.11為利用此方法合成不同形狀的奈米銀結構。
[PVP]/[Ag]=1.5,[AgNO3]=0.25 M,time=45 min
PVP]/[Ag]=1.5,[AgNO3]=0.25 M,time=30 min [PVP]/[Ag]=15,[AgNO3]=0.25 M
26 [Ag]/[Br]=8.4 × 105,Time=2.5 hr 圖 2.11 polyol process方法合成不同形狀的奈米銀結構 [30-33]。 2-4.2 光還原法 (photoreduction) 利用特定波長能量的光源,照射這些金屬離子或奈米金屬顆粒使 其還原成長的一種方法,稱為光還原 [34-36],其還原方法如示意圖 2.12 [34]。 圖 2.12 光還原金屬機制示意圖。 利用使用不同介面活性劑、硝酸銀濃度、反應時間等因素,還原 銀成奈米顆粒。Rongchao Jin等人利用化學還原法製造出球型奈米顆 粒,利用40W螢光照射這些銀奈米顆粒,隨著時間增長,幾乎都由球 型轉變為三角形。
27
圖 2.13 40W螢光下照射不同時間之TEM圖,A)照射前,B)照射40hr, C)照射55hr,D)照射70hr銀奈米粒子 [34]。
而By Yong Zhou等人利用PVA為保護劑,改變不同硝酸銀濃度, 利用30W汞燈還原銀奈米顆粒,成長成樹狀奈米銀結構。如圖2.14 所 示 [35]。
3wt% PVA,10-3M AgNO3 3wt% PVA,10-2M AgNO3
圖 2.14 不同硝酸銀濃度於30W汞燈還原之TEM圖譜。
2-4.3 晶種還原法 (seed-mediated growth)
晶種還原法主要有兩個步驟:
a) 合成晶種:通常利用citrate水溶液為包覆劑,加入低濃度金屬鹽
28 nm小晶種奈米顆粒。 b) 成長步驟:將晶種加入成長溶液中,成長溶液通常包含介面活 性劑(CTAB)、弱還原劑(ascorbic acid)、金屬鹽類、NaOH來調 節pH值。界面活性劑通常可形成較大微胞 (micelle)或柱狀結構 [37-38]。圖2.15 為晶種成長法之示意圖 [39]。 圖 2.15 晶種成長法之示意圖 [39]。 其成長機制為界面活性劑於適當濃度下形成柱狀結構,稱為 軟性的模板 (soft template) ,小顆粒的晶種銀顆粒於模板內成長成 棒狀結構,其成長機至示意圖如圖2.16所示 [40]。 圖 2.16 晶種成長法棒狀結構成長示意圖 [40]。
29 晶種成長法的優點為製作方式快速簡單、可在室溫下進行反 應與適合在實驗室進行等。圖2.17 為不同條件下合成銀奈米棒之 TEM圖譜。 不同晶種體積、酸鹼值合成銀奈米線 圖 2.17 不同合成條件下利用晶種成長法合成銀奈米棒之TEM圖 譜[37-38]。 2-4.4 水熱法 (hydrothermal) 利用水為介質,外加適當的溫度,在密封的壓力容器中內產生壓 力,進行反應。目前利用水熱法製備奈米等級產物的方法已經引起研 不同酸鹼值下合成銀奈米棒之TEM圖 a) pH=6.95 b) pH=12.24 不同溫度下合成銀奈米棒之TEM圖 a)20℃ b) 30℃
30 究者高度的興趣,其特點為純度高、粒徑易控制、分佈均勻、顆粒團 聚輕等優點。其合成步驟,是先把反應物與水放入反應器內,再把反 應器旋緊、密閉後,放入加熱爐中反應。反應後過濾取出產物,再清 洗烘乾即可。依反應溫度可分為中溫高壓型:溫度範圍由100℃-275℃ 間,以鐵氟龍為內容器材質;高溫高壓型:大於275℃。當處於高溫 高壓狀態下,水的性質會產生變化,所以處於臨界狀態下晶體生長速 度增加。 可以藉由控制界面活性劑、AgNO3、還原劑等濃度與反應的時間、 溫度,還原成不同形狀的奈米銀顆粒。如下圖2.18 所示,利用不同 合成條件下利用水熱法合成不同形狀結構奈米銀 [41-44]。
31
AgNO3=0.003 M ,NaCl=0.003 M,glucose=0.006 M;Temp=180℃,18 hr [41]。
AgNO3=43 mM,PVP=210 mM ,Temp=160℃,
time=4 hr 。[42]
AgNO3=17.3 mM,PVP=210 mM
Temp=140℃,time=6 hr [42]。
32 triangle:
Ag(NH3)2+=5.8 mM,CTAB=8.3 mM glucose=4.16 mM Temp=120℃;time=8hr [44]。
Ag(NH3)2+=6.5 mM,CTAB=8.3 mM glucose=4.16 mM Temp=120℃;time=8hr [44]。
33
第三章 實驗方法與流程
3-1 實驗藥品與設備
3-1.1 實驗藥品與氣體 1.硝酸銀(silver nitrate):SHOWA,純度 99.8 wt% 2.檸檬酸鈉(sodium citrate):Riedel-deHaen,純度 99.5 wt% 3.硼氫化鈉(sodium borohydride):Sigma-Aldrich,純度>98.5 wt% 4.溴化十六烷三甲基銨(hexadecyltrimethylammonium bromide CTAB):ACROS,純度>99 wt%
5.氫氧化鈉(sodium hydroxide):SHOWA,純度 96 wt%
6.抗壞血酸(ascorbic acid):Kanto Chemical co.,純度 99.5 wt% 7.氫氧化鉀(potassium hydroxide):SHOWA,純度 85 wt% 8.氨水(ammonia solution):SHOWA,純度 28 wt%
9.葡萄糖(glucose):Merck,純度 99 wt%
10.硝酸鑭(lanthanium (Ⅲ) nitrate hexahydrate):Alfa Aesar,純度 99.9 wt%
11.硝酸鈷(cobolt(Ⅱ) nitrate hexahydrate):SHOWA,純度 98 wt% 12.硝酸鈣(calcium (Ⅱ)nitrate tetrahydrate):Riedel-deHaen,純度 99 wt%
13.氧化釕(ruthenium(Ⅳ) oxide):SHOWA,純度 99.9 wt%
34 15.BP2000(black pearl 2000):台灣電力公司綜合研究所提供 16.聚乙烯醇(polyvinyl alcohol):長春化工,5 wt% 17.碳布:中科院提供 18.氮氣(nitrogen):建仁股份有限公司,99.95% 19.氧氣(oxygen):建仁股份有限公司,99.95% 20.空氣(air):建仁股份有限公司 3-1.2 實驗設備 1.電子天帄:Precisa,model XS 225A 2.加熱攪拌器:COFNING 3.超音波震盪器:TOHAMA D200H 4.超高溫電器管狀爐:YOKOGAWA,model UP350E 5.小型滾壓機:怡生機械股份有限公司 6.去離子水機:SUNTEX RM-220 7.高壓釜:宸昶企業-內容量 100 mL 8.離心機:HSIANGTAI 9.電化學系統:Solartron,SIC 1287 10.掃描式電子顯微鏡:Hitachi JSM 6500 11.穿透式電子顯微鏡:Philip TECNAI20
35 13.x-ray 繞射儀:Siemens D5000 14.UV-VIS:AGILENT 8453
3-2 實驗流程:
3-2.1 全部實驗流程: 圖 3.1 全部實驗流程圖。36 3-2.2 鈣鈦礦氧化物合成
37 3-2.3 棒狀奈米銀合成
38 3-2.4 線狀奈米銀合成
39 3-2.5 立方體奈米銀合成
40
3-3 催化劑合成
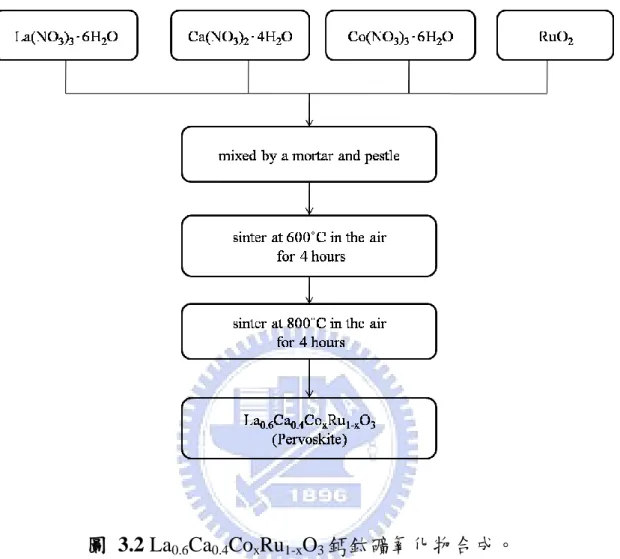
3-3.1 製備 La0.6Ca0.4CoxRu1-xO3 催化劑-固態燒結法
a) 依莫爾比例分別秤取硝酸鑭、硝酸鈣、硝酸鈷、氧化釕粉末,利
用研缽研磨均勻混合。製備不同 Ru 比例之 La0.6Ca0.4CoxRu1-xO3 鈣
鈦礦氧化物(La0.6Ca0.4CoO3、La0.6Ca0.4Co0.8Ru0..2O3、
La0.6Ca0.4Co0.6Ru0.4O3、La0.6Ca0.4Co0.5Ru0.5O3、La0.6Ca0.4Co0.4Ru0.6O3、 La0.6Ca0.4Co0.2Ru0.8O3)粉末。 b) 置於爐管中燒結,升溫速率:5℃/min,加溫至 600℃,在空氣氣 氛下持溫 4 小時,爐冷至室溫。 c) 得到黑色粉末,再利用研缽研磨。 d) 再置於爐管中燒結,升溫速率:5℃/min,加溫至 800℃,在空氣 氣氛下持溫 4 小時,爐冷至室溫。 e) 得到黑色粉末,研磨後儲存於藥膏盒中,即製備完成 La0.6Ca0.4CoO3、 La0.6Ca0.4Co0.8Ru0..2O3、La0.6Ca0.4Co0.6Ru0.4O3、La0.6Ca0.4Co0.5Ru0.5O3、 La0.6Ca0.4Co0.4Ru0.6O3、La0.6Ca0.4Co0.2Ru0.8O3粉末。 f) 電極製作:分別秤取 0.3 g 鈣鈦礦氧化物催化劑、0.3 g BP2000 碳 材、0.5 g 的 60 wt% 聚四氟乙烯(PTFE)與 2 g 的 5 wt% 聚乙烯醇 (PVA) ,其重量比為 3:3:3:1。利用均質機均勻分散於 10 mL 去離子水中,攪拌 5 分鐘後,得到均勻混合之漿料;混合均勻後,
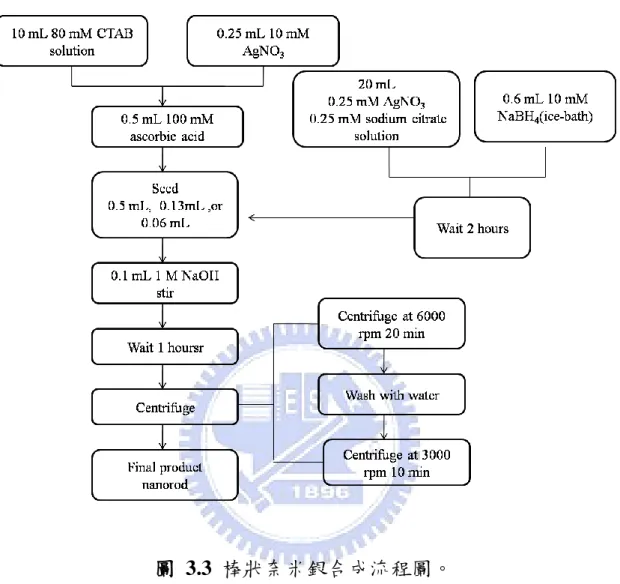
41 塗佈於異能科技公司生產的氣體擴散層;此外,為了去除殘餘的 溶劑,將電極放入 350 °C 的高溫箱型爐中,熱處理 30 分鐘。熱處 理後,將製作好電極滾壓成約 0.3 μm 的膜厚,即為電化學量測時 的工作電極 (working electrode)。 3-3.2 棒狀奈米銀、線狀奈米銀結構合成-晶種成長法 a) 晶種合成 (3-5 nm Ag nanoparticle):
1. 配製含 0.25 mM AgNO3和 0.25 mM sodium citrate 水溶液 20 mL
2. 於冰浴條件下新鮮配製 10 mM NaBH4 還原劑水溶液,取 0.6
mL NaBH4 水溶液加入步驟 1 溶液中,劇烈攪拌混合。
3. 靜置 2 小時,即合成晶種溶液。晶種溶液配製完成 5 小時後不 可使用。
b) 成長步驟 (nanorod):
1. 分別配製 80 mM CTAB (hexadecyltrimethylammonium bromide)
界面活性劑水溶液、10 mM AgNO3 水溶液、100 mM ascorbic
acid 還原劑水溶液。
2. 取 80 mM 10 mL CTAB、10 mM 0.25 mL AgNO3、100 mM 0.5 mL ascorbic acid 水溶液均勻混合,重複此步驟,配製三瓶相同的溶 液。
42 3. 分別加入不同量的晶種溶液(0.5 mL、0.13 mL、0.6 mL)於步驟 2 溶液中,製備不同深寬比(aspect ratio)的棒狀奈米銀結構。 4. 最後再加入 0.1 mL 1M NaOH 水溶液於上述溶液中,均勻混合。 1-10 分鐘內即產生顏色變化,不同晶種體積分別產生不同顏色 變化。 5. 静置 1 小時。以離心速度 6000 rpm 離心 20 min,丟棄上層液, 再以 3000 rpm 水洗 離心 10 min,收集沉澱物。即合成不同深 寬比之奈米棒狀銀結構。 c) 成長步驟 (nanowire):
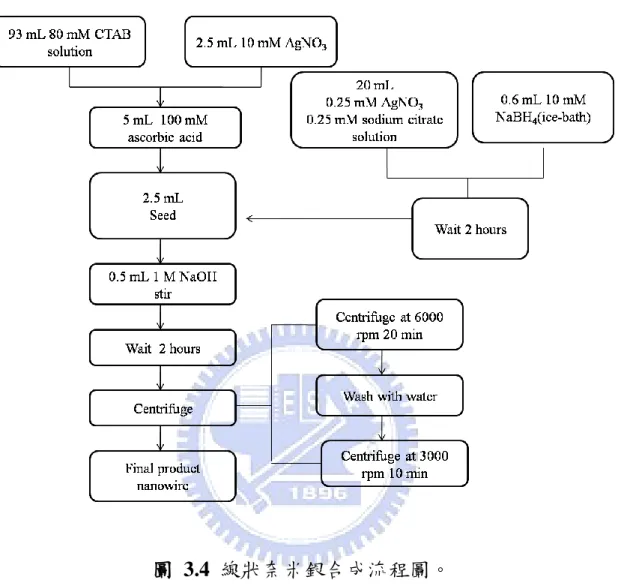
1. 分別配製 80 mM CTAB (hexadecyltrimethylammonium bromide)
界面活性劑水溶液、10 mM AgNO3 水溶液、100 mM ascorbic acid 還原劑水溶液。 2. 取 80 mM 93 mL CTAB、10 mM 2.5 mL AgNO3、100 mM 5 mL ascorbic acid 水溶液均勻混合。 3. 加入 2.5 mL 晶種溶液於上述步驟之溶液中。 4. 最後加入 0.5 mL 1M NaOH 水溶液於上述溶液中,均勻混合。 5. 静置 2 小時。以離心速度 6000 rpm 離心 20 min,丟棄上層液, 再以 3000 rpm 水洗 離心 10 min,收集沉澱物。即合成線狀奈 米銀結構。
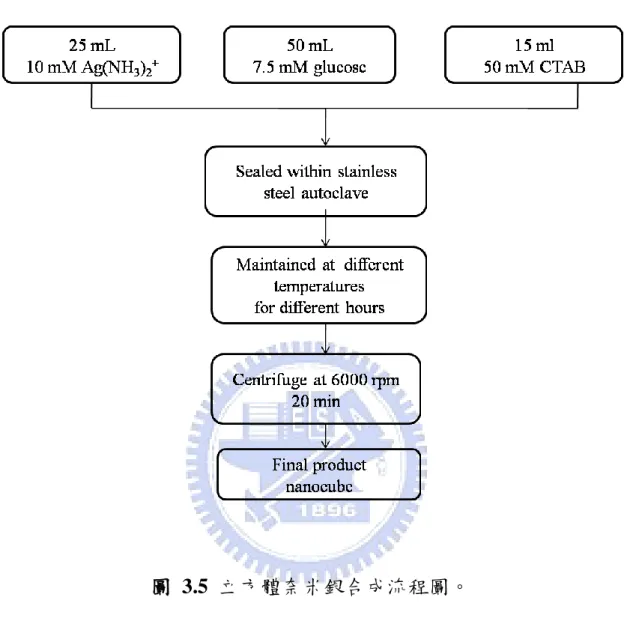
43 d) 電極製作:將棒/線狀奈米銀直接塗佈於2 × 2 cm 經疏水處理過的 碳布上,烘乾製成。 3-3.3 立方體奈米結構銀合成-水熱法 a) 配製 10 mM 銀氨錯離子水溶液: 1. 秤取 0.1699 g AgNO3溶於 20 mL 水溶液中。 2. 緩慢滴入 1 M NH3 水溶液,溶液由澄清變為土黃色,繼續滴入 NH3 水溶液直到再次澄清為止。 3. 將上述溶液稀釋至 100ml 為止,即配製完成 10mM [Ag(NH3)2]+ 水溶液。 b) 分別配製 7.5 mM glucose 水溶液與 50 mM CTAB 水溶液。 c) 取 10 mM 25 mL [Ag(NH3)2]+水溶液、7.5 mM 50mL glucose 水溶液、 50mM 15ml CTAB 水溶液將其放置於容量為 100 mL 的鐵氟龍瓶中, 均勻混合。 d) 將鐵氟龍瓶放入高壓釜中密封,放置於烘箱中,加熱至 120-160℃, 8-18 小時。 e) 待其自然冷卻至室溫,取出以 6000 rpm 轉速離心 20 分鐘。蒐集 沉澱物即合成正方體結構奈米銀。 f) 電極製作:將立方體奈米銀直接塗佈於2 × 2cm 經疏水處理過之碳
44 布上,烘乾製成。
3-4 分析儀器
3-4.1 定性分析 a) X 光繞射分析儀(XRD) [45]: 試樣在受到單色 X 光 (波長為 λ)照射時,當試樣中某些粒子之面 間 距 d 中 之 格 子面 (hkl)測 物 之 , 對 入 射 X 光 剛 好 傾 斜 θ 角 (Bragg’angle),符合布拉格 (Bragg’s)公式:nλ=2dsinθ 時,此時入射 之 X 光會產生繞射。將所得結果與 JCPDS 軟體對照,找出觸媒之晶 體結構資料。本實驗所使用之 X 光繞射分析儀為 Siemens D5000,使 用Cu Kα (λ=1.5418 Å)作為激發源,使用 θ-2θ 模式,掃描角度為 20~80 度,掃描速率為 2.4°/min。 b) 穿透式電子顯微鏡 (TEM): 穿透式電子顯微鏡具有極高的穿透能力及高解析度,已成為材料 科學研究上極有效的工具之一。藉由穿透式電子顯微鏡進行材料分析 主要可得到以下三種訊息:(1)擷取穿透物質的電子 (transmittedelectron)或彈性散射電子 (elastic scattering electron)而成像;(2)由電
子繞射圖樣 (diffraction pattern),作為微細組織和晶體結構之研究;(3)
45
loss spectroscope, EELS)作化學成份分析。本實驗採用之 TEM 為 Philip TECNAI20 利用 TEM 觀察探討經催化劑的結構與形貌,有助於與電 化學量測結果做更進一步的探討 [45]。 c) 掃描式電子顯微鏡 (SEM): SEM 主要利用電子槍產生電子束,經過三個電磁透鏡所組成的 電子光學系統,聚集成一微小電子束照射至試片的表面。試片表面原 子受到入射電子的撞擊,會產生二次電子、反射電子、歐傑電子等等。 而試片產生出的二次電子,其能量小於 50 eV 的低能量電子,只有在 試片深度 50-500 Å 才可能逃離試片表面而受到偵測,且其數量也會 受到試片表面起伏所影響。故藉由觀察二次電子的影像可以瞭解試片 表面之形貌特徵。實驗中使用之掃描式電子顯微鏡型號為 JEOL JSM-6500,操作加速電壓為 15 kV。成份之定性及定量分析則使用其 所附加之 EDX 進行。 d) 比表面積測定儀(BET) [47]: 在氮氣正常沸點(-195.8℃ or 77K),低於 1atm 下的數個壓力而 達到帄衡時,量取氮氣在物質表面上的吸附量,並根據 Brunauer-Emmett-Teller (BET)公式來求取比表面積,吸附原理如下: 0 0 1 1 1 1 P P C W C C W P P W m m
46 將 1 1 0 P P W 對 o P P 作圖可得到一直線,斜率 S 和截距 i 分別為 和 利用吸附氣體重量 Wm與分子吸附係數和每個分子的投影 面積 Acs可得到總表面積 St e) 可見光-紫外光吸收光譜儀 (UV-VIS): 利用金屬奈米粒子在不同型態下會對不同波長產生表面電漿共 振,因此可藉由觀察金屬奈米粒子吸收波段不同來判別其所產生之變 化。利用可見光-紫外光吸收光譜,可以測量並分析溶液的吸收度, 樣品的吸收度會與濃度呈現性關係,可由Beer’s law 表示 A = logP0 P = εbc A:樣品吸收度; P0:入射光強度; P:穿透光強度 ε:樣品吸收系數 ; b:cell 的長度 ;c:樣品濃度 3-4.2 電化學分析 a) 電流對電壓極化曲線 (galvanodynamic test): 電流對電壓極化曲線為在測試中逐漸增加電流輸出量(1 mA/sec), C W C S m 1 C W i m 1 M NA W S m cs t
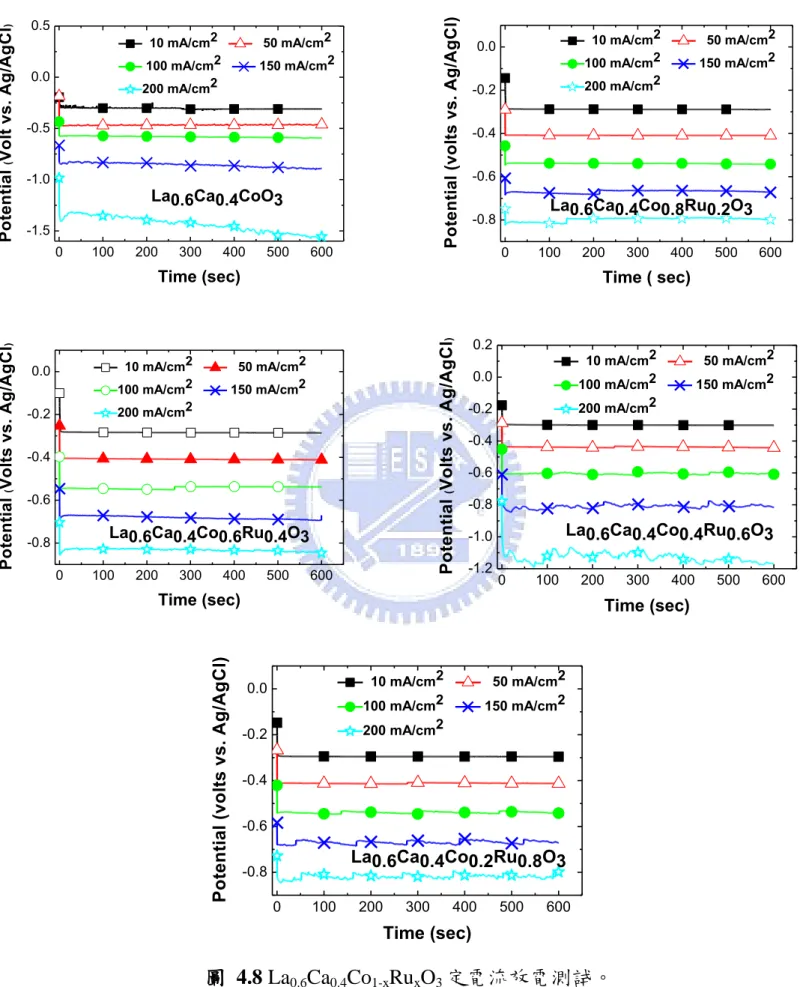
47 測量相對輸出電壓的改變,以此電流與電壓之變化量做圖。在曲線中, 極化現象為偏離理想電極特性行為的表現,且可藉此判斷觸媒電極特 性優劣。 雙效觸媒電極研究中,使用三電極的半電池反應;其中以氣體擴 散陰極為工作電極,電極反應面積為 3 cm2;輔助電極為鍍上氧化釕 與氧化銥的鈦網;以 Ag/AgCl 標準電極為參考電極,電解質為 30 wt% 氫氧化鉀 (KOH)。整體的電化學裝置可見於圖 3.6。 圖 3.6 電化學裝置。 b) 定 電 流 放 電 ( 氧 氣 還 原 ) 或 充 電 ( 氧 氣 生 成 ) 測 試 (galvanostatic test): 同樣採用三電極的半電池反應,定電流放電實驗中,量測陰極在
48 給予固定電流時的充電和放電能力,測試條件範圍為 10~200 mA/cm2, 觀測其電壓輸出值之穩定性,實驗裝置如圖 3.6。 c) 長時間充放電測試: 採用三電極的半電池反應,給予固定 30 mA/cm2電流放電 100 小 時,再給予 30 mA/cm2電流充電 100 小時,測試觀測其電壓輸出值之 穩定性,以此可得知實際電池運作時的穩定度與耐久度,實驗裝置圖 同上。 奈米棒狀銀研究中,使用三電極的半電池反應,將催化劑塗佈於 以經過疏水處理後的碳布做為工作電極;輔助電極為鍍上氧化釕與氧 化銥的鈦網;以 Ag/AgCl 標準電極為參考電極,電極反應面積為 1cm2, 電解質為 30 wt%氫氧化鉀 (KOH)。整體的電化學裝置可見於圖 3.7。 圖 3.7 電化學裝置圖
49 d)電流對電壓極化曲線 (potentiodynamic test): 電流對電壓極化曲線為在測試中逐漸增加電壓輸出量(1 mV/sec), 測量相對輸出電流的改變,以此電流與電壓之變化量做圖。在曲線中, 極化現象為偏離理想電極特性行為的表現,且可藉此判斷觸媒電極特 性優劣。 立方體奈米銀研究中,亦使用三電極的半電池反應,其中以 2 × 2 cm2塗佈奈米銀觸媒於經過疏水處理之碳布上做為工作電極; 輔助電極為 8 cm2鉑片;參考電極為 Ag/AgCl 標準電極,電極反應面 積為 1 cm2;電解質為 1 M 氫氧化鉀 (KOH)。實驗裝置圖如圖 3.8 圖 3.8 電化學裝置 [47]。
50
第四章 實驗結果與討論
4-1 La
0.6Ca
0.4Co
xRu
1-xO
3鈣鈦礦氧化物催化劑對氧氣還原反
應催化活性探討
4-1.1 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物 XRD 分析 圖4.1為不同Ru比例的La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物XRD比 較圖。由圖可以發現利用固態燒結法,經由600℃燒結4小時後,研磨 均勻再以800 ℃同樣條件下燒結4小時,所合成的催化劑(不同Ru比例 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物)可以明顯看出有La0.6Ca0.4CoO3 鈣 鈦礦氧化物 (Perovskite)的晶相生成,其為菱形六面體 (Rhombohedral) 結構,將圖4.1 之XRD圖與標準資料庫JCPDS中的La0.6Ca0.4CoO3比對, 發現經由此方法的合成的鈣鈦礦氧化物與標準資料(JCPDS-36-1389) 的 晶 相 完 全 相 符 , 明 確 顯 現 出 成 功 的 合 成 鈣 鈦 礦 氧 化 物 (La0.6Ca0.4CoxRu1-xO3) , 而 其 中 僅 有 La0.6Ca0.4RuO3此 樣 品 中 發 現 有 RuO2的特性繞射峰產生,顯示有RuO2的晶相生成。換句話說,除了 La0.6Ca0.4RuO3催化劑,其他比例的La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物皆成功的將Ru+4取代了Co+3於鈣鈦礦氧化物中的位置,將Ru參雜
(doping)至La0.6Ca0.4CoO3中。仔細比對JCPDS發現La0.6Ca0.4CoO3的特 性繞射峰有少許位移現象發生,繞射峰有些微的往小角度的方向位移, 推測其原因是由於將較大的Ru(0.68 Å )離子取代了相對上較小的
51
Co(0.61 Å )離子,改變了La0.6Ca0.4CoO3氧化物的晶格常數,使得繞射
峰角度產生位移。 由圖4.2可以發現晶格常數 (lattice parameter)的改變與Ru參雜的 量成正相關,隨著Ru參雜的量變多,晶格常數也隨著變大,確認成 功的將Ru參雜至鈣鈦礦氧化物中。 表4.1為不同Ru比例的La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物之晶格 常數與體積值。可以發現隨著參雜Ru的量變多,a-axis和c-axis值皆隨 著變大,使得單位晶格體積也隨之變大。 20 30 40 50 60 70 80 0 500 1000 1500 2000 La0.6Ca0.4RuO3 La2O3 RuO2 (0 1 2 ) (2 1 4 ) (0 2 4 ) (0 0 6 ) (2 0 2 ) (1 0 4 ) (1 1 0 ) In te n s it y ( a rb . u n it s ) 2 (degree) La0.6Ca0.4CoO3 La0.6Ca0.4Co0.8Ru0.2O3 La0.6Ca0.4Co0.6Ru0.4O3 La0.6Ca0.4Co0.4Ru0.6O3 La0.6Ca0.4Co0.2Ru0.8O3 圖 4.1 不同比例 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物之 XRD 比較圖。
52 0.0 0.2 0.4 0.6 0.8 5.36 5.38 5.40 5.42 5.44 5.46 5.48 5.50 5.52 5.54 5.56 L a tt ic e c o n s ta n t Stoichiometric Ru 圖 4.2 不同 Ru 比例鈣鈦礦氧化物與晶格常數 (a-axis)之趨勢圖。 表 4.1 不同比例 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物之晶格常數。 樣品 Lattice parameter (Å ) Unit-cell volume(Å3) a c V La0.6Ca0.4CoO3 5.397 13.178 332.47 La0.6Ca0.4Co0.8Ru0.2O3 5.431 13.294 339.64 La0.6Ca0.4Co0.6Ru0.4O3 5.517 13.389 352.98 La0.6Ca0.4Co0.4Ru0.6O3 5.531 13.711 359.10 La0.6Ca0.4Co0.2Ru0.8O3 5.500 13.480 353.13 4-1.2 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物 SEM 形貌與 EDX 分析 圖 4.3 為不同 Ru 比例的 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物之 SEM 圖。利用固態燒結法合成的鈣鈦礦氧化物因經過長時間燒結使其顆粒 大小大約 100-300 nm 左右。 以 La0.6Ca0.4Co0.2Ru0.8O3鈣鈦礦氧化物樣品為例,測試其元素組
53 成,其元素莫爾比為 La:Ca:Co:Ru=0.64:0.40:0.21:0.74,其 趨勢大致與當初合成時比例相符,但有些微誤差的存在,推測可能由 於機器量測造成誤差。其元素組成比例可由表 4.2 獲得。 更進一步分析,經由 EDX 之 mapping 的結果發現四種元素均均 勻分散於 La0.6Ca0.4Co0.2Ru0.8O3鈣鈦礦氧化物,由此印證確實成功的 將 Ru 參雜至此鈣鈦礦氧化物中。圖 4.4、圖 4.5 La0.6Ca0.4Co0.2Ru0.8O3 鈣鈦礦氧化物的 EDX 分析結果。 La0.6Ca0.4CoO3 La0.6Ca0.4Co0.8Ru0.2O3 La0.6Ca0.4Co0.6Ru0.4O3 La0.6Ca0.4Co0.4Ru0.6O3 b) a) c) d)
54 La0.6Ca0.4Co0.2Ru0.8O3 圖 4.3 La0.6Ca0.4CoxRu1-xO3 之 SEM 影 像 , a) La0.6Ca0.4CoO3 b) La0.6Ca0.4Co0.8Ru0.2O3 c) La0.6Ca0.4Co0.6Ru0.4O3 d) La0.6Ca0.4Co0.4Ru0.6O3 e) La0.6Ca0.4Co0.2Ru0.8O3 SEM 影像。 圖 4.4 La0.6Ca0.4Co0.2Ru0.8O3 SEM 影像與 EDX 結果。 e)
55
圖 4.5 La0.6Ca0.4Co0.2Ru0.8O3 EDX 之 mapping 結果。
表 4.2 元素組成。
Element Weight(%) Atomic(%)
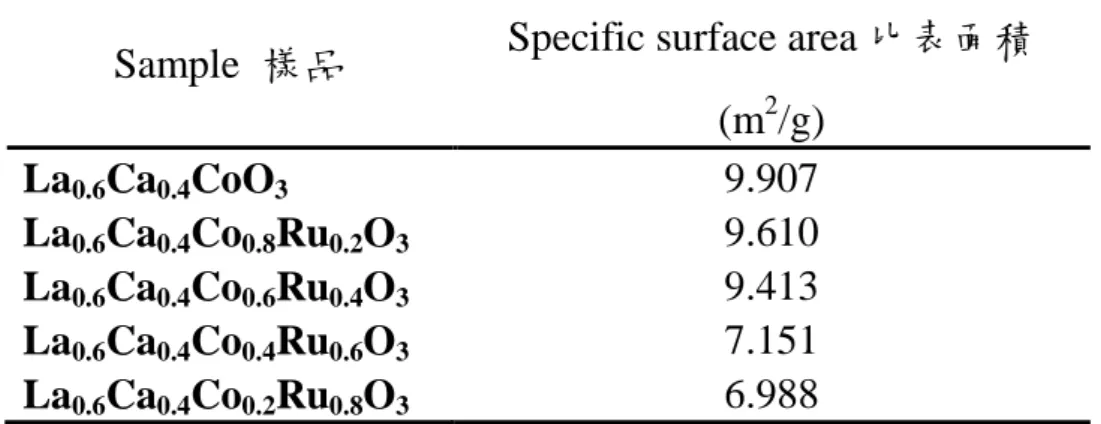
La 46.42 32.25 Ca 8.38 20.18 Co 6.45 10.56 Ru 38.75 37.01 Total 100 100 4-1.3 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物比表面積測試 將合成不同 Ru 比例的 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物做比表 面積測試,其結果如下表 4.3 顯示,由表可以得知不同 Ru 比例的 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物比表面積範圍大約介於 7 m2/g 到 10 m2/g,其表面積大小差異並不大,推測利用固態燒結法合成的不同 Ru 比例的 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物其電催化活性大小與表 面積大小較無直接相關性,推測應與催化劑本身性質較為相關。
56
表 4.3 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物比表面積。
Sample 樣品 Specific surface area 比表面積
(m2/g) La0.6Ca0.4CoO3 9.907 La0.6Ca0.4Co0.8Ru0.2O3 9.610 La0.6Ca0.4Co0.6Ru0.4O3 9.413 La0.6Ca0.4Co0.4Ru0.6O3 7.151 La0.6Ca0.4Co0.2Ru0.8O3 6.988 4-1.4 La0.6Ca0.4CoxRu1-xO3鈣鈦礦氧化物催化劑之對氧氣還原反應催 化活性比較 a) 極化曲線測試 (galvanodynamic test): 空氣陰極上的氧氣還原反應效能可以利用極化曲線來判定, 圖 4.6 為不同 Ru 比例 La0.6Ca0.4CoxRu1-xO3氧化物之極化曲線圖。 由圖可以明顯發現有參雜 Ru 組成的鈣鈦礦氧化物其催化效果皆 比 La0.6Ca0.4CoO3來的好,以在電流密度為 200 mA/cm 2 下,Ru 比 例為 0.2 其輸出電壓為-0.83 V;Ru 比例為 0.4 其輸出電壓為-0.79 V; 比例為 0.6 輸出為-0.73 V;Ru 比例為 0.8 則為-0.81 V;相較於純 La0.6Ca0.4CoO3 鈣鈦礦氧化物催化劑輸出電壓只有 -0.98 V,與 La0.6Ca0.4CoO3 相比其輸出電壓值均有顯著的提升,顯示參雜 Ru 組成於 La0.6Ca0.4CoO3鈣鈦礦氧化物中有明顯增進氧氣還原的效果, 其不同電流密度下的輸出電壓差異值可見表 4.4。而在參雜不同
57 Ru 比例的鈣鈦礦氧化物中又以 La0.6Ca0.4Co0.4Ru0.6O3有最好的電催 化表現。舉例來說,在 100 mA/cm2 高電流密度下還可以保有 -0.49 V 電壓。由極化曲線圖可以明顯發現 La0.6Ca0.4Co0.4Ru0.6O3具 有良好的氧氣還原之催化效能。 0 30 60 90 120 150 180 210 240 270 -1.2 -1.0 -0.8 -0.6 -0.4 -0.2 0.0 P o te n ti a l ( v o lt s v s . A g /A g C l)
Current density ( mAcm-2)
La0.6Ca0.4CoO3/BP2000 (1) La0.6Ca0.4Co0.8Ru0.2O3/BP2000 (2) La0.6Ca0.4Co0.6Ru0.4O3/BP2000 (3) La0.6Ca0.4Co0.4Ru0.6O3/BP2000 (4) La0.6Ca0.4Co0.2Ru0.8O3/BP2000 (5) 圖 4.6 La0.6Ca0.4CoxRu1-xO3氧氣還原 IV-極化曲線圖。 表 4.4 不同電流密度下的輸出電壓差異值。
i-V curve / Potential (volt)
sample Current density (mA/cm2)
50 100 150 200 La0.6Ca0.4CoO3 -0.469 -0.628 -0.796 -0.981 La0.6Ca0.4Co0.8Ru0.2O3 -0.423 -0.557 -0.696 -0.833 La0.6Ca0.4Co0.6Ru0.4O3 -0.385 -0.511 -0.643 -0.787 La0.6Ca0.4Co0.4Ru0.6O3 -0.372 -0.490 -0.614 -0.732 La0.6Ca0.4Co0.2Ru0.8O3 -0.415 -0.534 -0.661 -0.806