excited-state intramolecular proton
transfer for o-nitrophenol: An
ab initio on-the-fly nonadiabatic
molecular dynamic simulation
Chao Xu
1, Le Yu
2,3, Chaoyuan Zhu
2,4, Jianguo Yu
1& Zexing Cao
4The 6SA-CASSCF(10, 10)/6-31G (d, p) quantum chemistry method has been applied to perform on-the-fly trajectory surface hopping simulation with global switching algorithm and to explore excited-state intramolecular proton transfer reactions for the o-nitrophenol molecule within low-lying electronic singlet states (S0 and S1) and triplet states (T1 and T2). The decisive photoisomerization mechanisms of
o-nitrophenol upon S1 excitation are found by three intersystem crossings and one conical intersection
between two triplet states, in which T1 state plays an essential role. The present simulation shows
branch ratios and timescales of three key processes via T1 state, non-hydrogen transfer with ratio 48%
and timescale 300 fs, the tunneling hydrogen transfer with ratios 36% and timescale 10 ps, and the direct hydrogen transfer with ratios 13% and timescale 40 fs. The present simulated timescales might be close to low limit of the recent experiment results.
The excited-state intramolecular proton transfer (ESIPT) reaction is considered to be one of the most fundamen-tal and important processes in chemistry, material and biology1–12. The ESIPT reactions have been studied from
both ab initio quantum chemistry calculations13–17 and trajectory-based nonadiabatic molecular dynamic
simu-lations18–22. The most of the ESIPT processes have been found to be taken place at a subpicosecond time scale20–22.
The photo-excited molecule from electronic ground singlet state (S0) to the excited singlet states can undergo
rapid internal conversion via conical intersections (CI) to the lower singlet states as well as intersystem crossings (ISC) via spin-orbital couplings (SOC) to the triplet states. By means of the radiative or nonradiative interaction with target molecule, the redistribution of electron density is essential for the transfer of a hydroxyl (or amino) proton to an oxygen or nitrogen acceptor atom on the excited states within a hydrogen bond already formed in the electronic ground state. Subsequently, the excited state molecule can decay to the ground state accompanied with a reverse proton transfer.
Fundamental mechanistic insight into photoinduced ESIPT reaction can be understood through studying prototypical representative of nitroaromatics o-nitrophenol molecule. This is because that o-nitrophenol owns two functional groups, namely nitro and the adjacent hydroxyl groups which show strong intramolecular hydro-gen bond. Thus, the aci-nitrophenol can be formed via the initial intramolecular hydrohydro-gen transfer from hydroxyl group to nitro group. Differing from the other aromatic systems, the experimental and theoretical studies demon-strate peculiar behavior of the photoinduced decay process for nitrated aromatic compounds. The tautomeric aci-nitrophenol isomers via photoisomerization has been confirmed by infrared spectroscopy in low-temperature
1College of Chemistry, Beijing Normal University, Beijing 100875, P. R. China. 2Institute of Molecular Science, Department of Applied Chemistry and Center for Interdisciplinary Molecular Science, National Chiao Tung University, Hsinchu 30010, Taiwan. 3Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, The College of Chemistry & Materials Science, Shaanxi key Laboratory of Physico-Inorganic Chemistry, Northwest University, Xi’an 710069, P. R. China. 4State Key Laboratory for Physical Chemistry of Solid Surfaces and Fujian Provincial Key Lab of Theoretical and Computational Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. Correspondence and requests for materials should be addressed to C.Z. (email: [email protected])
Received: 08 March 2016 Accepted: 09 May 2016 Published: 25 May 2016
dynamics simulation in the present study, we actually found one new S0/T1 ISC which governs decay process for
non hydrogen transfer. The previous computational and experimental study has proposed two possible relaxation pathways29; one is via the ISC following (O)H-O(NO) stretch and the other is via the S
0/S1 CI after overcoming
potential energy barrier induced by hybrid torsion of HONO group and (O)H-O(NO) stretch. Actually, the new S0/T1 ISC is located near Frank-Condon region while S0/S1 CI is located in configuration after hydrogen transfer.
The present simulation has shown that the new S0/T1 ISC is very effective pathway and there is almost no
trajec-tory going via S0/S1 CI. We can expect that there must have strong intersystem crossing-branched ESIPT. Besides,
the existence the certain potential barriers on S1 and T1 states along ESIPT coordinate can add tunneling effects as
well. The present study should reveal entire picture of photoisomerization reaction and the photo-decay mecha-nisms including ESIPT for o-nitrophenol molecule.
Trajectory-based ab initio nonadiabatic molecular dynamics simulations have been successfully applied to photoisomerization and photoreaction processes involving intersystem crossings and conical intersections31–48.
Recently, within Tully’s fewest switching algorithm in diabatic representation dynamic simulations have been carried out for intersystem crossings43–48. We have developed trajectory-based ab initio nonadiabatic molecular
dynamics simulations without involving calculation for nonadiabatic couplings37, and it has been applied for
multiple-state azobenzene photoisomerization via conical intersections38. Now we extend our analytically global
switching probability method to include intersystem crossing with spin-orbital couplings. Trajectories are cal-culated on on-the-fly ab initio singlet or triplet potential energy surfaces and then by applying global switching algorithm we treat trajectory surface hopping in a unified way for internal conversion and intersystem cross-ing processes. Intersystem-crosscross-ing dynamic simulation with global switchcross-ing surface hoppcross-ing algorithm is not really new with Landau-Zener switching probability; the spin-diabatic and spin-adiabatic dynamic simulations31
were performed for the model system of spin-orbital couplings that are used as diabatic coupling parameter in Landau-Zener formula, and moreover good agreement with experimental observation for the triplet to singlet branching ratio has been achieved with Landau-Zener formula for performing real dynamic simulation of O(3P)
+ ethylene32. We utilize improved Landau-Zener formula, namely Zhu-Nakamura to treat intersystem-crossing
dynamic simulation in the present report. Moreover, tunneling effects are treated by one-dimensional semilcas-sical method along ESIPT coordinate49.
Results
Global switching algorithm.
Global switching algorithm makes trajectory hopping at the time t where= − −
d t( ) U t2( ) U t1( ) /V t2( ) V t1( ) reaches local maximum (U2 and U1 are adjacent two adiabatic potential
energy surfaces, while V2 and V1 are corresponding two diabatic potential energy surfaces). Analytical switching
probability can be generally expressed in terms of d(t) as50 δ δ δ δ − − → − d d sinh[( 1) ]
sinh( ) exp( ) exp( 2 ), (1)
d
2 2
1
in which δ is estimated from two potential energy surfaces and its gradients at hopping spot along on-the-fly running trajectory. In the case of d 1, it basically goes to Landau-Zener or improved version Zhu-Nakamura formula51,52, δ δ = − − −
p exp( 2 ) bewteen two adiabatic potentials
1 exp( 2 ) bewteen two diabatic potentials, (2) where δ= − π + ± a b b 8 2 1 (3) 2 2 4
µ = − = − − a F F F F V b E E F F F F V 2 (2 ) and ( t X) (2 ) (4) 2 2 2 1 2 1 123 2 2 1 2 1 12
where F1 and F2 are forces on two diabatic potential energy surfaces, V12 is diabatic coupling, μ is reduced mass
and EX is energy at crossing point and Et is potential energy plus kinetic energy component in direction of
hop-ping direction. All those quantities in equation 4 are calculated along on-the-fly running trajectory at hophop-ping spot37 and its details are also given in the Supplementary Note 1. The hopping direction in the present theory is
defined based on sort of the local modes and this agrees with the normal modes constructed from the regularized diabatic states53. Global switching algorithm has been extensively compared with the Tully’s fewest switching
algorithm37,40 and two algorithms are basically similar for highly averaged quantities like quantum yields and
lifetimes. In the present on-the-fly simulation, along three consecutive time steps we detect minimum energy separation between two singlet-states (S1 and S0) or two triplet-states (T2 and T1) and this gap is considered as
dia-batic coupling 2V12 in equation 4. In this case, the switching is considered as in between two adiabatic potential
energy surfaces. On the other hand, we detect the local maximum d(t) between the singlet and triplet states. The diabatic coupling is considered as spin-orbital coupling as V12 = SOC in equation 4 as proposed in ref. 43., and in
fact, singlet state V1 and triplet state V2 can be transformed into adiabatic representation according to
= + ± − + . U V V V V V 2 1 2 ( ) 4 (5) 1,2 1 2 1 22 122
However, in this case, we can do the switching directly between the singlet and triplet states as the spin-orbital coupling is known.
Intersystem crossings with ab initio dynamics.
We have previously performed ab initio quantum chemistry calculations at 6SA-CASSCF(10, 10)/6-31G (d, p) level and have compared with MRCI/cc-pVDZ energy corrections for two singlet states (S0 and S1) and two triplet states (T1 and T2). It is well-known that theCASSCF method is lack of dynamic correlation. Therefore, we carried out energy correction by multi-reference configuration interaction (MRCI) calculation at CASSCF optimized geometries. Both vertical excitation energies (see Fig. S1(a) at CASSCF and Fig. 1S(b) at MRCI given in Supplementary Note 2) and adiabatic energies (see Fig. S2(a) at CASSCF and Fig. 2S(b) at MRCI given in Supplementary Note 2) at all key geometries show the same energy sequences. Relative energy differences between CASSCF and MRCI are mostly smaller than 0.1 eV, espe-cially for energy gaps at six intersystem crossings and one conical intersection being even smaller (see Table S1). Therefore, we think that the present on-the-fly dynamical simulation based on CASSCF level should present the almost same results as it is based on MRCI level. We have confirmed 6SA-CASSCF(10, 10)/6-31G (d, p) is suitable choice for the present dynamic simulation.
We found there are two intersystem crossings between S1 and T2 (S1T2-IC1 and S1T2-IC2) around which SOC
can be almost considered as constant 10 cm−1, one between S
1 and T1 (S1T1-IC) around which SOC is 40 cm−1,
three between S0 and T1 (S0T1-IC1, S0T1-IC2 and S0T1-ICX) around which SOCs are close to 40 cm−1. The
S0T1-ICX is newly found in the present study by trajectory surface hopping dynamic simulation, and the other
five were optimized previously30. Furthermore, the present dynamic simulation shows that only three of six are
active for ESIPT reaction, namely S1T2-IC1, S0T1-ICX, and S0T1-IC1 as shown in Fig. 1. The S1T1-IC is less active,
and S1T2-IC2 and S0T1-IC2 are not active at all (key geometries and Cartesian coordinates for all six intersystem
crossings are given in Supplementary Note 2 and Note 4). Figure 1 shows that the dihedral angels C4C5O11H12
are zero for S0T1-ICX and S1T2-IC1, and the O11H12 bond lengths are 0.945 Å and 0.955 Å, respectively. However,
unlike to the planar geometry of S1T2-IC1, the nitro group of S0T1-ICX and S0T1-IC1 show a similarity although
the H12 bonded to O11 in S0T1-ICX instead of O15 in S0T1-IC1. The nitro group of them is out of the aromatic
skel-eton. Those geometry differences essentially govern the three intersystem crossings undergoing distinct ESIPT reaction pathways. Interestingly, the structure of S0T1-ICX is close Franck-Condon geometry and the structure of
S0S1-CI is close to S0T1-IC1 geometry (see Fig. S3 given in Supplementary Note 2). The S0T1-ICX is responsible for
Figure 1. Key geometries for three active intersystem crossings in trajectory surface hopping dynamics (also for atomic numbering).
relaxing pathway of non-hydrogen transfer, while the S0T1-IC1 responsible for relaxing pathway of after-hydrogen
transfer.
The initial condition of trajectories is started from Franck-Condon region of o-nitrophenol. We perform fre-quency calculation at the ground state equilibrium geometry of S0 to obtain the normal mode coordinates. Initial
normal-mode coordinates and velocities of trajectory are selected according to the Wigner distribution on S0
state. Finally, these initial normal-mode coordinates and velocities are converted into Cartesian coordinates and velocities plus vertical excitation energy to excited S1 state. The thermal kinetic energy with T = 300K is added
to all sampling trajectories with randomly distributing into initial Wigner velocities. However, such equally dis-tributed initial conditions are not suitable for stimulating ESIPT reaction, there are the certain vibronic modes enhanced more than the others as molecule absorbed light to be excited to S1 state. We found that four normal
modes (O11H12 stretch (4172 cm−1), C5O11H12 bend (1473 cm−1), and two C5O11H12-C4N13O15 scissor (408cm−1
and 310 cm−1)) involved in the ESIPT reaction path have stronger vibronic couplings than the others. Therefore,
we added extra kinetic energy (equivalent to T = 500 K) to these four normal modes. Along an on-the-fly run-ning trajectory, the nuclear coordinates and velocities in Cartesian coordinates are propagated by numerically integrating the Newtonian equation of equation of motion with the velocity-Verlet method54. We determine the
minimum potential energy gap between S1 and S0 states, and between T1 and T2 states at the conical intersection
zones, and determine maximum d (t) between singlet and triplet states at intersystem crossing zones, at which we compute the effective coupling parameter a2 and the effective collision energy b2. We found SOC varies very
slowly at intersystem crossing zones, so that we choose constants accordingly 10 and ~40 cm−1 in simulation
(when the energy gap is smaller than 0.25 eV, we start to check attempted trajectory surface hopping). We have run several expensive on-the-fly SOC trajectories in comparison with fixed SOC trajectories and results show small difference. We have checked for four active intersystem crossings, relative spin-orbital coupling variations (δ SOC/SOC) are about less than 2% around crossing zones in which trajectory hops take place. This fixed SOC technique was also utilized for performing real dynamic simulation of O(3P) + ethylene32.
Due to the present dynamics involving OH stretch vibration (over 3500 cm−1) along ESIPT reaction path,
we made test runs with time steps of 0.05 fs, 0.1 fs and 0.15 fs, and finally we set up the 0.1 fs time step from the beginning to the end for the entire dynamic simulation. The dynamics simulation time is set up as 500 fs and as we increase up to 1000 fs, there is no notable difference. All the quantum chemical calculations of on-the-fly potential energy surfaces and its gradients are carried out at 6SA-CASSCF(10, 10)/6-31G (d, p) level by using the quantum chemistry package MOLPRO 2009.1.55 and the dynamic simulation is carried by our own code.
On-the-fly trajectory analysis.
We have run total 280 trajectories and the outcomes show that 6 stay on S1 state regarded as resonance, 6 go via S1T1-IC as direct hydrogen transfer via T1 state, and 268 go via S1T2-IC1initially as shown in Fig. 2 (this is basically Fig. 7 in ref. 30). After going via S1T2-IC1, 265 trajectories hop to T1
states via conical intersection between T1 and T2 states, and 3 go direct hydrogen transfer via T2 state. Further
three bifurcations take place for the 265 trajectories on T1 state, 101 regarded as resonance on T1 state, 133 going
via S0T1-ICX as non-hydrogen transfer, and 31 as direct hydrogen transfer via T1 state. This is overview how
trajectories decay through competing nonadiabatic pathways in the singlet and triplet excited-state manifold. The percentage distribution of various nonadiabatic transition pathways is shown in Fig. 3a. The present ab initio dynamic simulation shows that the S1 → T2 → T1→ S0 (61.29%) and S1 → T2 → T1 (34.77%) are the dominant
processes, while the S1 → T1→ S0 (0.72%) and S1 → T1 (1.43%) are kind of rare cases. Initially starting from
Frank-Condon region on S1 state, the majority sampling trajectories run in the electronic configuration where
three excited states (S1, T2 and T1) are energetically close together, T2 state always keeps in between the S1 and T1
Figure 2. Potential profiles along ESIPT reaction coordinates. Four intersystem coursings (S1T2-IC1(SOC =
10 cm−1), S
1T1-IC(SOC = 40 cm−1), S0T1-IC1(SOC = 40 cm−1) and S0T1-ICX(SOC = 40 cm−1)) are approxi
mately along this path and the other two (S0T1-IC2(SOC = 40 cm−1) and S1T2-IC2(SOC = 10 cm−1)) are far way.
Number in parentheses associated with each intersystem crossing represents trajectories going via it. The small widow stands for potential energy profiles for newly found S0T1-ICX. The Cartesian coordinates of six ICs are
states for a while. On the other hand, the present ab initio dynamic simulation confirms that intersystem cross-ing zone between the S1 and T2 states with small SOC (10 cm−1) are much wider than intersystem crossing zone
between the S1 and T1 states with large SOC (40 cm−1). Therefore, without performing ab initio dynamic
simu-lation, we could not realize that S1→ T2 intersystem crossing (ISC-S1T2) channel is paramount relaxing pathway.
Actually, similar situation is found that the efficient ultrafast ISC dynamic can still be taken place with a rather small spin-orbit coupling due to the counterbalance mechanism56,57. Further detailed distribution is shown in
Fig. 3b for the sampling trajectories passing through the isomerization of the o-nitrophenol and aci-nitrophenol. In the present simulations, we defined the aci-nitrophenol isomers as aci-isomer1 and aci-isomer2, respectively. For aci-isomer1, it still exhibits intramolecular hydrogen bond with ortho oxygen atom of benzene and while for aci-isomer2, the hydrogen rotates with nitro group and far away from the ortho oxygen atom. Diverse dis-tributions are shown, for instance, that o-nitrophenol overcomes barrier of hydrogen transfer and reaches the aci-isomer1 (13.33%), after getting over the hydrogen transfer barrier it passes to aci-isomer1 and then back to
o-nitrophenol (42.22%), the o-nitrophenol converts to aci-isomer1 and then continue to produce aci-isomer2
with the inversion of O15H12 (13.33%), the o-nitrophenol changes to aci-isomer1and then regenerated again after
going via aci-isomer2 (13.33%), and so on. Starting from Frank-Condon region on S1 state, among 280 sampling
trajectories there are 3, 37, and 5 trajectories undergoing direct hydrogen transfer on T2, T1 and S0 states,
respec-tively. However there are 6 and 101 trajectories undergoing tunneling hydrogen transfer on S1 and T1 states,
respectively. Although direct hydrogen transfer on T2 is rare case, it presents a new perspective for general ESIPT
reactions. In most of situation, the T2 state looks more like a “hub”, trajectories stop on it for a moment and then
quickly decay to the reactive T1 state where is major channel for direct hydrogen transfer. The barrier for
hydro-gen transfer on T1 state is lower than that of on S1 state as shown in Fig. 2. On the other hand, the present dynamic
simulation shows that kinetic energy from the four vibartional normal modes responding for hydrogen transfer easily dissipates to the other vibrational modes on S1 state much faster than on T1 state. This is part of reason
that direct hydrogen transfer does not occur on S1 state. Hydrogen transfer has a peculiar property on S0 state
which differs from that of on T1 state, trajectory reaches to S0 state via multi-steps continuous hops in a relatively
short period of time and then it runs on the ground state, eventually the hydrogen transfer reaction occured and aci-isomer formed. It should be noted that the aci-isomer can undergo backward hydrogen transfer reaction to o-nitrophenol due to very low barrier (nearly barrier less). This is reason that hydrogen transfer occurs on S0 state
not very often. The newly found intersystem crossing S0T1-ICX has its geometry close to S0-NP, so that 133 out
of 280 go via it and those trajectories relax to ground state by non-hydrogen transfer pathway. In brief summary, we conclude that S0T1-ICX serves as the center player of intersystem crossing for three competing processes, 45
trajectories results in direct hydrogen transfer, 107 in tunneling hydrogen transfer, and 128 in non-hydrogen transfer.
Although every trajectory has its own lifetime when surface hopping occurs after photoexcitation, and the final result is estimated by the average assemble of all sampling trajectories. Figure 4 shows average population decay with respect of time from excited states S1, T2 and T1, respectively. A single exponential function curve
fitting is applied to calculate average lifetimes on those excited states. The lifetime of S1 state was estimated to be
about 8 fs as an ultrafast decay process and population is almost diminished after 25 fs. This is understandable in common sense that the first excited state decay more likely undergoes via intersystem crossing rather than vibrational cooling58,59. The decaying process is also an ultrafast on T
2 state and the lifetime of T2 state is estimated
to be 14 fs. The population on T2 state is quickly raised first and it reaches maximum at 10 fs and than it almost
diminishes after 40 fs. The most of population via T2 state transfers very quickly to T1 state via conical intersection
between T1 and T2 states. The population on T1 state reaches its maximum (∼ 93%) after 40 fs and the lifetime of
T1 state is estimated to be around 1000 fs. This is the most likely to contribute to the long-lived excited species that
Figure 3. (a) Distribution of multiple nonadiabatic transition pathways among the lowest four electronic states (S1, T2, T1 and S0). (b) Detailed distribution of trajectories passing through the isomers of the o-nitrophenol and
should be observed in both gas and liquid phase because the population on T1 state decays very slowly. However,
the real lifetimes on excited states from experimental observation might be longer than the present estimation due to some extra kinetic energy being initially put in four vibrational modes which are enhancing hydrogen transfer dynamics.
The triplet T1 state plays an essential role for intersystem crossing-branched ESIPT photoisomerization
dynamics in which 97% sampling trajectories bifurcate into three major distinct decay channels. The first branch is the non-hydrogen transfer that counts for 47% sampling trajectories, and timescale is about 100 fs to 500 fs (average is about 300 fs). For trajectories running on T1 state, once they form out-of plane synchronous motion
by O11H12 vibrating around C5O11 bond and the nitro group vibrating around C4N13 bond simultaneously,
trajec-tories can reach S0T1-ICX to decay to ground S0 state. The present simulation shows if this out-of plane
synchro-nous motion form coherent motion before trajectories reach S0T1-ICX (we checked those trajectories from 500 fs
to 1000 fs), they stay on T1 state as resonance trajectories. This is the second branch as the tunneling hydrogen
transfer that counts for 36% sampling trajectories. We have roughly estimated thermal and microcanonical rate constants of nonadiabatic chemical reaction along ESIPT coordinate as shown in Fig. 2 49,
∫
π β = − k Z E P E dE 1 2 r exp( ) ( ) (6)in which β is thermal energy (temperature is set up T = 300 K), Zγ is reactant partition function and P(E) is
non-adiabatic transmission probability for the tunneling trajectories. The average tunneling timescale estimated from equation 6 is about 10 ps. This tunneling timescale is about the same as tunneling proton transfer reaction for tropolone molecule60. The third branch is the direct hydrogen transfer that counts for 13% sampling trajectories,
and timescale is from 20 fs to 100 fs (average is about 40 fs). In this case, trajectories can go the direct hydrogen transfer before the out-of plane synchronous motion occurs. Then, the trajectories continue to run on the T1
state until they reach S0T1-IC1 accompanying with O14N13O15H12 group out-of-plane rotating and finally hop
to S0 state.
Typical trajectories.
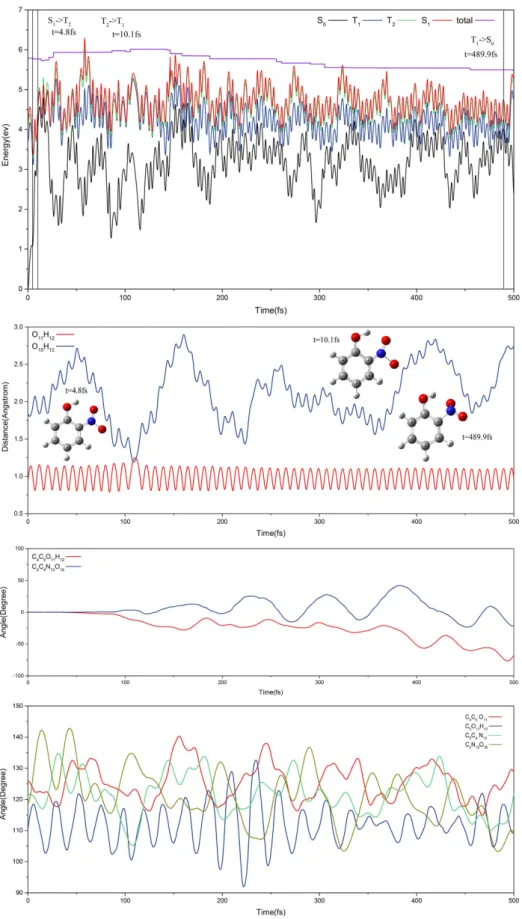
Figure 5 shows the non-hydrogen transfer for a trajectory that hops to the S0 state viaS0T1-ICX at 478.2 fs (this is trajectory staying the longest time on T1 state among all non-hydrogen transfer
sam-pling trajectories). Staring from Frank-Condon region on S1 state, the trajectory experiences the following
con-secutive processes, it hops from the S1 to T2 state via S1T2-IC1 at the 4.8 fs and from the T2 to T1 state via conical
intersection at the 10.1 fs. During the evolution period of time, O11H12 and O15H12 have small and large vibrations,
respectively as shown in the second panel of Fig. 5. Two dihedral angles oscillate smoothly while four bond angles oscillate drastically as shown in the third and fourth panels of Fig. 5, respectively. This trajectory propagates a long time on the T1 state accompanying with the twists around the C4C5O11H12 and C5C4N13O15 dihedral angles.
At 478.2 fs, it hops to ground S0 state. In the overall process, the out-of-plane synchronous motion by O11H12
vibrating around C5O11 bond and the nitro group vibrating around C4N13 bond and this reduces the possibility
of hydrogen migration. We show the tunneling hydrogen transfer on T1 state for a typical trajectory in Fig. S5 of
Supplementary Note 3. This case is similar to non-hydrogen transfer as shown in Fig. 5 except that O11H12 and
O15H12 bonds oscillate coherently which differs from the non-hydrogen transfer case.
Figure 6 shows the direct hydrogen transfer on T1 state for a trajectory that takes the aci-nitrophenol
isomer-ization in the ground state finally. This trajectory hops from the S1 to T2 state via S1T2-IC1 at the 16.6 fs and from
the T2 to T1 state via conical intersection at the 18.2 fs. Sequentially, the hydrogen migration is occurred around
Figure 4. Relative population distribution on the S1, T2 and T1 states as a function of time (small window is
Figure 5. Typical trajectory. The first panel is for potential energy profiles along non-hydrogen transfer channel against the time. The second pane is evolution for bond lengths O11H12 and O15H12, the third is for
dihedral angles C4C5O11H12 and C5C4N13O15 and the fourth is for bond angles C4C5O11, C5O11H12, C5C4N13, and
36 fs on the T1 state accompanied by a shortening of the O15H12 bond and an elongation of the O11H12 bond. After
135.0 fs, this trajectory hops from the T1 to S0 state via S0T1-IC1. In the initial stage of evolution on the S0 state,
this trajectory seems attempted back hydrogen transfer, but the reverse process is not successfully taken place. The aci-nitrophenol isomerization reaction appears in the rest of evolution time. The dihedral angle of O14N13O15H12
Figure 7. The same as Fig. 5 except for the direct hydrogen transfer trajectory on T1 state and back
rotates slightly around the C4N13 bond and the transferred H12 turns to away from the original O11, finally the
aci-isomer2 is observed in this trajectory.
Figure 7 shows that the trajectory makes the hydrogen transfer on the T1 and back hydrogen transfer on S0
state. Staring from Frank-Condon region on S1 state, the trajectory hops from the S1 to T2 state via S1T2-IC1 at
the 7.5 fs and from the T2 to T1 state via conical intersection at the 9.1 fs. Then, the hydrogen transfer is complete
and the aci-nitrophenol species is formed at 35 fs on T1 state. During trajectory evolution, the distance between
O15 and H12 is shortened gradually until bond forming, and bond angles of C5O11H12 and C5C4N13 change
dras-tically while dihedral angles of C4C5O11H12 and C5C4N13O15 fluctuate smoothly. At 86.1 fs, trajectory decays to
S0 state via S0T1-IC1. Soon after 130.0 fs, the back hydrogen transfer takes place on S0 state and regenerates the o-nitrophenol. In the rest of evolution, the trajectory shows a periodic fluctuation with H12 vibrates around the
O11H12 bond. We have plotted more cases in Supplementary Note 3 for various more complicated fast and slow
hydrogen transfer processes.
The present trajectory simulation indicates that the aci-nitro isomers are mostly generated on the triplet states, especially on T1 state and this confirms the previous assumption in which the HONO split-off motion is taken
place in the triplet manifold29,61,62. The present simulation also indicates that the o-NP dynamic decay via ISC
ESIPT is mostly in time scale of subpicosecond (expect tunneling ESIPT) and this is in close agreement with the observations for a large number of nitrated polycyclic aromatic compounds63–65. The present simulation shows
that there excites the wide Franck-Condon region with very small energy gap between the first excited singlet S1
and the second triplet T2 states, and this can facilitate the fast ISC radiationless process. Therefore, trajectories
hop from S1 to T2 state along the (O)H-O(NO) barrierless stretching pathway and in the tunneling zone the
ESIPT pathways can generally have lower barrier via triplet states than via S1 state. The present simulation shows
that the (O)H-O(NO) stretch related vibrational modes can enhance ESIPT and the torsion motion of nitro group would hinder the hydrogen transfer reaction.
Conclusion
By applying ab initio on-the-fly nonadiabatic molecular dynamic simulation for intersystem crossing-branched ESIPT for o-nitrophenol with use of global switching trajectory surface hopping algorithm, we have estimated reaction branch ratios and timescales of various ESIPT processes. As is summarized in Fig. 8, there are three decay branches, the first one is non-hydrogen transfer via newly found S0T1-ICX with ratio 47.5% and average
timescale 300 fs, the second branch is tunneling hydrogen transfer with ratios 36.08% and 2.14% taken place on T1 and S1 states, respectively and timescale 10 ps, and the third branch is direct hydrogen transfer with ratios
13.21% and 1.07% taken place on T1 and T2 states, respectively and timescale 40 fs. However, the real lifetimes
on excited states from experimental observation might be longer than the present simulated timescales due to some initial extra kinetic energy maybe enhance hydrogen transfer dynamics29. Finally, we believe that the
pres-ent trajectory-based on-the-fly nonadiabatic molecular dynamic simulation method can be generally applied to ultrafast photophysical and photochemical processes in a unified way involved in conical intersections and intersystem crossings for large-scale simulation.
Figure 8. One-slide summary of simulated branch ratios and its time scales for intersystem crossing-branched ESIPT reactions of o-nitrophenol.
9. Kwon, J. E. & Park, S. Y. Advanced organic optoelectronic materials: Harnessing excited-state intramolecular proton transfer (ESIPT) process. Adv. Mater. 23, 3615–3642 (2011).
10. Crespo-Otero, R., Mardykov, A., Sanchez-Garcia, E., Sander, W. & Barbatti, M. Photo-stability of peptide-bond aggregates: N-methylformamide dimers. Phys. Chem. Chem. Phys. 16, 18877–18887 (2014).
11. Crespo-Otero, R., Kungwan, N. & Barbatti, M. Stepwise double excited-state proton transfer is not possible in 7-azaindole dimer.
Chem. Sci. 6, 5762–5767 (2015).
12. Tseng, H.-W. et al. Excited-state intramolecular proton-transfer reaction demonstrating anti-Kasha behavior. Chem. Sci. 7, 655–665 (2016).
13. Sobolewski, A. L., Domcke, W. & Hättig, C. Tautomeric selectivity of the excited-state lifetime of guanine/cytosine base pairs: The role of electron-driven proton-transfer processes. Proc. Natl. Acad. Sci. USA 102, 17903–17906 (2005).
14. Sobolewski, A. L. & Domcke, W. Computational studies of the photophysics of hydrogen-bonded molecular systems J. Phys. Chem.
A, 111, 11725–11734 (2007).
15. Shemesh, D., Sobolewski, A. L. & Domcke, W. Efficient excited-state deactivation of the gly-phe-ala tripeptide via an electron-driven proton-transfer process. J. Am. Chem. Soc. 131, 1374–1375 (2009).
16. Li, Q., Migani, A. & Blancafort, L. Irreversible phototautomerization of o-phthalaldehyde through electronic relocation. Phys. Chem.
Chem. Phys. 14, 6561–6568 (2012).
17. Zhao, J., Yao, H., Liu, J. & Hoffmann, M. R. New excited-state proton transfer mechanisms for 1,8-Dihydroxydibenzo[a,h]phenazine.
J. Phys. Chem. A 119, 681–688 (2015).
18. Leyva, V. et al. A non-adiabatic quantum-classical dynamics study of the intramolecular excited state hydrogen transfer in ortho-nitrobenzaldehyde. Phys. Chem. Chem. Phys. 13, 14685–14693 (2011).
19. Daengngern, R. et al. Excited-state intermolecular proton transfer reactions of 7-azaindole (MeOH)n (n = 1–3) clusters in the gas phase: On-the-fly dynamics simulation. J. Phys. Chem. A 115, 14129–14136 (2011).
20. Cui, G., Lan, Z. & Thiel, W. Intramolecular hydrogen bonding plays a crucial role in the photophysics and photochemistry of the GFP chromophore. J. Am. Chem. Soc. 134, 1662–1672 (2012).
21. Spörkel, L., Cui, G. & Thiel, W. Photodynamics of schiff base salicylideneaniline: trajectory surface-hopping simulations. J. Phys.
Chem. A 117, 4574–4583 (2013).
22. Xia, S.-H., Xie, B.-B., Fang, Q., Cui, G. & Thiel, W. Excited-state intramolecular proton transfer to carbon atoms: nonadiabatic surface-hopping dynamics simulations. Phys. Chem. Chem. Phys. 17, 9687—9697 (2015).
23. Nagaya, M., Kudoh, S. & Nakata, M. Infrared spectrum and structure of the aci-nitro form of 2-nitrophenol in a low-temperature argon matrix. Chem. Phys. Lett. 427, 67–71 (2006).
24. Wei, Q., Yin, H.-M., Sun, J.-L., Yue, X.-F. & Han, K.-L. The dynamics of OH channel in the 266 and 355 nm photodissociation of 2-nitrophenol. Chem. Phys. Lett. 463, 340–344 (2008).
25. Orozco-Gonzalez, Y., Coutinho, K., Peon, J. & Canuto, S. Theoretical study of the absorption and nonradiative deactivation of 1-nitronaphthalene in the low-lying singlet and triplet excited states including methanol and ethanol solvent effects. J. Chem. Phys.
137, 054307–054314 (2012).
26. Rafiq, S., Yadav, R. & Sen, P. Femtosecond excited-state dynamics of 4-nitrophenyl pyrrolidinemethanol: Evidence of twisted intramolecular charge transfer and intersystem crossing involving the nitro group. J. Phys. Chem. A 115, 8335–8343 (2011). 27. Vogt, R. A., Reichardt, C. & Crespo-Hernández, C. E. Excited-state dynamics in nitro-naphthalene derivatives: Intersystem crossing
to the triplet manifold in hundreds of femtoseconds. J. Phys. Chem. A 117, 6580–6588 (2013).
28. Takezaki, M., Hirota, N. & Terazima, M. Nonradiative relaxation processes and electronically excited states of nitrobenzene studied by picosecond time-resolved transient grating method. J. Phys. Chem. A 101, 3443–3448 (1997).
29. Ernst, H. A. et al. Ultrafast dynamics of o-nitrophenol: An experimental and theoretical study. J. Phys. Chem. A 119, 9225–9235 (2015).
30. Xu, C., Yu, L., Zhu, C. & Yu, J. Photoisomerization reaction mechanisms of o-nitrophenol revealed by analyzing intersystem crossing network at the MRCI level. J. Phys. Chem. A 119, 10441–10450 (2015).
31. Granucci, G., Persico, M. & Spighi, G. Surface hopping trajectory simulations with spin-orbit and dynamical couplings. J. Chem.
Phys. 137, 22A501–22A509 (2012).
32. Hu, W., Lendvay, G., Maiti, B. & Schatz, G. C. Trajectory surface hopping study of the O(3P) + Ethylene reaction dynamics. J. Phys.
Chem. A 112, 2093–2103 (2008).
33. Xu, X., Zheng, J., Yang, K. R. & Truhlar, D. G. Photodissociation dynamics of phenol: Multistate trajectory simulations including tunneling. J. Am. Chem. Soc. 136, 16378–16386 (2014).
34. Weingart, O., Lan, Z., Koslowski, A. & Thiel, W. Chiral pathways and periodic decay in cis-azobenzene photodynamics. J. Phys.
Chem. Lett. 2, 1506–1509 (2011).
35. Fazzi, D., Barbatti, M. & Thiel, W. Modeling ultrafast exciton deactivation in oligothiophenes via nonadiabatic dynamics. Phys.
Chem. Chem. Phys. 17, 7787–7799 (2015).
36. Barbatti, M. Photorelaxation induced by water–chromophore electron transfer. J. Am. Chem. Soc. 136, 10246–10249 (2014). 37. Yu, L., Xu, C., Lei, Y., Zhu, C. & Wen, Z. Trajectory-based nonadiabatic molecular dynamics without calculating nonadiabatic
coupling in the avoided crossing case: Trans ↔ cis photoisomerization in azobenzene. Phys. Chem. Chem. Phys. 16, 25883–25895 (2014).
38. Yu, L., Xu, C. & Zhu, C. Probing the π → π * photoisomerization mechanism of cis-azobenzene by multi-state ab initio on-the-fly trajectory dynamics simulation. Phys. Chem. Chem. Phys. 17, 17646–17660 (2015).
39. Nanbu, S., Ishidab, T. & Nakamura, H. Future perspectives of nonadiabatic chemical dynamics. Chem. Sci. 1, 663–674 (2010). 40. Tapavicza, E., Tavernelli, I., Rothlisberger, U., Filippi, C. & Casida, M. E. Mixed time-dependent density-functional theory/classical
trajectory surface hopping study of oxirane photochemistry. J. Chem. Phys. 129, 124108 (2008).
constant and scattering matrix: The Landau–Zener case. J. Chem. Phys. 97, 8497–8514 (1992).
52. Zhu, C. & Nakamura, H. The two-state linear curve crossing problems revisited. III. Analytical approximations for Stokes constant and scattering matrix: Nonadiabatic tunneling case. J. Chem. Phys. 98, 6208–6222 (1993).
53. Köppel, H., Gronki, J. & Mahapatra, S. Construction scheme for regularized diabatic states. J. Chem. Phys. 129, 2377–2388 (2001). 54. Verlet, L. Computer “experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 159,
98–103 (1967).
55. Werner, H.-J. et al. MOLPRO, version 2009.1, a package of ab initio programs, 2009. See http://www.molpro.net.
56. Parkera, D. S. N., Minnsa, R. S., Penfoldb, T. J., Worthb, G. A. & Fieldinga, H. H. Ultrafast dynamics of the S1 excited state of benzene.
Chem. Phys. Lett. 469, 43–47 (2009).
57. Marian, C. M. Spin–orbit coupling and intersystem crossing in molecules. WIREs Comput. Mol. Sci. 2, 187–203 (2012).
58. Hare, P. M., Crespo-Hernández, C. E. & Kohler, B. Solvent-dependent photophysics of 1-cyclohexyluracil:Ultrafast branching in the initial bright state leads nonradiatively to the electronic ground state and a long-lived 1nπ * State. J. Phys. Chem. B 110, 18641–18650
(2006).
59. Hare, P. M., Crespo-Hernández, C. E. & Kohler, B. Internal conversion to the electronic ground state occurs via two distinct pathways for pyrimidine bases in aqueous solution. Proc. Natl. Acad. Sci. USA 104, 435–440 (2007).
60. Jose, D. & Datta, A. Tunneling governs intramolecular proton transfer in thiotropolone at room temperature. Angew. Chem. Int. Ed.
51, 9389 –9392 (2012).
61. Bejan, I. et al. J. The photolysis of ortho-nitrophenols: a new gas phase source of HONO. Phys. Chem. Chem. Phys. 8, 2028–35 (2006). 62. Cheng, S. B., Zhou, C. H., Yin, H. M., Sun, J. L. & Han, K. L. OH produced from o-nitrophenol photolysis: a combined experimental
and theoretical investigation. J. Chem. Phys. 130, 234311–234319 (2009).
63. Zugazagoitia, J. S., Almora-Diaz, C. X. & Peon, J. Ultrafast intersystem crossing in 1-nitronaphthalene. An experimental and computational study. J. Phys. Chem. A 112, 358–365 (2008).
64. Collado-Fregoso, E., Zugazagoitia, J. S., Plaza-Medina, E. F. & Peon, J. Excited-state dynamics of nitrated push-pull molecules: the importance of the relative energy of the singlet and triplet manifolds. J. Phys. Chem. A 113, 13498–13508 (2009).
65. Plaza-Medina, E. F., Rodriguez-Cordoba, W. & Peon, J. Role of upper triplet states on the photophysics of nitrated polyaromatic compounds: S1 lifetimes of singly nitrated pyrenes. J. Phys. Chem. A 115, 9782–9789 (2011).
Acknowledgements
This work is supported by Ministry of Science and Technology of the Republic of China under grant no. 103-2113-M-009 -007-MY3. C.X. thanks support from visiting student fellowship by National Chiao Tung University. L.Y. thanks support from Postdoctoral Fellowship by Ministry of Science and Technology of the Republic of China under grant no. 103-2811-M-009 -048. C.Z. thanks the MOE-ATU project of the National Chiao Tung University for support.
Author Contributions
C.X., L.Y. and C.Z. designed the simulations and the framework for trajectory surface hopping involving in both internal conversions and intersystem crossings. C.X. and L.Y. made the code for dynamic part and C.X. performed the simulations. C.X., L.Y., C.Z., J.Y. and Z.C. interpreted the data and analyzed excited-state intramolecular proton transfer for o-Nitrophenol. C.X. and C.Z. wrote the paper.
Additional Information
Supplementary information accompanies this paper at http://www.nature.com/srep Competing financial interests: The authors declare no competing financial interests.
How to cite this article: Xu, C. et al. Intersystem crossing-branched excited-state intramolecular proton transfer for o-nitrophenol: An ab initio on-the-fly nonadiabatic molecular dynamic simulation. Sci. Rep. 6, 26768; doi: 10.1038/srep26768 (2016).
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/