Urinary bladder cancer is among the most common
cers. In 2008, an estimated 69000 new cases of bladder
can-cer were diagnosed in the United States (51000 in men and
18000 in women) and approximately 14000 deaths from
bladder cancer were reported.
1)Its prevalence in the U.S.A.
and worldwide is approximately 490000 and over one
mil-lion, which makes bladder cancer a significant global public
health issue.
2)Approximately 70% of all urothelial bladder
cancer cases are classified as superficial bladder cancer
(SBC), i.e., non-muscle-invasive.
3)The current strategy for treating superficial bladder cancer
is transurethral tumor resection followed by intravesical
adju-vant treatment (chemo- and/or immunotherapy) to reduce
re-currence risk.
4,5)Although Bacillus Calmette-Guerin (BCG)
immunotherapy is currently the most potent topical
treat-ment, it often has local or systemic adverse effects, and 30%
of high-risk patients who respond poorly to BCG still
ulti-mately require cystectomy.
6,7)Therefore, more active
chemotherapy agents are needed for patients who respond
poorly to BCG.
Gemcitabine, a novel deoxycytidine analogue, is an
in-hibitor of DNA synthesis with a broad spectrum of antitumor
activity. This agent, which has overall response rates ranging
from 22.5 to 28%, is highly effective and well tolerated as a
first- or second-line single-agent therapy for treating
metasta-tic transitional cell carcinoma.
8,9)Moreover, gemcitabine
in-duces apoptosis through Fas upregulation without activating
nuclear factor-kappa B (NF-
kB). Hence, it may be more
ef-fective than other anticancer drugs such as doxorubicin,
mit-omycin C and cisplatin for reducing undesirable side effects
such as proliferation, migration, immortality, and inhibition
of apoptosis. Therefore, gemcitabine is a strong candidate for
intravesical therapy in SBC patients who are refractory to
BCG.
10—13)However, after intravesical administration of high
doses (40 mg/ml) of gemcitabine in saline solution,
signifi-cant systemic absorption can still cause gastrointestinal,
bladder, and bone marrow toxicity, which limits its clinical
value.
14,15)Therefore, this study attempted to develop a
deliv-ery vehicle, which can increase gemcitabine accumulation in
bladder tissue and/or decrease systemic exposure for
effec-tive intravesical administration with minimal side effects.
Microemuslions are dispersions of oil in water (o/w) or of
water in oil (w/o) that are thermodynamically stable due to
the significant reduction of interfacial tension by adsorption
of surface amphiphiles. Microemulsions have been studied
intensively in recent years because of their ease of production
and their unique properties, including thermodynamic
stabil-ity, drug solubilstabil-ity, and drug permeability.
16—18)Previous
studies
19,20)pointed that microemulsions formulated with
non-irritant components such as non-inonic surfactant with a
very low topical LD
5021)
can be applied throughout the body,
which enables significant epidermal localization of the drug.
Therefore, the microemulsion system proposed in this study
was designed to be an intravesical vehicle for delivering
gemcitabine to the bladder. Microemulsions were prepared
using a mixture of polyoxyethylene sorbitan monooleate
(Tween) and sorbitan monolaurate (Span) as surfactant,
ethanol as cosurfactant, isopropyl myristate (IPM) as oil
phase, and distilled water as aqueous phase. The
physico-chemical properties of the microemulsions, including their
electrical conductivity, droplet size, and viscosity as well as
in vitro drug release properties were evaluated. In vivo
stud-ies including drug concentration in plasma, drug
accumula-tion in bladder tissue, and histological changes in tissue were
performed in a rat model to evaluate the effectiveness and
safety of the microemulsion delivery system.
Experimental
Materials Gemcitabine hydrochloride was purchased from Scinopharm (Taiwan). Sorbitan monolaurate (Span) was from Tokyo Chemical Industry (Japan). Polyoxyethylene sorbitan monooleate (Tween) was acquired from Showa Corporation (Japan). Sodium pentanesulfonic acid was from Wako Pure Chemical (Japan). Isopropyl myristate (IPM), perchloric acid and sodium phosphate were obtained from Merck Chemicals (Germany). Aceta-minophen and rhodamine B base were purchased from Sigma-Aldrich (U.S.A.). Tetrahydrouridine was obtained from Calbiochem (U.S.A.). All other chemicals and solvents were analytical reagent grade.
Preparation of Gemcitabine or Rhodamine Microemulsion
Formula-Microemulsions for Intravesical Delivery of Gemcitabine
Yi-Hung T
SAI,
aYi-Hang H
SIEH,
bYaw-Bin H
UANG,
aJui-Sheng C
HANG,
bChi-Te H
UANG,
band
Pao-Chu W
U*,baGraduate Institute of Clinical Pharmacy, Kaohsiung Medical University; and bSchool of Pharmacy, Kaohsiung Medical
University; 100 Shih-Chuan 1st Road, Kaohsiung 80708, Taiwan.
Received May 22, 2010; accepted August 13, 2010; published online August 13, 2010
The objective of this work was to develop a safe and effective delivery vehicle for topical treatment of gem-citabine. The physicochemical properties, drug release rate, drug level in plasma and bladder, and histological changes of tissue after drug administration were investigated. The electrical conductivity, mean size, and viscos-ity of drug-loaded microemulsions were 0.8—102.0mS/cm, 116.8—322.5 nm, and 42.9—105.0 cpsⴛ103, respec-tively. Gemcitabine loaded microemulsions showed a slower and sustained release. After intravesical administra-tion of aqueous control and microemulsions treated, the drug concentraadministra-tions in plasma were 15.11mg/ml and 2.81—12.82mg/ml, respectively, and the accumulation in bladder were 18.27 mg and 9.12—64.16 mg, respectively. Microemulsions slightly decreased the systemic absorption and significantly enhanced the accumulation in blad-der tissue. Moreover, the preliminary toxicity studies revealed no overt adverse histological changes or tissue irri-tation by the microemulsion application. Therefore, the microemulsions were suggested to be a promising drug carrier for intravesical chemotherapy.
Key words gemcitabine; microemulsion; intravesical administration
© 2010 Pharmaceutical Society of Japan ∗ To whom correspondence should be addressed. e-mail: pachwu@kmu.edu.tw
tions The component rations of microemulsion formulations are listed in Table 1. The aqueous phase consisted of double-distilled water containing 40% of ethanol (cosurfactant) was prepared. The surfactant mixture of Tween/Sapn⫽3/2 and IPM was mixed well. Then the aqueous phase was added to the oily phase drop by drop. The clear and transparent microemul-sions were obtained under a vortex shaken at room temperature. Gemc-itabine and rhodamine were dissolved in the final microemulsion formula-tions to obtain concentraformula-tions of 1% and 0.5%, respectively.
Microemulsion Characterization The electrical conductivity of the microemulsions was measured by a handheld conductivity meter (WTW Cond 315i, SUNTEX, Germany) at 25⫾2 °C. Average particle sizes of gem-citabine microemulsions were determined by photo correlation spectroscopy by laser light scattering (Zetasizer 3000HSA, Malvern, U.K.) using a he-lium-neon laser with a l of 633 nm. Samples were loaded into 1 cm2
cylin-drical cuvettes and placed in a thermostated scattering chamber. Light scat-tering was monitored at a fixed angle of 90° and a fixed temperature of 25 °C.
The viscosity of the microemulsions was measured using a cone-and-plate viscometer (Brookfield, Model LVDV-II, U.S.A.) maintained at 37 °C. The x was read 30 s after y was measured, at which time the level of z had stabi-lized. The sample was sheared at a rate of 20 rpm. All experiments were re-peated three times, and the average results were recorded.
In-Vitro Gemcitabine Release Gemcitabine release rates from the mi-croemulsions were measured through a cellulose membrane (CelluSep®T2
with a molecular weight cutoff of 6000—8000, Sartorious, Goettingen, Ger-many). Franz diffusion cells with a diffusion area of 3.46 cm2and 20 ml of
receptor volume of pH 7.4 phosphate-citrate buffer were used. One milliliter of drug-loaded microemulsion was dosed in the donor compartment. The system was kept in a temperature-controlled water bath to maintain the donor compartment temperature at 37 °C, and the receptor phase was stirred continuously at 600 rpm. At predetermined time intervals, 0.5 ml samples were taken and replaced by the same volume of fresh preheated receptor medium. Gemcitabine concentrations were determined by HPLC. Each ex-periment was done in triplicate.
Cumulative release of gemcitabine was plotted against square root of time:
Q(t)⫽K⫻t(1/2)
where Q(t) is the cumulative amount (mg/cm2) of gemcitabine released in
time t (⬍60%), K (mg/(h1/2cm2)) is the kinetic constant indicating
gemc-itabine release rate, and t(1/2)is square root of time.
In Vivo Intravesical Administration of Gemcitabine Sprague-Dawley female rats weighing 200—250 g were used in this study according to the care and use protocol for experimental animals approved by the Institutional Review Board at this institution. Animals were housed in a temperature-con-trolled room with free access to food and water until use.
Each animal was anesthetized by isoflurane. The residual urine was evac-uated by pressing the lower abdomen. A polyurethane catheter (25 gauge, BD Angiocath Plu®, Becton Dickinson Korea, Gyeongbuk, Korea) was
in-serted into the bladder through the urethra. The bladder was washed twice with 0.5 ml normal saline. An 0.8 ml quantity of gemcitabine microemulsion or saline solution (as control) was then instilled into the bladder and main-tained for 1 h by ligating the urethra orifice using a cotton thread under isoflurane anesthesia. One hour after the instillation, the cotton thread was
cut off and the isoflurane was moved out. The animals will promptly recover consciousness, and then evacuate the residual formulation by urine mic-turate. The animals were euthanized at 0.5, 1, and 2 h after drug instillation, and blood samples were collected in heparinised tubes containing 10ml tetrahydrouridine (1 mg/ml saline) to prevent ex vivo degradation of the gemcitabine by cytidine deaminase in the serum. Blood samples from the jugular vein were examined for systemic exposure of gemcitabine during and after intravesical administration of gemcitabine. The heparinised tubes were centrifuged for 10 min at 4000 rpm at 4 °C. A pipette was used to trans-fer the top 0.2 ml plasma layer into another tube containing 0.05 ml internal standard of acetaminophen 200mg/ml and 0.1 ml of 1M perchloric acid.
After 5 s vortex, the mixture was incubated in ice bath for 10 min then cen-trifuged at 16000 g for 5 min at 4 °C. The 0.02 ml of clear supernatant was analyzed by HPLC method as reported previously with some modifica-tions.22)
At the end of the intravesical administration experiment, the gemcitabine accumulation in the bladder was also determined by a homogenization method. After wash, the excised bladder cut to small pieces and place into a glass tube containing 2 ml saline in an ice bath. The sample was homoge-nized at 17800 rpm for 1 min. The homogenizer probe was washed with 2 ml of saline to recover residual adhering tissues. The two saline fractions were combined, and then shaken horizontally for 10 min. The resulting solution was centrifuged at 3000⫻g for 5 min. The supernatant was used for the assay by HPLC.22)
HPLC Analysis The HPLC analysis was performed using an Agilent 1200 series HPLC system. A Merck Lichrospher®C18 column (250⫻4 mm
i.d., particle size 5mm) was used. The mobile phase was a mixture of aque-ous phase containing 3 mMpentanesulfonic acid and 50 mMsodium phos-phate (adjusted to pH 3.0 by phosphoric acid) and acetonitrile at a 95 : 5 ratio and flow rate was 0.9 ml/min. The UV detection was performed at 278 nm. The detection limits for drug concentration in plasma and drug ac-cumulation in the bladder were 25 ng/ml and 250 ng/bladder, respectively.
Penetration Depth Measurement by Confocal Laser Scanning Mi-croscopy (CLSM) Permeation of bladder tissue by rhodamine-loaded mi-croemulsions was investigated using CLSM (FV 500, Olympus, Tokyo, Japan). Rats were sacrificed at 1 h after drug administration, and bladders were removed intact. Connective tissue, lipoid tissue, and drug residues were removed from the bladder walls. The bladders were then sectioned into 1 mm2specimens to compare the penetration depths of the microemulsion
formulations. Bladder wall thickness was measured by CLSM through the z axis at ca. 20mm increments. Optical excitation was performed using a 500 nm argon laser, and fluorescence emission was detected at 540 nm. Two different sites were evaluated in each bladder. Fluorescence emissions were measured in darkness to avoid errors caused by ambient light.
Histopathological Evaluation Rats were sacrificed at 1 h after blank microemulsion instillation was performed. The bladders were removed in-tact, cleaned to remove connective and lipoid tissue from around the wall, and weighed to test for presence of edema. The bladders were then fixed in 10% buffered formaldehyde for 24 h. Each bladder was cut into three equal sections from the dome to the bottom. Each piece was dehydrated using ethanol and embedded in paraffin. At least three cross sections 20mm thick were taken from each section of bladder for hematoxylin–eosin staining.
Statistical Analysis Group comparisons were performed using analysis of variance (ANOVA) tests. A p value less than 0.05 was considered statisti-Table 1. Composition, Particle Size, Polydispersity Index (PI), Electrical Conductivity (EC) and Viscosity of Blank and Gemcitabine Loaded Microemul-sions
IPM W S Particle size
PI EC Viscosity (%) (%) (%) (nm) (mS/cm) (cps⫻103) Aa) 13 35 52 299.7⫾22.1 0.78⫾0.06 28.6 42.2⫾0.7 Ba) 50 10 40 122.7⫾1.8 0.37⫾0.01 0.5 44.3⫾0.5 Ca) 20 20 60 323.8⫾27.7 0.79⫾0.09 2.3 108.8⫾3.4 Ab) 13 35 52 176.1⫾10.7 0.62⫾0.04 102.0 42.9⫾1.0 Bb) 50 10 40 322.5⫾9.6 0.75⫾0.06 0.8 45.1⫾1.0 Cb) 20 20 60 116.8⫾2.5 0.30⫾0.01 6.6 105.0⫾1.7 Ac) 13 35 52 163.7⫾13.7 0.68⫾0.11 103.6 49.8⫾0.4 Bc) 50 10 40 323.4⫾10.6 0.68⫾0.05 0.9 43.9⫾3.5 Cc) 20 20 60 120.3⫾3.8 0.45⫾0.04 6.3 102.3⫾2.4
Microemulsion composited of water (W) containing 40% cosurfactant, isopropyl myristate (IPM), and mixture surfactant (S) of Tween/Span⫽3/2. a) Blank microemul-sions. b) Gemcitabine loaded microemulsions. c) After 2 months storage of gemcitabine loaded microemulsions.
cally significant. Tukey test was then performed to analyze two groups con-secutively.
Results and Discussion
Physicochemical Characterization of Microemulsions
Translucent and stable microemulsions were formed by
mix-ing different components of the oil phase with the aqueous
phase containing cosurfactant and surfactant. The
physico-chemical parameters of blank and drug loaded
microemul-sions are shown in Table 1. The electrical conductivity of all
blank microemulsions was 0.5 to 28.6
mS/cm, which
ex-ceeded the electrical conductivities of oil phase (0.0
mS/cm),
aqueous phase with cosurfactant (0.4
mS/cm), and surfactant
(0.40
mS/cm). The results were consistent with a previous
re-port
23)that microemulsions, even w/o type, can increase the
electrical conductivity of formulations. The w/o
microemul-sion B with higher content of oil had lower electrical
conduc-tivity than the o/w type microemulsions A and C with lower
content of oil. As expected, addition of gemcitabine
hy-drochloride significantly increased the electrical conductivity
of both type microemulsions. Electrical conductivity of the
w/o type microemulsion B increased from 0.5 to 0.8
mS/cm,
a 1.9 fold increase, whereas those of o/w type
microemul-sions A and C increased from 2.3—28.6
mS/cm to 6.6—
102.0
mS/cm, a 2.9—15.5 fold increase. These data indicate
that gemcitabine dissolution was fastest in aqueous phase.
Moreover, electrical conductivity of both blank and
drug-loaded microemulsions correlated with that of proportion of
aqueous phase in formulation. The increased electrical
con-ductivity may have been caused by increased dissociation of
surfactant as a function of water content.
24)Table 1 shows that, at 37 °C, blank and drug-loaded
mi-croemulsions exhibited viscosities of 42.2—108.8 cps
⫻10
3and 42.9—105.0 cps
⫻10
3, respectively. Viscosity was
unaf-fected by drug incorporation (p
⬎0.05), whereas increased
surfactant correlated with increased viscosity (A vs. C,
p
⬍0.05).
Mean droplet size was small in all blank and drug-loaded
microemulsions (122.7—323.8 nm and 116.8—322.5 nm,
re-spectively). Notably, the drug-loaded formulations had the
smallest droplet size in o/w microemulsions and the largest
droplet size in w/o microemulsions. The phenomenon might
be attributed to that drug is solubilized at the interface of
mi-croemulsion droplets and shrinks the droplets by interacting
with the surfactant.
25)After 2 months storage at room temperature, none of the
microemulsions in the current study revealed changes in
clar-ity, phase behavior, or particle size (Table 1). All
microemul-sions exhibited gemcitabine concentrations above 98.0
⫾
1.5%, which indicated that no degradation occurred.
In Vitro Gemcitabine Release
It is well known that
mi-croemulsion type, internal structure, size, and viscosity
might influence the drug release from microemulsions.
26)In
this study, two type microemulsions with different
physico-chemical properties (Table 1) were investigated. Figure 1
shows their release profiles. Gemcitabine release from saline
was studied as a control. Figure 1 shows that drug release
from microemulsions was slower than that from saline (78%
release in 1 h), which indicates the potential effectiveness of
microemulsions as drug delivery vehicles for controlled
re-lease.
27)To facilitate comparison of different formulations,
the release rate constant was calculated from the slope of the
linear portion of the plots of cumulative drug quantity
re-leased against t
1/2and expressed in
mg/(cm
2h
1/2). The release
rates of microemulsions A, B, and C were 630.1
⫾55.0,
390.3
⫾96.2, and 141.1⫾9.6
mg/(cm
2h
1/2), respectively,
indi-cating that the ratio of ingredients in the microemulsions was
an important factor for modulating drug release. Generally,
increased emulsion viscosity tends to decrease drug release
rate by increasing structural rigidity and droplet size while
reducing total surface area.
28—30)The current study revealed
a strong correlation (r
⫽0.886) between viscosity and release
rate, but a weak correlation (r
⫽0.291) between droplet size
and release rate. In addition, surfactant quantity
corre-sponded positively with viscosity but negatively with release
rate and droplet size (A vs. C, p
⬍0.05). The result might be
due to a decreased thermodynamic activity of the drug in
mi-croemulsion with higher concentrations of surfactant.
31)An-other possibility was that drug diffusion through the double
layer microemulsion might be a rate-determining step, as the
viscosity plays an important role in controlling the release of
the drug into the receptor.
32)In Vivo Intravesical Instillation of Gemcitabine
The
rats were given intravesical doses of 0.8 ml gemcitabine
saline solution (control) or drug-loaded microemulsions, and
the doses were maintained in the bladder for 1 h by ligating
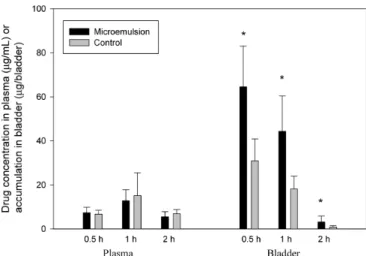
the urethral orifice. Figures 2 and 3 show the plasma
concen-trations and gemcitabine accumulations in the bladder, which
were determined at 0.5, 1, and 2 h after drug instillation and
used to evaluate the safety and efficacy of the
microemul-sions. Formulations were ranked by measuring the plasma
drug concentration at 1 h after drug instillation as control
⬎
A
⬎B⬎C. In terms of drug accumulation in the bladder, they
were ranked as A
⬎B⬎control⬎C. Strong correlations were
noted between release rate and bladder accumulation
(r
⫽0.9996) and between release rate and plasma
concentra-tion (r
⫽0.9994), which indicated that the permeation and
ac-cumulation of gemcitabine delivered by microemulsion
de-pended on the release rate of drug from the formulation.
Moreover, plasma concentrations corresponded positively
with gemcitabine accumulations in the bladder. These
labora-tory results agree with earlier canine studies,
14)which
re-ported significant dose-dependent systemic absorption of
Fig. 1. In Vitro Release–Time Profiles of Gemcitabine from
gemcitabine after intravesical administration.
Compared to controls, subjects treated with
microemul-sion C showed lower plasma concentration (p
⬍0.05) and less
bladder accumulation (p
⬎0.05). In subjects treated with
mi-croemulsion B, the plasma concentration and bladder
accu-mulation were comparable to those of controls (p
⬎0.05).
Compared to controls, subjects treated with microemulsion
A, the plasma concentration was slightly decreased (p
⬎0.05)
and the bladder accumulation was significantly increased
(p
⬍0.05). The comparison results showed that the bladder
accumulation was increased without a corresponding
in-crease in plasma drug concentration when compared to the
control. The result was consistent with previous reports
20,33)which pointed that microemulsions can efficiently promote
localization without concomitantly increasing systemic side
effects.
In addition, plasma concentration and bladder
accumula-tion of gemcitabine were also evaluated at 0.5 and 2 h after
instillation of microemulsion A and control. Figure 3 shows
that, in both groups of microemulsion and control,
gemc-itabine was absorbed and distributed to the bladder wall by
0.5 h. After 2 h, most of the drug was eliminated, which
indi-cated its short half-life after intravesical administration,
pos-sibly due to urine maturation and excretion. Previous dog
studies
14)have also reported a short half-life after intravesical
administration of 350 mg gemcitabine. However, as expected,
bladder accumulation and plasma concentration at 0.5 and
2 h, respectively, were higher in subjects treated with
mi-croemulsions than in controls.
In Vivo CLSM Analysis
To clarify the permeation
depth of microemulsion in the bladder, superficial
distribu-tion of rhodomine-loaded microemulsion was analyzed by
CLSM. Figure 4 depicts the optical scanning results for the
superficial layer at ca. 20
mm increments for 16 fragments
from the surface to the muscle (left to right, top to bottom).
After intravesical administration of microemulsion A, only
pale signals were detected in the first image (ca. 20
mm) and
in the 9—16th images (180—300
mm), and most signals
were in the 140—160
mm range.
Histological Examination of Bladder Tissue
Safety is
a key factor in delivery vehicle formulation. For preliminary
safety evaluation of the experimental formulation,
histologi-cal photographs (Fig. 5) were compared between controls
treated with saline for 1 h and the group treated with blank
microemulsion A for 1 h. Mild signs of inflammatory
re-sponse (subepithelial leukocyte infiltration) and epithelial
cellular nuclear enlargement was seen in drug solution
Fig. 3. Plasma Concentration and Accumulation in Bladder at 0.5, 1 and 2 h after Gemcitabine Microemulsion A and Aqueous Control Solution In-travesical Administration in Rats (n⫽6)
∗ p⬍0.05 compared with control group.
Fig. 4. Confocal Laser Scanning Microscopic (CLSM) Micrographs of the Rhodamine Intensity in Bladder after Intravesical Administration of Mi-croemulsion A for 1 h
The full thickness was divided into 16 fragments from the surface of the bladder mu-cosa (left to right, top to bottom). Images below the photographs of the 16 segments are the sum of all segments.
Fig. 2. Plasma Concentration and Accumulation in Bladder at 1 h after Gemcitabine Microemulsions and Aqueous Solution Intravesical Adminis-trations in Rats (n⫽6)
∗ p⬍0.05 compared with saline control group.
treated group. The photographs revealed no bladder wall
damage in the microemulsion-treated groups, and their
mor-phologies were similar to those of controls.
Conclusion
This study evaluated the potential use of microemulsions
as vehicles for topical delivery of gemcitabine. The results
suggest that microemulsions efficiently promote gemcitabine
localization into the bladder wall. By enhancing gemcitabine
accumulation in the bladder wall, the microemulsions may be
useful for optimizing drug delivery without concomitantly
increasing systemic side effects.
Acknowledgment This work was supported by the National Science Council of Taiwan (NSC 95-2320-B-037-022).
References
1) Jemal A., Siegel R., Ward E., Hao Y., Xu J., Murray T., Thun M. J., CA
Cancer J. Clin., 58, 71—96 (2008).
2) Lerner S. P., Urol. Oncol., 23, 275—279 (2005).
3) Kirkali Z., Chan T., Manoharan M., Algaba F., Busch C., Cheng L., Kiemeney L., Kriegmair M., Montironi R., Murphy W. M., Sesterhenn I. A., Tachibana M., Weider J., Urology, 66, 4—34 (2005).
4) Mugabe C., Hadaschik B. A., Kainthan R. K., Brooks D. E., So A. I., Gleave M. E., Burt H. M., BJU Int., 103, 978—986 (2009).
5) Shen Z., Shen T., Wientjes M. G., O’Donnell M. A., Au J. L., Pharm.
Res., 25, 1500—1510 (2008).
6) Nseyo U. O., Lamm D. L., Semin. Surg. Oncol., 13, 342—349 (1997). 7) Nadler R. B., Catalona W. J., Hudson M. A., Ratliff T. L., J. Urol., 152,
367—373 (1994).
8) Lorusso V., Pollera C. F., Antimi M., Luporini G., Gridelli C., Frassineti G. L., Oliva C., Pacini M., De Lena M., Eur. J. Cancer, 34, 1208—1212 (1998).
9) Moore M. J., Tannock I. F., Ernst D. S., Huan S., Murray N., J. Clin.
Oncol., 15, 3441—3445 (1997).
10) Serretta V., Galuffo A., Pavone C., Allegro R., Pavone-MacAluso M.,
Urology, 65, 65—69 (2005).
11) Gardmark T., Carringer M., Beckman E., Malmstrom P. U., Urology, 66, 527—530 (2005).
12) Dalbagni G., Russo P., Bochner B., Ben-Porat L., Sheinfeld J., Sogani P., Donat M. S., Herr H. W., Bajorin D., J. Clin. Oncol., 24, 2729— 2734 (2006).
13) Mohanty N. K., Nayak R. L., Vasudeva P., Arora R. P., Urol. Oncol., 26, 616—619 (2008).
14) Cozzi P. J., Bajorin D. F., Tong W., Nguyen H., Scott J., Heston W. D., Dalbagni G., Clin. Cancer Res., 5, 2629—2637 (1999).
15) Hendricksen K., Witjes J. A., Curr. Opin. Urol., 16, 361—366 (2006). 16) Paolino D., Ventura C. A., Nistico S., Puglisi G., Fresta M., Int. J.
Pharm., 244, 21—31 (2002).
17) Peltola S., Saarinen-Savolainen P., Kiesvaara J., Suhonen T. M., Urtti A., Int. J. Pharm., 254, 99—107 (2003).
18) Yuan Y., Li S. M., Mo F. K., Zhong D. F., Int. J. Pharm., 321, 117— 123 (2006).
19) Grundmann-Kollmann M., Behrens S., Peter R. U., Kerscher M.,
Pho-todermatol. Photoimmunol. Photomed., 15, 87—89 (1999).
20) Baroli B., Lopez-Quintela M. A., Delgado-Charro M. B., Fadda A. M., Blanco-Mendez J., J. Controlled Release, 69, 209—218 (2000). 21) Kibbe A. H., “Handbook of Pharmaceutical Excipients,” 3rd ed.,
Phar-maceutical Press, London, 2000.
22) Kirstein M. N., Hassan I., Guire D. E., Weller D. R., Dagit J. W., Fisher J. E., Remmel R. P., J. Chromatogr. B Analyt. Technol. Biomed. Life
Sci., 835, 136—142 (2006).
23) Bumajdad A., Eastoe J., J. Colloid Interface Sci., 274, 268—276 (2004).
24) Baker R. C., Florence A. T., Ottewill R. H., Tadros T. H. F., J. Colloid
Interface Sci., 100, 332—349 (1984).
25) Spernath A., Aserin A., Ziserman L., Danino D., Garti N., J.
Con-trolled Release, 119, 279—290 (2007).
26) Madhusudhan B., Rambhau D., Apte S. S., Gopinath D., J. Drug
Tar-get., 15, 154—161 (2007).
27) Nornoo A. O., Osborne D. W., Chow D. S., Int. J. Pharm., 349, 108— 116 (2008).
28) Chung H., Kim T. W., Kwon M., Kwon I. C., Jeong S. Y., J. Controlled
Release, 71, 339—350 (2001).
29) Spernath A., Aserin A., Adv. Colloid Interface Sci., 128—130, 47—64 (2006).
30) Wu H., Ramachandran C., Bielinska A. U., Kingzett K., Sun R., Weiner N. D., Roessler B. J., Int. J. Pharm., 221, 23—34 (2001). 31) Rhee Y. S., Choi J. G., Park E. S., Chi S. C., Int. J. Pharm., 228, 161—
170 (2001).
32) Ho H. O., Hsiao C. C., Sheu M. T., J. Pharm. Sci., 85, 138—143 (1996).
33) Grundmann-Kollmann M., Behrens S., Krahn G., Leiter U., Ochsendorf F., Kaufmann R., Peter R. U., Kerscher M., Arch.