Cardiovascular Research
Promoter polymorphism G-6A, which modulates angiotensinogen gene expression, is
associated with non-familial sick sinus syndrome
--Manuscript Draft--Manuscript Number:
Full Title: Promoter polymorphism G-6A, which modulates angiotensinogen gene expression, is
associated with non-familial sick sinus syndrome
Short Title: Angiotensinogen gene polymorphism and non-familial SSS
Article Type: Original Article
Keywords: promoter activity, gene polymorphism, non-familial sick sinus syndrome.
Corresponding Author: Jan-Yow Chen, M.D.
China Medical University Hospital
Taichung, TAIWAN, PROVINCE OF CHINA Corresponding Author Secondary
Information:
Corresponding Author's Institution: China Medical University Hospital Corresponding Author's Secondary
Institution:
First Author: Jan-Yow Chen, M.D.
First Author Secondary Information:
All Authors: Jan-Yow Chen, M.D.
Ying-Ming Liou, PhD Hong-Dar Isaac Wu, PhD Kuo-Hung Lin, M.D.
Kuan-Cheng Chang, M.D., PhD All Authors Secondary Information:
Abstract: Aims It is well-known that familial sick sinus syndrome (SSS) is caused by functional
alterations of ion channels and gap junction. Limited information is available on the mechanism of age-related non-familial SSS. Although evidence shows a close link between arrhythmia and the renin-angiotensin system (RAS), it remains to be determined whether the RAS is involved in the pathogenesis of non-familial SSS. Methods and results In this study, 113 patients with documented non-familial SSS and 125 controls were screened for angiotensinogen (AGT) and connexin 40 (Cx40) promoter polymorphisms by gene sequencing, followed by an association study. A luciferase assay was used to determine the transcriptional activity of the promoter polymorphism. The interaction between nuclear factors and the promoter
polymorphism was characterized by an electrophoretic mobility shift assay (EMSA). Association study showed the Cx40 -44/+71 polymorphisms are not associated with nonfamilial SSS; however, it indicated that four polymorphic sites at positions 6, 20, -152, and -217 in the AGT promoter are linked to non-familial SSS. Compared to controls, SSS patients had a lower frequency of the G-6A AA genotype (OR 2.88, 95% CI 1.58-5.22, P = 0.001) and a higher frequency of the G allele at -6 position (OR 2.65, 95% CI 1.54-4.57, P = 0.0003). EMSA and luciferase assays confirmed that nucleotide G at position -6 modulates the binding affinity with nuclear factors and yields a lower transcriptional activity than nucleotide A (P<0.01).
Conclusion G-6A polymorphism, which modulates the transcriptional activity of the AGT promoter, may contribute to non-familial SSS susceptibility.
Suggested Reviewers: San-Jou Yeh, M.D.
Opposed Reviewers: Hilma Holm, M.D.
deCODE genetics, Sturlugata, Iceland
Kari Stefansson, M.D.
Promoter polymorphism G-6A, which modulates angiotensinogen gene expression, is associated with
non-familial sick sinus syndrome
Jan-Yow Chen, Ying-Ming Liou, Hong-Dar Isaac Wu, Kuo-Hung Lin, Kuan-Cheng Chang
Abstract
Aims It is well-known that familial sick sinus syndrome (SSS) is caused by functional alterations of ion
channels and gap junction. Limited information is available on the mechanism of age-related non-familial
SSS. Although evidence shows a close link between arrhythmia and the renin-angiotensin system (RAS), it
remains to be determined whether the RAS is involved in the pathogenesis of non-familial SSS.
Methods and results In this study, 113 patients with documented non-familial SSS and 125 controls were
screened for angiotensinogen (AGT) and connexin 40 (Cx40) promoter polymorphisms by gene sequencing,
followed by an association study. A luciferase assay was used to determine the transcriptional activity of the
promoter polymorphism. The interaction between nuclear factors and the promoter polymorphism was
characterized by an electrophoretic mobility shift assay (EMSA). Association study showed the Cx40-44/+71
polymorphisms are not associated with non-familial SSS; however, it indicated that four polymorphic sites at
positions -6, -20, -152, and -217 in the AGT promoter are linked to non-familial SSS. Compared to controls,
SSS patients had a lower frequency of the G-6A AA genotype (OR 2.88, 95% CI 1.58-5.22, P = 0.001) and a
higher frequency of the G allele at -6 position (OR 2.65, 95% CI 1.54-4.57, P = 0.0003). EMSA and
luciferase assays confirmed that nucleotide G at position -6 modulates the binding affinity with nuclear
factors and yields a lower transcriptional activity than nucleotide A (P<0.01).
Conclusion G-6A polymorphism, which modulates the transcriptional activity of the AGT promoter, may
Dear Dr. Piper:
On behalf of all my co-authors, I would like to submit a manuscript entitled, “Promoter polymorphism G-6A, which modulates angiotensinogen gene
expression, is associated with non-familial sick sinus syndrome”, for consideration
of publication under the section, original articles in the Cardiovascular Research. The
importance of this work is briefly described as follows.
Growing evidence has shown that genetic mutations may lead to familial sick sinus
syndrome (SSS). In contrast to the extensive knowledge available on familial SSS,
limited information is available on the mechanisms underlying age-related
nonfamilial SSS. In this submitting MS, we showed that polymorphism in the
angiotensinogen (AGT) promoter, which might modulate AGT synthesis, is closely
associated with non-familial SSS. The results obtaining with this study may provide
useful information in the prevention and management of the age-related non-familial
SSS.
The contribution of each author was listed as followings:
Jan-Yow Chen, MD: (1) Conception and design (2) Analysis and interpretation of data (3) Drafting of the manuscript.
Ying-Ming Liou, PhD: (1) Conception and design (2) Analysis and interpretation of *Cover Letter/Declaration
the manuscript submitted.
Hong-Dar Isaac Wu, PhD: (1) Analysis and interpretation of data (2) Revising it critically for important intellectual content.
Kuo-Hong Lin, MD: (1) Conception and design (2) Analysis and interpretation of data.
Kuan-Cheng Chang, MD, PhD: (1) Conception and design (2) Analysis and interpretation of data.
The manuscript, or part of it, has neither been published nor is currently under consideration for publication by any other journal
The co-authors have read the manuscript and approved its submission to Cardiovascular Research.
We agree to pay for the cost of printing for colour figures.
No conflict of interest in connection with the submitted article to disclose.
Thank you for your time and consideration.
Jan-Yow Chen, MD
Department of Life Science, National Chung-Hsing University
Division of Cardiology and Department of Medicine, China Medical University Hospital, Taichung, Taiwan
Promoter polymorphism G-6A, which modulates angiotensinogen gene expression, is
associated with non-familial sick sinus syndrome
Jan-Yow Chen,1,2,3 Ying-Ming Liou,1,4 Hong-Dar Isaac Wu,5 Kuo-Hung Lin,2,3 Kuan-Cheng
Chang2,3,
1
Department of Life Sciences, National Chung Hsing University, Taichung, Taiwan
2
Division of Cardiology, Department of Medicine, China Medical University and Hospital, Taichung, Taiwan
3
School of Medicine, China Medical University, Taiwan
4
Graduate Institute of Basic Medical Science, China Medical University, Taichung, Taiwan
5
Department of Applied Mathematics and Institute of Statistics, National Chung-Hsing University, Taichung, Taiwan
Running title: Angiotensinogen gene polymorphism and non-familial SSS
Corresponding author: Ying-Ming Liou
Department of Life Sciences, National Chung Hsing University, 250, Kuo Kuang Road,
Taichung, Taiwan Tel: +886-4-22840416
Email: [email protected] *Manuscript
Abstract
Aims It is well-known that familial sick sinus syndrome (SSS) is caused by functional
alterations of ion channels and gap junction. Limited information is available on the
mechanism of age-related non-familial SSS. Although evidence shows a close link between
arrhythmia and the renin-angiotensin system (RAS), it remains to be determined whether the
RAS is involved in the pathogenesis of non-familial SSS.
Methods and results In this study, 113 patients with documented non-familial SSS and 125
controls were screened for angiotensinogen (AGT) and connexin 40 (Cx40) promoter
polymorphisms by gene sequencing, followed by an association study. A luciferase assay was
used to determine the transcriptional activity of the promoter polymorphism. The interaction
between nuclear factors and the promoter polymorphism was characterized by an
electrophoretic mobility shift assay (EMSA). Association study showed the Cx40 -44/+71
polymorphisms are not associated with non-familial SSS; however, it indicated that four
polymorphic sites at positions -6, -20, -152, and -217 in the AGT promoter are linked to
non-familial SSS. Compared to controls, SSS patients had a lower frequency of the G-6A AA
genotype (OR 2.88, 95% CI 1.58-5.22, P = 0.001) and a higher frequency of the G allele at -6
position (OR 2.65, 95% CI 1.54-4.57, P = 0.0003). EMSA and luciferase assays confirmed
that nucleotide G at position -6 modulates the binding affinity with nuclear factors and yields
a lower transcriptional activity than nucleotide A (P<0.01).
Introduction
Sick sinus syndrome (SSS), including profound sinus bradycardia, sinus arrest,
sino-atrial exit block, and tachy-bradycardia, is a group of abnormal heart rhythms
presumably caused by a malfunction of the sinus node.1,2 The syndrome is prevalent in 1 out
of every 600 individuals over the age of 65 years, and accounts for approximately 50% of
pacemaker implantations.1,2 Growing evidence has shown that genetic mutations in the
hyperpolarization-activated cyclic nucleotide-gated cation channel (HCN-4), the cardiac
sodium channel (SCN5A), and gap junction protein (connexin) may lead to familial SSS.3,4,5
In contrast to the progress in illustrating the mechanism for familial SSS, limited information
is available regarding the mechanism of age-related non-familial SSS.6,7
Gap junctions composed of connexin (Cx) molecules are responsible for the electrical
coupling of cardiac myocytes. In the human heart, there are 3 cardiac connexin isotypes,
Cx40, Cx 43 and Cx45. Cx40 is the major isotype expressed in the atrium. In Cx40 knockout
mice, increased atrial vulnerability has been shown to cause arrhythmogenesis.8 In addition,
Cx40 promoter polymorphism has been linked to congenital atrial standstill and atrial
arrhythmia.4,9 However, it is still unclear whether alterations in gap junction proteins would
contribute to non-familial SSS.
It is generally accepted that the renin-angiotensin system (RAS) can modulate the
functions of the sinus node and cardiac conduction system.10-12 Angiotensin II is known to
receptor in the myocardium, which was lethal, was associated with myocyte hyperplasia,
heart block, and sinus bradycardia.14 However, the role of RAS in the pathogenesis of
age-related non-familial SSS remains to be determined.
Studies analyzing the association between angiotensinogen (AGT) promoter
polymorphism and stroke have indicated that polymorphic alterations of the AGT promoter
modulate its transcriptional activity and cause cerebral vascular diseases.15,16 In addition, a
study using AGT knockout mice showed a feedback mechanism for regulating the expression
of RAS molecules and the AT1 receptor.17 Thus, we hypothesized that AGT promoter
polymorphism modulates the expression of RAS molecules and thereby influences sinus node
function. Here, the data reported indicates a possible relationship between age-related
non-familial SSS and the AGT promoter polymorphism. Apparently, polymorphic variations
in the AGT promoter might contribute to non-familial SSS susceptibility by modulating the
Methods
Study population
A total of 113 consecutively eligible patients with documented SSS were studied. SSS
was diagnosed by symptomatic bradycardia with evidence of sinus node dysfunction.
The criteria for inclusion were symptomatic bradycardia with a documented sinus
pause of greater than 3 seconds or sinus bradycardia of less than 40 beats/min for
more than 1 min while awake.18,19 Other supporting evidences were provided by a
cardiac electrophysiological study to determine the prolonged corrected sinus nodal
recovery time or sinoatrial conduction time. Long sinus pauses and profound sinus
bradycardia were also examined by using a series of electrocardiograms (ECG) and
ambulatory ECG. All SSS patients met the indications for permanent pacemaker
implantation. Patients with a history of familial SSS, severe systemic disease, acute
coronary syndrome, neurogenic or drug-induced bradycardia, or bradycardia with
reversible cause were excluded from this study. The control group consisted of 125
age- and sex-matched unrelated volunteer patients who were free of SSS and
underwent clinical follow-up in the cardiovascular outpatient department of the same
hospital. Informed consent was obtained from each patient. The study protocol was
approved by the institutional review board of the China Medical University Hospital.
Helsinki.
Genotyping and association study
Blood samples from patients were prepared, and genomic DNA was isolated
using a DNA extraction kit (IllustraTM, GE Healthcare). Polymerase chain reactions
(PCRs) were performed with 100 ng genomic DNA, 2-6 pmol of selected primers, 1X
Taq polymerase buffer, and 0.25 units of AmpliTaq GoldTM polymerase (Roche) in a
final reaction volume of 50 L using a programmable thermal cycle (GeneAmp PCR
system 2700, Applied Biosystems, CA). The primers for the AGT promoter were
5’-CCTCTTGGGGGTACATCTCC-3’ (forward) and
5’-TCCTAGCCCACAGCTCAGTT-3’ (reverse). The primers for the Cx40 promoter
were 5’-AGGCTACGAGGAGGTGGA-3’ (forward) and
5’-AACTCACAGGTAGAAAGAAAGAGC-3’ (reverse). The gene sequences of the PCR products were subsequently determined by using a gene sequencing analyzer
(ABI 3730 XL DNA Analyzer, Applied Biosystems). An association study between
gene promoter polymorphisms and SSS was performed to measure the frequency of
the genotypes and alleles of the Cx40 and AGT promoters in SSS and control groups.
Construction of expression vectors, transfection, and luciferase activity
The association study showed that polymorphic sites in the AGT promoter were
located within the proximal region of the promoter. Therefore, this region (position −290 to +35 relative to transcription starting site) was amplified by PCR from the genomic DNA of homozygous patients. Primers to amplify AGT polymorphisms
were designed to contain the restriction sites of MluI and BglII (for cloning) and
polymorphic sites for AGT promoter: forward primer, 5’-
ACCGACGCGTAGATGCTCCCGTTTCTGG -3′ (artificial MluI restriction site
underlined); reverse primer, 5′-CGGAAGATCTTCTGCTGTAGTACCCA-3′
(artificial BglII restriction site underlined) (Figure 1). After digestion with MluI and
BglII, PCR products were ligated into the corresponding restriction sites of the pGL3
plasmid containing the luciferase reporter gene according to the manufacturer’s instructions (Promega). The promoter-luciferase constructs containing the -6G and
-6A polymorphic sites are defined as P(-6G) and P(-6A), respectively.
The constructs were transiently transfected into HepG2 (cell line-derived from
human hepatoma; HB-8065; ATCC) and cultured in Dulbecco’s Modified Eagle
Medium (DMEM) without serum using a transient liposome (Lipofetamine2000;
Invitrogen, Carlsbad, CA) cotransfection method. A control vector containing the
beta-galactosidase gene (Promega; 0.2 g) was used as an internal control of
System (PerkinElmer).
Electrophoretic mobility shift assay
Electrophoretic mobility shift assays (EMSA) were performed by using the EMSA “Gel Shift” Kit (Panomics, Fremont, CA, USA). To determine the essential role of the specific position at -6 at the proximal segment of the AGT promoter, 2 different
lengths of oligonucleotides containing the nucleotide A or G at -6 of the AGT
promoter were designed for the longer oligonucleotide, G33 or A33, and for the
shorter oligonucleotide, G23 or A23. The sequences for these double-stranded
oligonucleotides are listed below with the polymorphic sites underlined: G23:5’-GTGACCCGGCCGGGGGAAGAAGC-3’, A23:5’-GTGACCCGGCCAGGGGAAGAAGC-3’,
G33:5’-AAATAGGGCATCGTGACCCGGCCGGGGGAAGAA-3’, A33:5’-AAATAGGGCATCGTGACCCGGCCAGGGGAAGAA-3’. These synthesized oligonucleotides were labeled with biotin at the 3’ end. The
biotinylated oligonucleotide (10 ng/µL) was added to nuclear extracts of HepG2 cells
after their nuclear proteins were incubated with poly (dI-dC) (1µg/µL) and binding
buffer for 5 minutes. The specific binding was evidenced by adding a 50- to 100-fold
excess of corresponding non-labeled oligonucleotide with nuclear extracts prior to the
mixtures were then incubated for 30 min at 15˚C. After electrophoresis, gels were
transferred to nylon membranes. For detection of bound oligonucleotides, membranes
were blocked using blocking buffers (Panomics EMSA Gel-Shift Kit) followed by the
addition of Streptavidin-HRP, and blots were developed by ECL according to the manufacturer’s instructions.
Resting ECG recordings
The patients resting in supine position for 10 minutes were performed ECG
recordings. Both heart rate and PR interval were taken into account as essential
factors affecting sinus node function and rhythmic conduction in the heart.1 The
control patients without SSS were divided into two groups with age and sex match
according to AGT genotype. Six patients with chronic atrial fibrillation or without
ability to maintain sinus rhythm during ECG recording were excluded.
Statistical analysis
Student’s t test was used when the continuous data were normally distributed; otherwise, the nonparametric Mann-Whitney U test was used. Categorical data were
compared by the conventional chi-square test if the observation numbers in all
categories were larger than 5; otherwise, the Fisher exact test was used. Numeric
variables for the promoter genotypes were compared using one-way analysis of
Hardy–Weinberg equilibrium (HWE) were assessed by using the conventional χ2
goodness-of-fit test. Haplotype profile analysis for the polymorphisms was estimated
by using Haploview software.20 Owing to short distances between each polymorphism
location on the AGT gene, polymorphisms probably did not separate by
recombination and had linkage disequilibrium (LD).21 Thus, pairwise measurement of
LD was performed to test the LD between the polymorphisms. D’ was used to
estimate LD. Because the magnitude of D’ strongly depends on the sample size, and it
is known to increase when a small number of samples or rare alleles are examined, we
also utilized the r2 value to confirm LD. Expectation-maximization (EM) based
haplotype frequency estimation with a permutation test was performed to determine
whether any specific haplotypes are associated with SSS on the basis of previous
reports.21,22 Statistical significance of LD was defined as r2>1/3 and D′>0.7, as
suggested by previous reports.23,24 A P-value <0.05 was considered to be statistically
Results
Patient characteristics
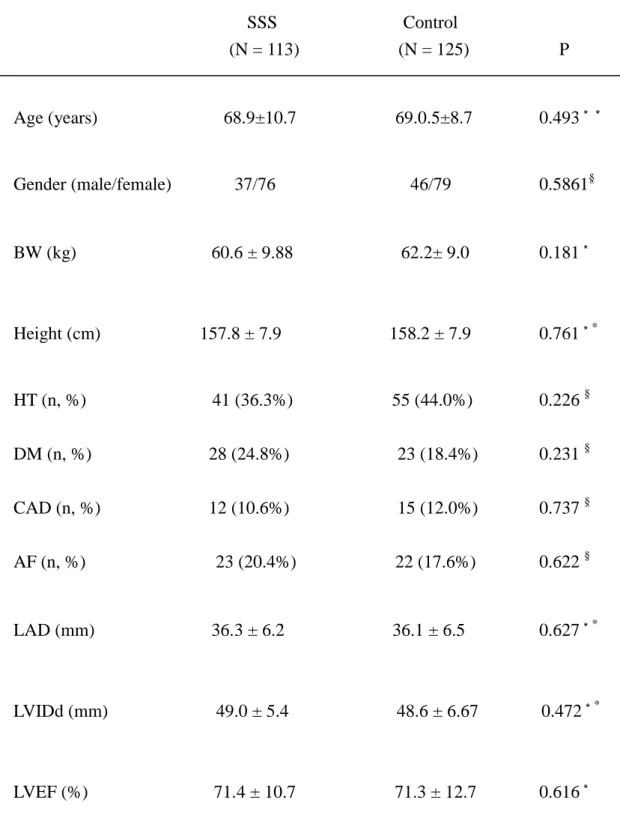
The clinical features of SSS patients and controls are summarized in Table 1.
There were no significant differences in age, gender, percentage of patients with
hypertension, diabetes mellitus (DM), coronary artery disease (CAD), atrial
fibrillation (AF), and left ventricle dysfunction between groups.
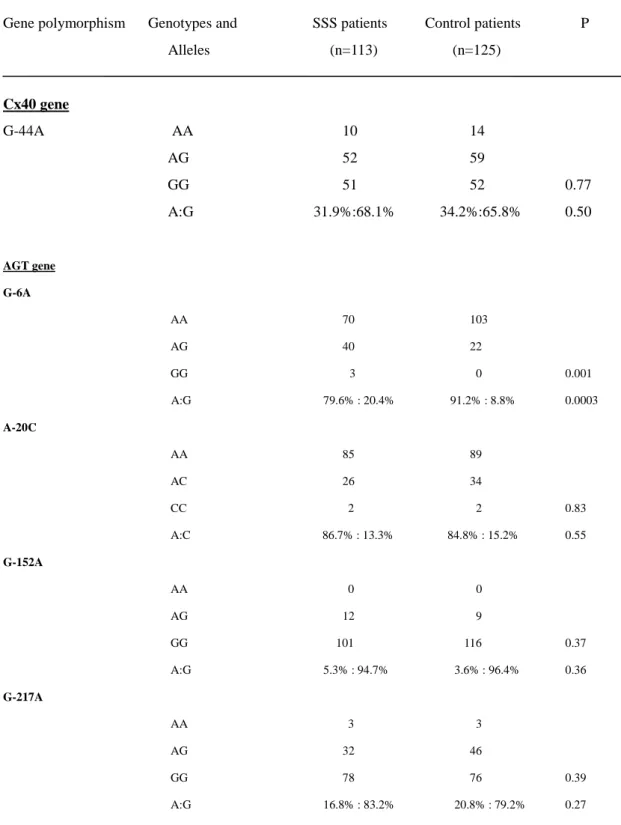
HWE tests and LD measurements
Four polymorphic sites were found at positions -6, -20, -152, and -217 within the
promoter region of the AGT gene (Figure 2 and see Supplementary material online,
Figure 1-3). The HWE genotype distributions were assessed for each AGT promoter
polymorphism G-6A, A-20C, G-152A, and G-217A by the conventional chi-squared
goodness-of-fit test. The P-value was 0.33, 0.65, 0.47, and 0.29, respectively. In
addition, the same test was also performed for the SSS and control groups separately.
The AGT genotype distribution in the SSS and control groups did not significantly
deviate from the HWE (P>0.2 in each polymorphism).
The pairwise linkage among these four polymorphic sites on the AGT promoter gene was evaluated by the LD test using D’ and r2. The D’ values of the loci pairs for
-6/-20, -6/-152, -6/-217, -20/-152, -20/-217, and -152/-217 were 1, 1, 1, 1, 0.463, and
0.005, and 0.001, respectively (Figure 3). The D’ values indicated a significant
linkage in the loci pairs of -6/-20, -6/-152, -6/-217 and -20/-152. However, the r2 values for these three loci pairs were low. This inconsistency between the D’ and r2
values may be due to the small sample size in this study.21 The high D’ values and the
low r2 values of these 4 loci pairs (-6/-20, -6/-152, -6/-217 and -20/-152) suggest an
incomplete linkage among these 4 loci pairs, which explains the wide range of
haplotypes for the AGT gene.
Two polymorphic sites were found at position of -44 and +71 in Cx40 gene (see
Supplementary material online, Figure 4). The Cx40 genotype distribution in total
population, SSS patients and control groups did not significantly deviate from the
HWE (P=0.46, 0.38 and 0.65, respectively). These two Cx40 polymorphisms were in
complete linkage disequilibrium. The patients with allele G at position -44,
consistently had A at -71 position and vice versa.
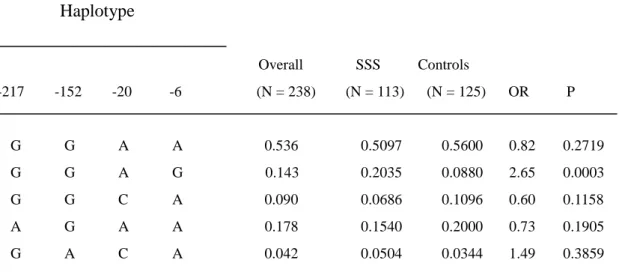
Relationship between the AGT promoter haplotypes and SSS
In the present study, five major haplotypes in the AGT promoter showing a
frequency of > 0.01 were identified, and their relationship with SSS was examined.
The GGAG haplotype (-217G, -152G, -20A, -6G) occurred with a significantly higher
frequency in the SSS group compared to the control group (haplotype frequency:
Single locus analysis of AGT promoter polymorphisms and SSS association
A significant difference was observed in the distribution of the genotypes at
position -6 between SSS and control subjects (P = 0.001). The AA genotype
frequency of G-6A was significantly lower in the SSS group than in the control group
(OR = 2.88, 95% CI: 1.58-5.22, P = 0.001). The G allele frequency of G-6A was
significantly higher in the SSS group than in the control group (20.4% vs. 8.8%, OR =
2.65, 95% CI: 1.54 - 4.57, P = 0.0003) (Table 3). Results of the haplotype analysis
and single locus analysis indicate a significant association between G-6A
polymorphism and SSS.
Genotypes and alleles distribution of Cx40 polymorphisms in SSS patients and
controls
Owing to the Cx40 polymorphisms at positions -44 and +71 were in complete
linkage disequilibrium, we only reported the results for Cx40 -44(GA)
polymorphism. There was no significant difference in the distribution of allele
frequency of -44G and -44A and genotype distribution of -44AA, -44AG and -44GG
between SSS and control patients (P = 0.50 and 0.77, respectively). These results
indicate no association between Cx40 -44 polymorphism and non-familial SSS (Table
3)
The effect of polymorphisms on AGT promoter activity was determined by
transiently transfecting plasmids containing AGT promoter polymorphisms upstream
of the luciferase gene into a hepatocyte (HepG2) cell line and measuring luciferase
activity. The AGT promoter containing G at -6 (p(-6G)) had a lower transcriptional
activity than the AGT promoter containing A at -6 (p(-6A)) in HepG2 cells ( P < 0.05)
(Fig. 4). This suggests that decreased AGT promoter activity in RAS is involved in
the pathogenesis of SSS.
EMSA
The luciferase assay showing that the AGT transcription rate is modulated by the
nucleotide substitution at position -6 of the proximal promoter reflects that
polymorphic substitution of nucleotide -6 may alter the interaction between
transcriptional factors and the proximal region of the AGT promoter, subsequently
altering the transcription rate. To test this possibility, the direct binding experiment of
EMSA was conducted to compare the formation of retarded complexes of
oligonucleotide G33 or A33 with nuclear extracts from HepG2 cells. The results that
were obtained showed that a stronger blot shift by nuclear proteins was observed for
the biotin-labeled oligonucleotide G33 than for oligonucleotide A33. This retarded
complex could be abolished by pretreatment of each sample preparation with each
oligonucleotide (G23 or A23), a stronger blot shift by nuclear proteins was also
observed for G23 than for A23 (Fig. 5).
Effects of AGT G-6A polymorphism on heart rate and PR interval in control
patients without SSS
To verify the functional association of G-6A polymorphism to non-familial SSS,
we examined whether ATG G-6A polymorphism in control patients without SSS
would affect their sinus node function. Both heart rate and PR interval were
considered as critical factors in association with sinus node function and rhythmic
conduction of the heart. The resting heart rate in the subjects with GA genotype is
significant lower than the subjects with AA genotype (GA vs. AA = 68.2 ± 12.2 vs.
74.4 ± 11.9 beats/min, P = 0.03) (Table 4). This might suggest that AGT G-6A
polymorphism might play a role in sinus rate control. In addition, the subjects with
GA genotype also showed a trend of longer PR interval than the subjects with AA
genotype (AG vs, AA = 164.9 ± 26.5 vs. 154.1 ± 18.2 ms, P = 0.07) (Table 4).
Consistently, the control patients with GA genotype have longer electrical conduction
for the atria to ventricles that might lead to impaired atrioventricular conduction. Thus,
these results suggested that the AGT G-6A polymorphism not only associates with
non-familial SSS, but also has effects on the sinus node function in the control
Discussion
It is well-known that malfunctions in HCN-4, SCN5A, and connexin are
genetically associated with familial SSS. In addition, sinus node fibrosis has been
reported to cause abnormal sinus node function.1 Moreover, the risk for
fibrosis-related SSS without genetic inheritance (non-familial SSS) is known to
increase with age.1,2 Only a few studies have analyzed the influence of genetic
characteristics on the development of fibrosis and age-related SSS. Since the
pathogenesis of non-familial SSS is quite different from that of familial SSS, it is of
interest to explore the underlying mechanism of non-familial SSS. In the present study,
we showed that AGT promoter polymorphism is highly associated with non-familial
SSS, possibly by modulating AGT expression. This result may provide useful
information in the prevention of age-related non-familial SSS.
RAS has been implicated in cardiac fibrosis and sinus node dysfunction.13,14 It
has been reported that angiotensin II mediates the proliferation of fibroblasts through
the mitogen-activated protein kinase signaling pathway.25,26 Immunohistochemical
studies with monoclonal antibody against the endogenous proteins of AGT in the
heart visualized their expression in the cardiac conduction system.11 Autoradiography
also showed that angiotensin II binding sites (AT1 receptors) are localized in the sinus
apoptosis via AT1 receptors in the conducting system.26 These studies suggest that
fibrosis-related non-familial SSS may be closely associated with alterations in RAS.
Single locus, haplotype analyses, and transcriptional activity of AGT gene
promoter polymorphisms
Although the present study showed four polymorphic sites at positions -6, -20,
-152, and -217 in the promoter region of the AGT gene (Table 3), only the position at
-6 with nucleotide substitution GA was found to be significantly different between
control and SSS groups. In the haplotype analysis, we found 5 major haplotypes of the
AGT promoter polymorphism in control and SSS patients (Table 2). The GGAG
haplotype (with a single AG nucleotide substitution at position -6) was found to be
associated with a significant risk for SSS in comparison to the common haplotype
GGAA. In addition, compared to the control group, the SSS group had a higher
frequency of the G allele and a lower frequency of AA genotype for the G-6A
polymorphism. Taken together, these results suggest that G-6A polymorphism is a
locus significantly associated with non-familial SSS.
The frequency of the G allele in our control Taiwan Chinese population was
8.8%, which is lower than that reported in the white European population (49.6%).27
The frequency of the AGT G-6A AA genotype in our control population was 61.9%,
Recently, a rare polymorphic site in MYH6 has been reported to be associated with
high risk of non-familial SSS66. Apparently, non-familial SSS is one of the complex
diseases associated with multiple susceptibility loci7 such as that different ethnic
population would appear the distinguished association pattern between genomic
polymorphism and incidence of this syndrome.
In the present study, the luciferase activity assay demonstrated that the AGT
promoter with nucleotide G at -6 had a lower transcriptional activity in comparison to
the promoter with nucleotide A at -6 (Figure 4). Results obtained with a competitive
binding assay on 2 different lengths of the oligonucleotide containing the specific
nucleotide A or G at the -6 position of the AGT promoter indicated that a
polymorphic site at the -6 position of the AGT promoter, regardless of having a length
spanning from 23 to 33 nucleotides, would affect the binding affinity of the specific
nuclear complex that modulates basal transcription of AGT gene expression. The
EMSA result also indicates a stronger binding affinity to nuclear protein extracts for
the oligonucleotide containing a nucleotide substitution for A to G at position -6.
Consistent with a previous study by Inoue et al.28, the present study strongly
suggested that G-6A polymorphism involved in modulation of the AGT promoter
activity is responsible for the RAS-induced fibrosis found in non-familial SSS.
associated with SSS.6 Based on their results, G-6A as a polymorphic site for
non-familial SSS found in this study was not included. This might be due to the
complex susceptibility of SSS determined by multiple susceptibility loci, included
ethnic population, and age differences. In our study, for seventy-year-old Taiwanese
patients (N=113) and controls (N = 125) the AGT promoter polymorphism G-6A was
found to be highly associated with non-familial SSS (Table 2 and 3). A variant G-6A
in AGT promoter appears to be a novel determinant associated with non-familial SSS
in aged Taiwanese patients. It will be of interest to investigate the disturbance of RAS
in the aged patients characterized with non-familial SSS in the future.
Effects of G-6A polymorphism on heart Rate and PR interval
In addition to affecting non-familial SSS patients (Tables 2 and 3), AGT G-6A
polymorphism clearly has substantial effect on sinus node function in control patients
without non-familial SSS (Table 4). Since the patients with homozygous GG
genotype examined in this study all appeared non-familial SSS (Table 3),
heterozygous GA control patients without SSS was compared to homozygous AA
control patients for evaluating the effect of G-6A polymorphism on the sinus rate in a
population without non-familial SSS. In this study, the heterozygous GA patients with
G allele at -6 position significantly have a 6.2 beats/min lower heart rate and a 10.8
functional association of AGT G-6A polymorphism to sinus node function and
rhythmic conduction in control and non-familial SSS patients.
Mechanism for G-6A AGT polymorphism is associated with SSS
Cardiac pacemaker activity is regulated by different classes of ion channels.2,5 A
recent study investigating the expression of ion channels in the human sinus node has
shown the role of various ion channels (e.g., HCN, SCN5A, K+ channels, and Ca++
channels) in generating pacemaking activity.2 Studies using transgenic knockout
animals for HCN4, Ca++ channels, Na+ channels, and Cx40 demonstrated that all these
ion channels, as well as gap junctions, are involved in the control of sinus node
pacemaking function.2,29 Contrary to familial SSS, which is due to the genetic control
of ion channels, non-familial SSS is believed to be closely associated with cardiac
fibrosis occurring during the aging process, and during which ion channels and gap
junctions may be modified by RAS disorders.1,2,30 A recent study using angiotensin
converting enzyme 8/8 transgenic mice with local overexpression in cardiac tissue
showed that RAS overexpression resulting in the downregulation of SCN5A and gap
junction proteins leads to low-voltage electrical activity and conduction delays in the
heart.31 It appears that cardiac-specific RAS dysregulation causing changes in SCN5A
and gap junction protein levels may be associated with non-familial SSS. In addition,
atrial myocytes derived from guinea pig hearts29 and acts as a modulator of L-type
calcium channels by activating protein kinase C via the AT1 receptor.31,32 These
studies validate the novel action of RAS in modulating ion channels and gap junctions
via the AT1 receptor, which results in a dysfunction of the pacemaking activity in the
fibrotic heart tissue.
It has been reported that overexpression of the AT1 receptor causes sinus
bradycardia.14 Upregulation of AT1 receptors was observed in AGT knockout mice.
These studies indicate that there is a feedback mechanism for the RAS and AT1
receptors in cardiovascular homeostasis.17 The present study consistently shows that
the SSS group, which has a higher frequency of G at position -6, has a lower AGT
transcriptional activity than the control group. This suggests a unique feedback
mechanism for the modulation of RAS expression, which may result in sinus node
fibrosis and in the downregulation of ion channels and gap junction proteins in the
heart. To our knowledge, this is the first study to demonstrate the association between
G-6A AGT promoter polymorphisms and non-familial SSS, and provides functional
results to clarify the underlying mechanism of non-familial SSS.
Study limitations
Based on our study results, we found that the G-6A polymorphism is associated
luciferase assay, to explain the underlying mechanism of the candidate locus that
affects the sinus node. However, the possible feedback mechanism of the RAS system
that affects the development of non-familial SSS via modulation of AGT gene
expression by G-6A polymorphism still needs additional in vivo studies for further
clarification. Another limitation of the study is that the study population is small. Our
results should be confirmed in a larger scale study.
Conclusions
Patients with SSS have a lower frequency of the AGT G-6A AA genotype and a
higher G allele frequency, suggesting a possible role for the AGT G-6A promoter
polymorphism in determining the risk of SSS. Results obtained with the EMSA assay
suggested that nucleotide substitution in AGT polymorphic site -6 would affect the
promoter binding affinity for the specific nuclear complex and transcription activity.
Taken together, the data reported in this study provide biological insight for the
possible mechanism of non-familial SSS. The results suggest that AGT promoter
G-6A polymorphisms act as modulators of the transcription activity of RAS
References
1. Mangrum JM, DiMarco JP. The evaluation and management of bradycardia. N
Engl J Med. 2000;342:703-709.
2. Dobrzynski H, Boyett MR, Anderson RH. New insights into pacemaker activity:
promoting understanding of sick sinus syndrome. Circulation 2007;115:1921-32.
3. Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D. Familial sinus
bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J
Med. 2006;354:151-7.
4. Makita N, Sasaki K, Groenewegen WA, Yokota T, Yokoshiki H, Murakami T, et al.
Congenital atrial standstill associated with coinheritance of a novel SCN5A mutation
and connexin 40 polymorphisms Heart Rhythm 2005;2:1128-34.
5. Ctieber J, Hofmann F, Ludwig A. Pacemaker channels and sinus node arrhythmia.
Trends Cardiovasc Med 2004;14:23-8.
6. Holm H, Gudbjartsson DF, Sulem P, Masson G, Helgadottir HT, Zanon C, et al. A
rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet
2011; 43: 316–320.
7. Stefan Herrmann, Andreas Ludwig. Not so fast! Sick sinus syndrome is a complex
and incompletely understood disease that might prove hard to model in animals:
8. Hagendorff A, Schumacher B, Kirchhoff S; Lu¨deritz B, Willecke K. Conduction
disturbances and increased atrial vulnerability in connexin40-deficient mice analyzed
by transesophageal stimulation Circulation. 1999;99:1508-1515.
9. Firouzi M, Ramanna H, Kok B, Jongsma HJ, Koeleman BPC, Doevendans PA, et
al. Association of human connexin40 gene polymorphisms with atrial vulnerability as
a risk factor for idiopathic atrial fibrillation Circ Res. 2004;95:e29-e33.
10. Vongvatcharanon U, Vongvatcharanon S, Radenahmad N, Kirirat P, Intasaro P,
Sobhon P, et al. Angiotensin II may mediate apoptosis via AT1-receptors in the rat
cardiac conduction system. J Renin Angiotensin Aldosterone Syst. 2004;5:135-40.
11. Sawa H, Tokuchi F, Mochizuki N, Endo Y, Furuta Y, Shinohara T, et al.
Expression of the angiotensinogen gene and localization of its protein in the human
heart. Circulation 1992;86:138-46.
12. Saito K, Gutkind JS, Saavedra JM. Angiotensin II binding sites in the conduction
system of rat hearts. Am J Phsyiol 1987;253:H1618-22.
13. Sadoshima J, Izumo S. Molecular characterization of angiotensin II–induced
hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Circ Res
1993;73:413-23.
14. Hein L, Stevens ME, Barsh GS, Pratt RE, Kobilka BK, Dzau VJ. Overexpression
phenotype associated with myocyte hyperplasia and heart block Proc Natl Acad Sci
USA 1997; 94:6391–6.
15. van Duijn CM, Schmidt R. Angiotensinogen gene promoter haplotype and
microangiopathy-related cerebral damage- results of the Austrian stroke prevention
study. Stroke 2001;32:405-12.
16. Schmidt H, Aulchenko YS, Schweighofer N, Schmidt R, Frank S, Kostner GM, et
al. Angiotensinogen promoter B-haplotype associated with cerebral small vessel
disease enhances basal transcriptional activity. Stroke 2004;35:2592-7.
17. Tamura1 K, Yokoyama1 N, Sumida1 Y, Fujita1 T, Chiba E, Tamura N, et al.
Tissue-specific changes of type 1 angiotensin II receptor and angiotensin-converting
enzyme mRNA in angiotensinogen gene-knockout mice. J Endocrinol
1999;160:401–8.
18. Lamas GA, Lee K, Sweeney M, Leon A, Yee R, Ellenbogen K, et al. The mode
selection trial (MOST) in sinus node dysfunction: design, rationale, and baseline
characteristics of the first 1000 patients. Am Heart J 2000;140:541–51.
19. Nielsen JC, Thomsen PE, Højberg S, Møller M, Vesterlund T, Dalsgaard D, et al.
A comparison of single-lead atrial pacing with dual-chamber pacing in sick sinus
syndrome.Eur Heart J 2011;32: 686–696
and haplotype maps. Bioinformatics 2005;21:263-5.
21. Tsai CT, Lai LP, Lin JL, Chiang FT, Hwang JJ, Ritchie MD, et al.
Renin-angiotensin system gene polymorphisms and atrial fibrillation. Circulation
2004;109:1640-6.
22. Fallin D, Cohen A, Essioux L, Chumakov I, Blumenfeld M, Cohen D, et al.
Genetic analysis of case/control data using estimated haplotype frequencies: application to APOE locus variation and Alzheimer’s disease. Genome Res 2001;11:143–51.
23. Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human
genome. Nat Rev Genet 2002;3:299–309.
24. Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The
structure of haplotype blocks in the human genome. Science 2002;296:2225–9.
25. Li D, Shinagawa K, Pan L, Leung TK, Cardin S, Wang Z, et al. Effects of
angiotensin-converting enzyme inhibition on the development of the atrial fibrillation
substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation
2001;104:2608-14.
26. Duff JL, Marrero MB, Paxton WG, Schieffer B, Bernstein KE, Ber BC. Angiotensin II
signal transduction and the mitogen-activated protein kinase pathway. Cariovasc Res
27. Hladikova M, Vašků A, Štourač P, Benešová Y, Bednařík J. Two frequent
polymorphisms of angiotensinogen and their association with multiple sclerosis progression
rate. Neurol Sci. 2011; 303:31-4.
28. Inoue I, Nakajima T, Williams CS, Inoue I, Nakajima T, Williams CS, et al. A
nucleotide substitution in the promoter of human angiotensinogen is associated with
essential hypertension and affects basal transcription in vitro. J Clin Invest
1997;99:1786–97.
29. Hagendorff A, Schumacher B, Kirchhoff S, Luderitz B, Willecke K. Conduction
disturbances and increased atrial vulnerability in connexin40-deficient mice analyzed by
transesophageal stimulation. Circulation 1999;99:1508-15.
30. Goette A, Lendeckel U: Electrophysiological effects of angiotensin II. Part I: signal
transduction and basic electrophysiological mechanisms. Europace 2008;10:238–41.
31. Kasi VS, Xiao HD, Shandg LL, Iravanian S, Langberg J, Witham EA, et al.
Cardiac-restricted angiotensin-converting enzyme overexpression causes conduction defects
and connexin dysregulation. Am J Physiol Heart Circ Physiol 2007:293: H182–92.
32. Zankov DP, Omatsu-Kanbe M, Isono T, Toyoda F, Ding WG, Matsuura H, et al.
Angiotensin II potentiates the slow component of delayed rectifier Kt current via the AT1
Figure Legends:
Figure 1. Construction of expression variants in pGL3 vector using
oligonucleotides for the proximal AGT promoter. The oligonucleotides containing the proximal promoter region of the AGT gene from position −290 to +35 were ligated in the pGL3 vector to produce the reporter construct. luc+, cDNA encoding
the modified firefly luciferase; Ampr , gene conferring ampicillin resistance; f1 ori,
origin of replication derived from filamentous phage; E, exon.
Figure 2. AGT G-6A polymorphism genotyping by direct sequencing. The arrows
indicated the polymorphic site of GG, AA and GA genotypes.
Figure 3. Linkage disequilibrium plot of AGT promoter polymorphisms.
Pairwise linkage disequilibrium analysis shows r2 (× 100) values. The intensity of
gray is proportional to r2.
Figure 4. Comparison of the transcriptional activities of reporter constructs
containing AGT proximal promoter polymorphisms in HepG2 cells.
Transcriptional activities are presented as a ratio of the activity of the p(-6G)
contain any AGT promoter sequence. Values are presented as means ± SE.
Figure 5. EMSA results of comparison of the binding affinities of biotinylated
oligonucleotides. (A) Lanes 1 and 2 indicating the mobilities of labeled, biotinylated
oligonucleotides (A33 and G33) with nuclear extracts. A stronger shift was observed
in the G33 in comparison to the A33 oligonucleotide. Lanes 3 and 4 showing the
competition experiments. The unlabeled oligonucleotides completely inhibited the
specific binding complex of biotin-labeled A33 and G33 probes to nuclear extract. (B)
Lanes 1 and 2 indicating the mobilities of labeled oligonucleotides (G23 and A23)
with nuclear extracts. A stronger binding complex was observed in G23 in
comparison with the A23 oligonucleotide. The unlabeled oligonucleotides completely
inhibited the specific binding complex of biotin-labeled G23 and A23 probes to
nuclear extract in lane 3 and 4. The arrow points to the specific nuclear complex,
which binds with the labeled, biotinylated oligonucleotides. C, competitor.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
their support of this study.
Conflict of interest: none declared.
Funding
This work was supported by the research project DMR-96-039 from China Medical
Table 1 General characteristics of patients included in the study SSS Control (N = 113) (N = 125) P Age (years) 68.9±10.7 69.0.5±8.7 0.493** Gender (male/female) 37/76 46/79 0.5861§ BW (kg) 60.6 ± 9.88 62.2± 9.0 0.181* Height (cm) 157.8 ± 7.9 158.2 ± 7.9 0.761** HT (n, %) 41 (36.3%) 55 (44.0%) 0.226 § DM (n, %) 28 (24.8%) 23 (18.4%) 0.231 § CAD (n, %) 12 (10.6%) 15 (12.0%) 0.737 § AF (n, %) 23 (20.4%) 22 (17.6%) 0.622 § LAD (mm) 36.3 ± 6.2 36.1 ± 6.5 0.627** LVIDd (mm) 49.0 ± 5.4 48.6 ± 6.67 0.472** LVEF (%) 71.4 ± 10.7 71.3 ± 12.7 0.616*
*Student t test; **Mann-Whitney U test; §χ2
test; SSS, sick sinus syndrome; HT,
hypertension; DM, diabetes mellitus; CAD, coronary artery disease; LAD, left atrial Table 1
Table 2 Haplotype frequency estimates of AGT gene in patients with sick sinus syndrome and controls
Haplotype Overall SSS Controls -217 -152 -20 -6 (N = 238) (N = 113) (N = 125) OR P G G A A 0.536 0.5097 0.5600 0.82 0.2719 G G A G 0.143 0.2035 0.0880 2.65 0.0003 G G C A 0.090 0.0686 0.1096 0.60 0.1158 A G A A 0.178 0.1540 0.2000 0.73 0.1905 G A C A 0.042 0.0504 0.0344 1.49 0.3859
* There results were confirmed by permutation test which also revealed that GGAG is the only significant candidate haplotype (P = 0.0007). Haplotypes are not listed if all the estimated frequencies are < 0.01 in patients with sick sinus syndrome, controls, and overall population.
Table 3 Distribution of genotypes and alleles of Cx40 and AGT in patients with and without sick sinus syndrome
Gene polymorphism Genotypes and SSS patients Control patients P Alleles (n=113) (n=125) Cx40 gene G-44A AA 10 14 AG 52 59 GG 51 52 0.77 A:G 31.9%:68.1% 34.2%:65.8% 0.50 AGT gene G-6A AA 70 103 AG 40 22 GG 3 0 0.001 A:G 79.6% : 20.4% 91.2% : 8.8% 0.0003 A-20C AA 85 89 AC 26 34 CC 2 2 0.83 A:C 86.7% : 13.3% 84.8% : 15.2% 0.55 G-152A AA 0 0 AG 12 9 GG 101 116 0.37 A:G 5.3% : 94.7% 3.6% : 96.4% 0.36 G-217A AA 3 3 AG 32 46 GG 78 76 0.39 A:G 16.8% : 83.2% 20.8% : 79.2% 0.27
SSS , sick sinus syndrome. P values obtained based on χ2test or Fisher’s exact test; the upper P value is for comparison of
genotype frequencies, and the lower is for allele frequencies.
Table 4 Effects of AGT G-6A polymorphism on heart rate and PR interval measured by ECG recordings in control patients without SSS
Genotypes
GA AA P (n = 22) (n = 97)
Heart rate (beats/min) 68.2 ± 12.2 74.4 ± 11.9 0.03
PR interval (ms) 164.9 ± 26.5 154.1 ± 18.2 0.07 Table 4
Figure 1
Figure 2
Figure 3
Figure 4
Figure 5