Its Role in Regulation of Signal Transduction and Its
Involvement in Human Cancers
Tzu-Ching Meng
1, Yi-Wei Lou, Yi-Yun Chen, Shu-Fang Hsu, and Yi-Fen Huang
Institute of Biological Chemistry, Academia Sinica and
Institute of Biochemical Sciences, College of Life Sciences,
National Taiwan University, Taipei, Taiwan [T.-C. Meng, Y.-W. Lo, S.-F. Hsu, Y.-F. Huang]; National Core Facility for
Proteomics Research, Academia Sinica, Taipei, Taiwan [Y.-Y. Chen]
Reactive oxygen species (ROS) are currently viewed as secondary messengers that
control signal transduction through the post-translational modifications of
Cys-oxidation in targeting proteins. The physiological levels of ROS are generated in
re-sponse to stimulation induced by extracellular ligands. Due to the labile feature of
ROS, the effect of protein oxidation is normally transient. Such a character is essential
for the precise control of redox-dependent signaling homeostasis. Nevertheless,
studi-es have shown that under pathological conditions, such as certain typstudi-es of cancer, the
cellular ROS are produced in a ligand-independent manner. Interestingly, the
genera-tion of ROS is required for the development of transformed phenotype and tumors,
suggesting that the constitutive oxidation of signaling modulators might contribute to
the uncontrolled signaling cascades, further leading to pathological consequences.
Emerging evidence has revealed that protein tyrosine phosphatases (PTPs), which
regulate tyrosine phosphorylation-dependent signal transduction through tyrosine
dephosphorylation of substrates, are highly susceptible to oxidation. Interestingly,
oxidation of catalytic Cys residue results in enzymatically inactivated form of PTPs,
concomitant with an enhanced tyrosine phosphorylation level in cells. The primary
fo-cus of this review is to comprehend recent studies that greatly advanced our
under-standing towards redox-dependent regulation of cell signaling through Cys oxidation of
endogenous PTPs. The rational implication of this mechanism in contributing to the
development and maintenance of cell transformation is discussed.
Journal of Cancer Molecules 2(1): 9-16, 2006.
Keywords:
Cys-oxidation protein tyrosine
phos-phatase protein tyrosine kinase reactive oxygen species
signal transduction
Introduction
At the early stage of studies on the biological role of redox regulation, attentions were focused on the bactericidal effect of reactive oxygen species (ROS2) that can be transiently
produced in a large amount by neutrophils at the site of in-flammation. This phenomenon was initially considered as an innate immune response against infection rather than a signaling event. The plasma membrane-associated NADPH oxidase (Nox) gp91phox, which, when assembled with other regulatory factors, generates a ROS burst in neutrophils [1]. The locally high concentration of ROS might disrupt the normal function of bacterial proteins, therefore killing those invaded microorganisms. Due to the fact that gp91phox is specifically expressed in phagocytes such as neutrophils Received 11/9/05; Revised 2/3/06; Accepted 2/4/06.
1Correspondence: Dr. Tzu-Ching Meng, Institute of Biological
Chemistry, Academia Sinica, 128 Academia Road Sec. 2, Taipei 115, Taiwan. Phone: 886-2-27855696. Fax: 886-2-27892161. Email: [email protected]
2Abbreviations: ROS, reactive oxygen species; Nox, NADPH oxidase; PI3-K, phosphatidylinositol 3-kinase; PTP, protein tyrosine phosphatase; PTK, protein tyrosine kinase; DUSP, dual specificity phosphatase; MKP, MAP kinase phosphatase.
and macrophages [1], it was thought that the ROS produc-tion was restricted to the inflammatory area. The biological function of ROS was therefore attributed to merely a self-defensive system.
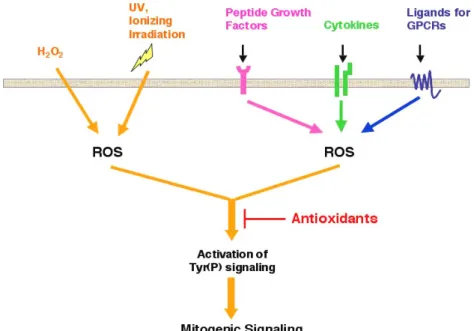
More recently, however, experimental evidence has been gradually accumulated from various studies that reassigned a novel role of ROS as second messengers [2-5]. It was first demonstrated that, in addition to the phagocytes, other cell types with various tissue origins were capable of generating a high level of ROS [6]. Interestingly, among the majority of cases reported so far, the ROS generation was concomitant with the activation of tyrosine phosphorylation-dependent signaling pathways in response to the action of extracellular stimuli [2,7,8], which include not only a variety of stress but also physiological ligands (Figure 1). With the application of ROS scavengers such as the chemical antioxidant N-acetyl cysteine, or the hydrogen peroxide-decomposing enzyme catalase, studies have shown that ligand-stimulated ROS production was required for growth factor-induced tyrosine phosphorylation signaling [8](Figure 1). Several important signaling effectors including cytosolic phosphatidylinositol 3-kinase (PI3-K)[9] and small G protein Rac 1 [10] were later identified to be essential for ligand-mediated increase of cellular oxidants. The role of ROS as second messengers was further demonstrated by the recent discovery that Nox
Figure 1: ROS function as second messen-gers that mediate cell signaling pathways in response to stimulation of various extracel-lular stimuli. Celextracel-lular ROS can be generated in response to various external stimuli. Treatment of cells with H2O2, which is diffusi-ble across the plasma membrane, increases the cellular ROS level instantly. It has been shown that UV and ionizing irradiation could also promote intracellular oxidant generation. Upon the activation of plasma membrane receptors through physiological ligands such as peptide growth factors, cytokines and ligands for G protein coupled receptor (GPCR), cellular oxidases are transactivated, thus producing ROS in a transient manner. As shown in the diagram, the tyrosine phos-phorylation signaling pathway, which is activated in response to the treatment of those stimuli, is ROS-dependent. An in-crease of cellular antioxidant levels signifi-cantly suppresses stress and physiological ligands-induced signal transduction.
isoforms, which constitute a growing family of enzymes, are broadly expressed in various non-phagocytic cells [6,11]. It has been shown that the transactivation of specific Nox en-zyme, for example, Nox1 or Nox4, accounted for the tran-sient production of cellular ROS in response to epidermal growth factor (EGF)[12] or insulin [13], respectively. Fur-thermore, the disruption of Nox expression by RNA interfer-ence (RNAi) technique led to the significant decrease of ROS production, the attenuation of tyrosine phosphorylation-dependent signaling and the suppression of physiological consequences in response to insulin stimulation [13].
The substantial body of evidence thus demonstrated that cellular ROS, whose production is tightly controlled by specific Nox enzymes in a precisely temporal and spatial manner, function as a key mediator in the regulation of vari-ous signaling pathways. It was therefore proposed that the aberrant levels of ROS might lead to the disordered signal-ing outcome. Perturbation of redox balance has been linked to several pathological consequences such as senescence and cell transformation. Furthermore, recent investigations have suggested that ROS play an essential role in the devel-opment of human diseases including ageing, cancers, athe-rosclerosis, restenosis and cardiac hypertrophy. Therefore, the understanding of molecular mechanisms by which sig-naling cascades are regulated by ROS will provide opportu-nities for therapeutic intervention.
Cysteine residue as a redox sensor
Since a decade ago a great number of studies have been focusing on the identification of missing links, which might connect the role of ROS with the biological response of es-sential effectors in signaling cascades. Those kinases, phosphatases, adaptors and scaffold proteins involved in the regulation of cell signaling pathways were the primary targets. It is now generally accepted that the ROS-mediated post-translational modifications play a critical role to the control of biological activity of proteins [2-5,14]. Moreover, the biochemical and structural data revealed that the Cys residue in protein might act as a redox sensor in response to proximal ROS [2,15]. Several examples have clearly demon-strated the key role of Cys oxidation that determines the signaling consequence controlled by a diverse array of func-tional proteins. It has been shown that the active site Cys residue in reducing enzymes peroxiredoxin [16] and thiol peroxidase [17] is highly susceptible to oxidation. The Cys
oxidation of those enzymes resulted in the formation of an intermolecular disulfide bond with other cellular components, such as stress-activated protein kinases [16] or transcrip-tional factors [17], thus activating downstream signaling cascades. Furthermore, a significant progress has been made that demonstrates the redox regulation of caspase family of enzymes [18-21], which are key initiators and ex-ecutors that control the apoptotic signaling cascades. All caspases have a single catalytic Cys residue, whose oxida-tion leads to enzymatic inactivaoxida-tion [22]. Extensive investi-gations have further revealed a common role of Cys oxida-tion that underlies funcoxida-tional regulaoxida-tion of various cellular components, including protease inhibitors [23], transcrip-tional factors [24-26] and protein tyrosine phosphatases (PTPs) [27].
In addition to ROS, the gaseous free radical nitric oxide (NO), which is generated by the nitric oxide synthase (NOS) family of enzymes, functions as second messengers that regulate a broad array of cell signaling pathways [28]. The soluble guanylate cyclase, which produces cGMP while its heme-iron group coupled with NO, has been viewed as a primary endogenous NO receptor [29]. Nevertheless, recent investigations revealed that NO may execute its biological function through cGMP-independent mechanisms. Interest-ingly, among those NO-mediated actions being described so far, the S-nitrosylation of Cys residue is the most important post-translational modification that influences various cel-lular functions. Accumulating evidence has demonstrated that the activity of a number of key enzymes, which play an essential role in the control of cell signaling cascades, is regulated by S-nitrosylation and denitrosylation. For exam-ples, the catalytic activity of caspases was suppressed in a NO-dependent manner through the S-nitrosylation of their active-site Cys residue [30]. In contrast, the S-nitrosylation of matrix metalloproteinases, another family of Cys-mediated proteases that are involved in the pathogenesis of neurode-generative disorders, resulted in the enzymatic activation [31]. Furthermore, it has been proposed that the catalytic Cys of PTPs is also highly susceptible to S-nitrosylation, leading to inhibition of phosphatase activity [32,33]. Those data thus suggest a critical role of Cys residue in signaling modulators, that functions as a redox sensor in response to both ROS- and NO-induced post-translational modifications.
Although a large number of important issues in the field deserve a thorough analysis, we will primarily focus our attention on the redox regulation of PTPs as these enzymes
Figure 2: Catalytic Cys residue of PTPs is susceptible to oxidation. Due to the low pKa character, the catalytic Cys of PTPs is in the thiolate anion format under the neutral pH, rendering this Cys residue highly susceptible to oxidation. Upon the increased concentration of environmental ROS, the catalytic Cys is initially converted to the sulfenic acid derivative, leading to catalytic inactivation of PTPs. Such a single oxygen-modified form of PTPs could be reduced through a disulfide intermediate upon reacting with thiol reductants such as glutathione (GSH), followed by enzymatic catalysis of thioltransferases. In contrast, additional oxygen molecules could be sequentially added onto the sulfenic acid form of catalytic Cys, resulting in the forma-tion of sulfinic acid (two-oxygen) or sulfonic acid (three-oxygen) derivatives.
Interesting-ly, those highly oxidized PTPs are permanently inactivated as the thiol reductants are unable to reduce sulfinic or sulfonic acid-modified Cys residue.
stand for their own right in the control of tyrosine phos-phorylation-dependent signal transduction. In addition, due to the fact that very limited information about S-nitrosylation of PTPs is known, we will only discuss the ROS-mediated actions on the regulation of PTP activity in the rest of this review.
Catalytic Cys residue is a redox switch of PTP activity
As mentioned above, the early studies have demonstrated that intracellular ROS generation is required for the activated tyrosine phosphorylation-dependent signal transduction in response to a broad array of extracellular stimuli. This gen-eral phenomenon was observed in various cell types, in-cluding neutrophils [7] and non-phagocytic epithelia [34] or fibroblasts [8]. Two hypotheses were initially proposed for interpreting the regulatory role of ROS. First, protein tyrosi-ne kinases (PTKs) might be activated through an oxidation-dependent manner. This possibility, however, was soon ruled out as ROS failed to enhance PTK activity in vitro [7]. Moreover, reducing reagents were unable to suppress the activity of endogenous PTKs isolated from H2O2-treated cells
[7], suggesting that ROS were not directly involved in the activation and inactivation of kinase. In contrast, it seemed to be a promising alternative hypothesis that cellular ROS might function to release negative constraints, thus facili-tating PTK-mediated protein tyrosine phosphorylation via an indirect way. The PTP family of enzymes, which have been regarded as antagonists of PTKs, were therefore brought into consideration as “oxidant receptors” in the context of redox-regulated tyrosine phosphorylation-dependent signal transduction.
The superfamily of PTPs consists of a large number of en-zymes, which include classical, phosphotyrosine-specific phosphatases and dual specificity phosphatases (DUSPs) that recognize phosphorylated serine/threonine as well as phosphotyrosine residues in protein substrates. The cata-lytic activity of PTPs is absolutely dependent upon the in-variant Cys residue, which is located within the conserved signature motif [I/V]HCxxGxxE[S/T] that defines the PTP superfamily [35,36]. Due to the unique environment sur-rounding the active site, this catalytic Cys residue displays an unusually low pKa (between 4.5 and 5.5, compared to pKa of a typical Cys being ~8.5), allowing its presence as the thiolate anion form at the neutral pH [37,38]. Biochemical analysis has clearly revealed that the nucleophilic attack on the phosphate group of substrates is the first step of reac-tion catalyzed by PTPs [39]. The negatively ionized Cys therefore enhances PTP-medicated phosphotyrosyl dephos-phorylation. Interestingly, the low pKa character also
renders the catalytic Cys of PTPs highly susceptible to oxi-dation in response to proximal ROS [27](Figure 2).
Although the primary product of activated Nox enzymes is superoxide (O2.-)[11], it is unlikely that such a negatively charged radical
would react with a free thiol at the catalytic Cys. Attentions have been focused on H2O2, which is spontaneously or
en-zymatically converted from superoxide, as a bioactive form of ROS. The uncharged H2O2 with relatively stable chemical
character is diffusible across cell membrane. While encoun-tering PTPs in cells, H2O2 can easily transfer a hydroxonium
moiety (HO+) to a nucleophilic center at the sulfur atom, thus
completing the key step of Cys oxidation. Interestingly, the oxidized Cys can longer function as a nucleophile, leading to enzymatic inactivation of PTPs. On the basis of such a no-tion, a novel hypothesis was postulated as following. In response to extracellular stimuli, which induce the produc-tion of active oxidants, endogenous PTPs are transiently inactivated. The lift of such negative constraints through a ROS-dependent inhibition of PTPs thus places tyrosine kinases in a predominant position, resulting in the full acti-vation of tyrosine phosphorylation signaling cascade.
Biochemical and structural basis of catalytic
Cys-oxidation in PTPs
The first piece of biochemical evidence that assigned re-dox regulation as a mechanism to the control of PTP activity came from a report by Denu and Tanner [40]. In their study, three members of phosphatases including PTP1 (a non-receptor classical PTP), LAR (leukocyte antigen-related, a receptor type classical PTP) and VHR (vaccinia H1-related, a non-receptor DUSP) in the form of recombinant enzymes, were tested. The results clearly showed that, at the presence of low micromolar concentrations of H2O2, these
enzymes were rapidly inactivated. Interestingly, when glu-tathione and other free thiol-containing reagents were added to the reaction, the PTP activity was fully restored but with a much slower rate than H2O2-induced inactivation. These
data suggested that the oxidative modification of the cata-lytic Cys in PTPs might occur efficiently even in the presence of physiological levels of endogenous thiols. A follow-up study reported by Barrett and colleagues subse-quently examined the biochemical mechanism of free thiol-mediated reactivation of oxidized PTPs [41]. By the applica-tion of mass spectrometry, this study revealed that a disulfi-de bond was formed between the active site Cys of recombi-nant PTP1B and glutathione. Importantly, such a disulfide intermediate could be further reduced by dethiolase enzyme glutaredoxin, which is broadly expressed in various types of cells. Those biochemical data together suggested a
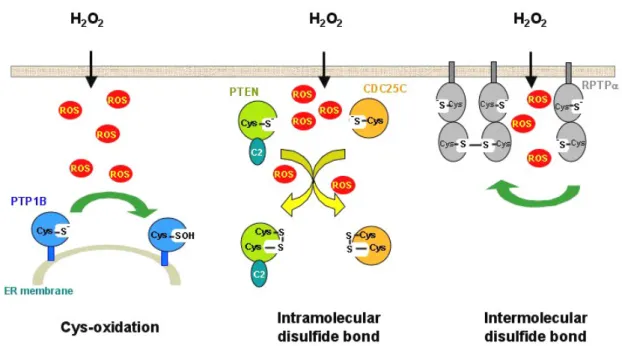
possi-Figure 3: Increased concentrations of intracellular oxidants lead to inactivation of endogenous PTPs through oxidation of catalytic Cys residue. Three forms of oxidized PTPs might be generated in cells under oxidative stress. First, endogenous PTP1B is inactivated, presumably through the formation of sulfenic acid derivatives, in response to H2O2 treatment in cells. Second, upon stimulation with H2O2, cellular PTEN and CDC25C quickly form an intramolecular disulfide bond between the catalytic Cys and the other unpaired Cys residue in the same molecule. Such a modifica-tion also leads to inactivamodifica-tion of phosphatases. Third, it was demonstrated that the catalytic Cys residue in D2 domain of cellular RPTPα is highly susceptible to oxidation, resulting in the formation of an intermolecular disulfide bond with the other unit of RPTPα molecule. It has been proposed that the dimer formation of receptor PTPs through oxidation-induced disulfide bond might have a profound influence in the enzymatic activity of D1 domain. However, the exact regulatory mechanism is yet to be identified.
ble in vivo regulatory process in which PTP was first rapidly oxidized to sulfenic acid-modified form (PTP-Cys-SOH) when encountered ROS, followed by a slow step of glutathiolation (PTP-Cys-S-S-G) that could be contributed by millimolar levels of intercellular GSH. Finally, with the catalysis by cellular glutaredoxin, the glutathiolated, originally oxidized form of PTPs, could be fully reversed back to the reduced and active state.
Such a proposed model established the basis of a novel mechanism through which the tyrosine phosphorylation-dependent signal transduction is regulated by reversible oxidation of PTPs. To further elucidate the exact status of oxidized PTPs, more direct evidence was sought by struc-tural analyses. Two recent papers published simultaneously by Barford’s [42] and Jhoti’s [43] group illustrated that the sulfenic acid-modified catalytic Cys of the oxidized PTP1B was rapidly converted to a cyclic sulfenamide intermediate through the formation of a covalent bond between the sulfur atom of the catalytic Cys and the main chain nitrogen atom of the adjacent Ser residue. This observation greatly ad-vanced our understanding on how the oxidized Cys is stabi-lized by such a distinct structure rather than continuously progressed to the irreversible sulfinic (Cys-SO2) or sulfonic
(Cys-SO3) form of modification. In the case of CDC25
phos-phatase, which functions as an essential regulator of cell cycle control, a different mode of oxidant-induced, unique structural feature was observed. An intracellular disulfide bond was formed between the catalytic Cys473 and a back door Cys426 when the crystals of the catalytic domain of CDC25B were under mildly oxidizing conditions [44]. It has been proposed that the formation of sulfenamide species or intracellular disulfide bond might provide excellent strategi-es to maintain oxidized PTPs in transient intermediatstrategi-es. In this way, upon the decrease of local oxidant concentrations, cellular reducing thiols, which are at millimolar levels, could efficiently restore the activity of PTPs, therefore terminating the extracellular stimuli-induced activation of tyrosine phos-phorylation signaling.
Oxidation and inactivation of endogenous PTPs in
response to extracellular oxidative stress
In addition to the data being illustrated above, more direct evidence was required in order to demonstrate that the re-versible oxidation is indeed a regulatory mechanism for the control of function and activity of endogenous PTPs. The most straightforward model came from the treatment of cells with H2O2, which freely diffuses into cells, thus mimicking
the situation where endogenous ROS are generated in re-sponse to external stimuli (Figure 3). Rhee’s group initially demonstrated that PTP1B was transiently inactivated in A431 human epidermoid cancer cells exposed to H2O2 [45].
Sub-sequent studies revealed that the active site Cys residue of various phosphatases, including PTEN which is a lipid phosphatase regarded as a tumor suppressor, receptor-like PTP-alpha (RPTPa), and CDC25C, was susceptible to oxida-tion in a cellular context. Interestingly, in response to H2O2
treatment, the oxidized form of endogenous phosphatases displayed unique biochemical and structural characters. In the case of PTEN, the oxidized form of catalytic Cys124 (pre-sumably sulfenic acid modification) quickly reacted with an unpaired Cys71 for developing an intracellular disulfide bond [46]. The formation of such oxidized therefore inacti-vated PTEN provided an opportunity for PI3-K to phos-phorylate the 3’-position of inositol, thus enhancing the ac-tivity of PKB through a 3’-phosphoinositol-dependent path-way [47]. Similarly, in CDC25C treated with H2O2, the
oxi-dized Cys377, which is also the catalytic site, was bridged with another invariant Cys residue at position 330 in the same molecule [48]. Data further suggested that the forma-tion of intramolecular disulfide bond triggered fast degrada-tion of CDC25C in cells [48]. The RPTPa, on the other hand, developed an intermolecular disulfide bond, presumably with the second unit of RPTPa, through the H2O2-induced,
oxidized form of catalytic Cys723 in D2 domain [49]. Never-theless, the biological significance of the oxidant-mediated formation of RPTPa dimers is yet to be delineated.
Figure 4: Transient production of ROS inactivates PTP1B and TC-PTP, thus facilitating insulin receptor-mediated signaling cascades. Under the resting state, the catalytic Cys residue of endogenous phosphatases, including PTP1B and TC-PTP, is mainly in the thiolate and reduced form. Such active forms of PTP are important for maintaining a basal tyrosine phosphorylation level of insulin receptor (IR). Upon insulin stimulation, the partially activated IR quickly induces ROS generation, presumably through the membrane-associated Nox enzymes. The cellular oxidants effec-tively react with catalytic Cys residue of endogenous PTPs, thus suppressing their enzymatic activity. It has been shown that the transient oxidation of PTP1B and TC-PTP plays an essential role for the optimal activation of IR-mediated signaling cascades. Interestingly, the cellular ROS levels are soon returned back to the normal condition ~30 min post-insulin stimulation, leading to the gradual reduction of PTP1B and TC-PTP. The reactivat-ed phosphatases, which function as negative constraints, start to dephosphorylate IR and possibly other downstream effectors, thus terminating insulin-induced signal transduction in cells.
Transient oxidation of PTPs in the regulation of cell
signaling
Results from the analyses of PTP1B, PTEN, RPTPa and CDC25C thus suggested a general regulatory mechanism by which the oxidation of active site Cys residue leads to the inactivation of endogenous PTPs in response to increased concentrations of oxidants in cellular contexts. This matter has been further explored under physiological conditions. As described above, a number of studies have shown that ligand-induced activation of plasma membrane receptors, such as receptor protein tyrosine kinases, is concomitant with the transient production of cellular ROS. On the basis that endogenous PTPs are susceptible to oxidation, the physiological ligand-induced ROS production has been pro-posed as a regulatory means for fine-tuning cell signaling through PTP inactivation. Several excellent studies demon-strated that the optimal activation of a ligand-mediated sig-naling pathway was in concurrence with the transient oxida-tion and therefore inactivaoxida-tion of a particular PTP, which, if present in its reduced form, functions as a negative con-straint to down-regulate that particular signaling pathway. Rhee’s group showed that, in response to EGF stimulation, A431 cells generated a ROS burst within 5-20 min post-treatment [45]. By the use of [14C]-iodoacetic acid, which
only reacts with the reduced Cys, but not with Cys in oxi-dized form or disulfide complex, their study demonstrated that the proportion of active PTP1B was temporarily de-creased in EGF-stimulated cells following a time-dependent manner [45]. This result thus suggested that the concurrent inhibition of PTP1B by ROS might be required for the full
activation of EGF receptor-induced tyrosine phosphorylation signaling. Goldstein’s group investigated the role of ROS-mediated PTP inactivation involved in the regulation of insu-lin signainsu-ling [50]. By the application of a method of enzy-matic activity assay under a strictly anaerobic condition, they showed that in both HepG2 and NIH3T3 cells, PTP1B was significantly inactivated in concurrence with a transient increase of cellular ROS levels upon the insulin stimulation [50]. In the presence of forced expression of catalase, insu-lin-induced inactivation of PTP1B was suppressed, sug-gesting that the ROS generation was required for inhibiting endogenous phosphatase activity [50].
A more systematic approach came from a series of reports presented by Tonks’ lab. In their studies, a technique based on in-gel phosphatase activity assay was employed that allows simultaneously tagging multiple PTPs undergoing reversible oxidation in response to any stimulus, which trig-gers cellular ROS production. The iodoacetic acid was used to permanently alkylate the free thiolate of lysate proteins, whereas those oxidized Cys residues were resistant to alky-lating reaction [51,52]. In the presence of reducing reagents, the reversibly oxidized PTPs were converted back to the active state, therefore displaying enzymatic activity in the format of in-gel phosphatase assay [51,52]. This novel ana-lysis provided the unambiguous evidence that cellular PTPs are indeed transiently oxidized in response to external oxida-tive stress [53]. Furthermore, in the investigation of specific signaling pathways, results showed that the SHP-2, a cyto-solic SH2 domain-containing phosphatase, was reversibly inactivated through a ROS-mediated mechanism in fibro-blasts treated with PDGF [53]. Moreover, in insulin-stimulated cells, the ROS generation led to the inactivation
of two cytosolic phosphatases, PTP1B and TC-PTP (T Cell-PTP)[54]. In both cases, the reversible oxidation of the specific PTP(s) being identified in turn plays an essential role in fine-tuning the signal pathway in response to the stimulation of that particular ligand [54](Figure 4).
Two recent studies further illustrated that the reversible PTP oxidation participates in the regulation of more diverse and complicated signaling networks. It was showed that the B cell antigen receptor (BCR), which exists as a membrane-bound multiprotein complex, transactivates cytosolic protein tyrosine kinases upon ligand antigen stimulation. In the study reported by Rao’s group, the data indicated that a rapid production of H2O2 and the mobilization of intracellular
Ca2+ worked synergistically through a positive feedback loop
for promoting the optimal activation of Lyn kinase, a key effector responsible for BCR-mediated physiological conse-quences [55]. Although the confirmed result was yet un-available, their preliminary data suggested that the reversi-ble oxidation of SHP-1, a cytosolic PTP, was essential for BCR-induced Ca2+
release from endoplasmic reticulum and Lyn activation [55]. The second study was reported by Karin’s group [56]. They investigated the effect of ROS gen-eration in response to TNF-α, which triggered sustained JNK MAP kinase activation, eventually leading to cell death in
NF-κB-deficient cells. The main effort was focused on
examin-ing whether the MAP kinase phosphatases (MKPs) in DUSP superfamily were the ROS targets in this signaling context. Their data showed that three MKPs (1, 3 and MKP-5), when overexpressed in IKKβ-/- fibroblasts exposed to TNF-α, aggregated as high molecular oligomers through the catalytic Cys-mediated intermolecular disulfide bond [56]. Interestingly, pretreatment of cells with antioxidant prevent-ed the aggregate formation, suggesting that ROS were in-volved in the inactivation of MKPs [56]. The authors thus proposed a novel regulatory mechanism, in which ROS-mediated oxidation and oligomerization of MKPs contributes to the sustained JNK phosphoyrlation, a key factor that de-termines the fate of either cell proliferation or cell death in response to TNF-α stimulation.
ROS as potential mediators of cell transformation and
human cancers
As summarized in Figure 4, our current understanding in-dicates that ROS-induced inactivation of PTPs plays an es-sential role for the maintenance of a normal response in a diverse array of signaling cascades. It is important to note that, under physiological conditions, the oxidation of PTPs is a specific and transient event. The Nature seems to desig-nate ROS-dependent regulation of PTPs in a precisely con-trolled manner. The unique localization of Nox isoforms [57], which might be transactivated by specific upstream signal-ing modulators, confines transiently produced ROS in par-ticular subcellular compartments. Therefore, only a subset of cellular PTPs could gain access to oxidants, suggesting that the redox-dependent regulation is tightly controlled. Soon after the ROS burst is terminated, oxidized PTPs are converted back to the active state via millimolar concentra-tions of GSH and other endogenous reductants [41], there-fore restoring the negative constraint for down-regulating tyrosine phosphorylation signals. Thus, the delicate redox-dependent regulation of PTPs is not only important for achieving an optimal response of physiological stimulation, but also critical for the maintenance of phosphotyrosyl ho-meostasis. Moreover, as mentioned above, other signaling molecules that have a catalytic Cys residue are also highly susceptible to the change of cellular redox potential. The same principle that controls the signal transduction through ROS-mediated regulation of PTPs should be broadly appli-cable to a variety of signaling cascades.
Obviously, the redox homeostasis is a prerequisite for maintaining the normal function of signaling networks. It is
therefore interesting to postulate that, if the cellular redox potential is altered, the perturbed balance of redox–sensitive switch in Cys-containing modulators might lead to profound consequences, including pathological conditions. Indeed, overproduction of ROS has been linked to the development of various human diseases. Data accumulated over last two decades suggested that the elevated ROS levels might pro-mote carcinogenesis, angiogenesis, atherosclerosis, cardiac hypertrophy and aging [58]. Clinical data further supported the pathological role of ROS. For examples, the application of antioxidants showed protective effect at least in the con-text of cancers and cardiovascular diseases [59]. These observations suggested that high levels of overproduced oxidants might severely oxidize cellular proteins, thus causing uncontrolled signaling cascades at the disease le-sions; whereas the restoration of normal redox potential via antioxidants might result in the reduced situation of ROS targets, further leading to more physiologically regulated signal transduction.
Recent research progress at the molecular and cellular levels has greatly advanced our understandings in the proc-ess of ROS-mediated cell transformation and angiogenesis, which are two key steps that lead to cancerous phenotype. It was initially observed that various human cancer cell lines constitutively generated large amounts of ROS, such as H2O2
[60]. The hypothesis at the time was focusing on the prob-ability that ROS function as potent mutagens, therefore in-ducing frequent genetic instability in cancer cells [61]. How-ever, developing progresses gradually revealed that the role of intrinsic ROS was far more complicated than merely DNA mutagens. The first evidence that linked ROS with signal transduction in the context of cell transformation came from a report by Goldschmidt-Clermont’s group. By the use of Ras-transformed fibroblasts as a model, their data clearly showed that the mitogenic signal was oxidant-dependent [62]. The authors therefore proposed that the elevated level of ROS was important for oncogene-induced cell transforma-tion. The causal relationship between oxidant production and cell transformation was then established by a series of studies from Lambeth’s lab. They successfully identified several genes that encode homologous enzymes of catalytic subunit of phagocyte-expressed Nox [6]. As indicated above, those enzymes, which were classified as members of the Nox family, could constitutively generate ROS when ex-pressed in normal fibroblasts. Strikingly, the overexpres-sion of Nox enzymes in fibroblasts led to the development of transformed phenotype, such as increased mitogenic rate, enhanced anchorage-independent growth, and formation of tumor when transplanted in animals [63,64]. Their study further demonstrated that the transformed characters were significantly reversed by introducing the H2O2-decomposing
enzyme catalase into cells [64]. Moreover, high levels of Nox expression also triggered the angiogenic switch as evi-denced by the increased vascularity of tumors and induced molecular markers of angiogenesis [65]. In a separated study, Kamata’s group investigated the role of ROS that might participate in the Ras-mediated mitogenic signaling. Using RNAi technique to knockdown the expression of Nox mRNA in transformed cells, their data demonstrated that indeed Nox enzymes are functionally required for Ras-induced transformation [66].
Closing remark: involvement of PTP oxidation in
hu-man cancers
Now that the pathological role of overproduced ROS has been established for developing and maintaining trans-formed phenotype, the task at hand is to identify ROS tar-gets, which are incapable of executing biological function if constitutively oxidized. Obviously, enzymes in the PTP
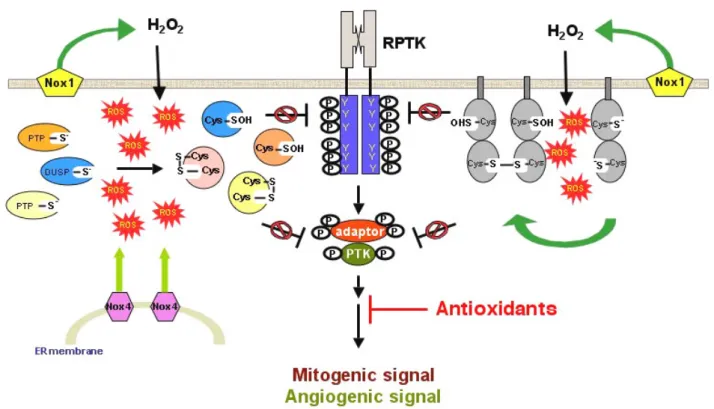
su-Figure 5: Constitutive oxidation and inactivation of endogenous PTPs might contribute to elevated phosphotyrosyl, mitogenic and angiogenic signals. It has been shown that certain types of cancer cells constantly generate ROS in a ligand-independent manner. Preliminary studies sug-gested that the membrane-associated Nox enzymes might be responsible for ROS production under such pathological conditions. The accumula-tion of cellular ROS might react with catalytic Cys residue of endogenous phosphatases, including those PTPs that funcaccumula-tion as tumor suppressors. On the basis of suppressed PTP activity, tyrosine kinases dominate the control of signal transduction, thus promoting tyrosine phosphorylation levels of cellular proteins. We propose that constitutive oxidation and inactivation of endogenous PTPs might play a key role for rapid mitogenic and angiogenic signals in response to cancer cell-associated intrinsic oxidative stress.
perfamily are candidates that deserve careful examinations. Several PTPs, including DEP-1 [67] and PTEN [68,69], have been viewed as tumor suppressors, suggesting that the ac-tive state of these enzymes is a prerequisite for maintaining the normal cellular functions. Interestingly, as described above, studies based on biochemical, structural, cellular and signaling analyses have pointed out PTPs as endogenous redox-sensitive switches. The oxidized PTPs lose their en-zymatic activity, therefore incapable of dephosphorylating substrates in a precise manner. The operating hypothesis is that the active site Cys of PTPs might be constitutively oxi-dized in those transformed or cancer cells that generate large amounts of ROS, presumably due to aberrantly activat-ed Nox enzymes (Figure 5). The oxidizactivat-ed therefore inactivat-ed PTPs are unable to antagonize tyrosine kinase-minactivat-ediatinactivat-ed signaling cascades, eventually leading to the uncontrolled tyrosine phosphorylation level, which is a hallmark of pathological conditions such as cancers [70]. While this hypothesis is waiting for experimental approval, we antici-pated that a great number of investigations will soon provide more insights into PTP oxidation for its role(s) involved in cancers and other human diseases. The long-term goal is to develop novel therapeutic strategies based on the under-standing of molecular processes that control complicated signaling networks in cells.
Acknowledgment
This work was supported by the grant NSC-93-2311-B-001-056 to T.-C. Meng from National Science Council, Taiwan. We also acknowledge the support from Academia Sinica, Taiwan.
References
1. Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys 397: 342-344, 2002.
2. Rhee SG, Bae YS, Lee SR, Kwon J. Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE 2000: PE1, 2000.
3. Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol 10: 248-253, 1998.
4. Finkel T. Redox-dependent signal transduction. FEBS Lett 476: 52-54, 2000.
5. Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol 15: 247-254, 2003.
6. Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 269: 131-140, 2001.
7. Brumell JH, Burkhardt AL, Bolen JB, Grinstein S. Endogenous reactive oxygen intermediates activate tyrosine kinases in hu-man neutrophils. J Biol Chem 271: 1455-1461, 1996.
8. Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 70: 296-299, 1995.
9. Bae YS, Sung JY, Kim OS, Kim YJ, Hur KC, Kazlauskas A, Rhee SG. Platelet-derived growth factor-induced H2O2 production re-quires the activation of phosphatidylinositol 3-kinase. J Biol Chem 275: 10527-10531, 2000.
10. Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, Irani K, Goldschmidt-Clermont PJ, Finkel T. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem J 318: 379-382, 1996.
11. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4: 181-189, 2004.
12. Park HS, Lee SH, Park D, Lee JS, Ryu SH, Lee WJ, Rhee SG, Bae YS. Sequential activation of phosphatidylinositol 3-kinase, beta Pix, Rac1, and Nox1 in growth factor-induced production of H2O2. Mol Cell Biol 24: 4384-4394, 2004.
13. Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an in-tegral role in insulin signal transduction. Mol Cell Biol 24: 1844-1854, 2004.
14. Rhee SG. Redox signaling: hydrogen peroxide as intracellular messenger. Exp Mol Med 31: 53-59, 1999.
15. Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Curr Opin Struct Biol 14: 679-686, 2004. 16. Veal EA, Findlay VJ, Day AM, Bozonet SM, Evans JM, Quinn J,
Morgan BA. A 2-Cys peroxiredoxin regulates peroxide-induced oxidation and activation of a stress-activated MAP kinase. Mol Cell 15: 129-139, 2004.
17. Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in ge-ne activation. Cell 111: 471-481, 2002.
18. Park HS, Huh SH, Kim Y, Shim J, Lee SH, Park IS, Jung YK, Kim IY, Choi EJ. Selenite negatively regulates caspase-3 through a redox mechanism. J Biol Chem 275: 8487-8491, 2000.
19. Ueda S, Nakamura H, Masutani H, Sasada T, Yonehara S, Taka-bayashi A, Yamaoka Y, Yodoi J. Redox regulation of caspase-3(-like) protease activity: regulatory roles of thioredoxin and cyto-chrome c. J Immunol 161: 6689-6695, 1998.
20. Borutaite V, Brown GC. Caspases are reversibly inactivated by hydrogen peroxide. FEBS Lett 500: 114-118, 2001.
21. Baker A, Santos BD, Powis G. Redox control of caspase-3 activ-ity by thioredoxin and other reduced proteins. Biochem Biophys Res Commun 268: 78-81, 2000.
22. Hampton MB, Stamenkovic I, Winterbourn CC. Interaction with substrate sensitises caspase-3 to inactivation by hydrogen per-oxide. FEBS Lett 517: 229-232, 2002.
23. Griffiths SW, King J, Cooney CL. The reactivity and oxidation pathway of cysteine 232 in recombinant human alpha 1-antitrypsin. J Biol Chem 277: 25486-25492, 2002.
24. Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, 3rd, Charrier V, Parsonage D. Protein-sulfenic acids: diverse roles for an un-likely player in enzyme catalysis and redox regulation. Bio-chemistry 38: 15407-15416, 1999.
25. Gorman MA, Morera S, Rothwell DG, de La Fortelle E, Mol CD, Tainer JA, Hickson ID, Freemont PS. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recogni-tion of extra-helical deoxyribose at DNA abasic sites. EMBO J 16: 6548-6558, 1997.
26. Rothwell DG, Barzilay G, Gorman M, Morera S, Freemont P, Hickson ID. The structure and functions of the HAP1/Ref-1 pro-tein. Oncol Res 9: 275-280, 1997.
27. Salmeen A, Barford D. Functions and mechanisms of redox regulation of cysteine-dependent phosphatases. Antioxid Redox Signalling 7: 560-577, 2005.
28. Lane P, Hao G, Gross SS. S-nitrosylation is emerging as a specific and fundamental posttranslational protein modification: head-to-head comparison with O-phosphorylation. Sci STKE 2001: RE1, 2001.
29. Davis KL, Martin E, Turko IV, Murad F. Novel effects of nitric oxide. Annu Rev Pharmacol Toxicol 41: 203-236 2001.
30. Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Fas-induced caspase denitrosyla-tion. Science 284: 651-654, 1999.
31. Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metallopro-teinases: signaling pathway to neuronal cell death. Science 297: 1186-1190, 2002.
32. Li S, Whorton AR. Regulation of protein tyrosine phosphatase 1B in intact cells by S-nitrosothiols. Arch Biochem Biophys 410: 269-279, 2003.
33. Yu CX, Li S, Whorton AR. Redox regulation of PTEN by S-nitrosothiols. Mol Pharmacol 68: 847-854, 2005.
34. Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hy-drogen peroxide. Role in EGF receptor-mediated tyrosine phos-phorylation. J Biol Chem 272: 217-221, 1997.
35. Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Structural and evolutionary relationships among protein tyrosi-ne phosphatase domains. Mol Cell Biol 21: 7117-7136, 2001. 36. Andersen JN, Jansen PG, Echwald SM, Mortensen OH, Fukada T,
Del Vecchio R, Tonks NK, Moller NP. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J 18: 8-30, 2004.
37. Zhang ZY, Dixon JE. Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry 32: 9340-9345, 1993.
38. Lohse DL, Denu JM, Santoro N, Dixon JE. Roles of aspartic acid-181 and serine-222 in intermediate formation and hydrolysis of the mammalian protein-tyrosine-phosphatase PTP1. Biochemis-try 36: 4568-4575, 1997.
39. Barford D, Das AK, Egloff MP. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu Rev Biophys Biomol Struct 27: 133-164, 1998.
40. Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37: 5633-5642, 1998.
41. Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phospha-tase 1B. J Biol Chem 274: 34543-34546, 2004.
42. Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phospha-tase 1B involves a sulphenyl-amide intermediate. Nature 423: 769-773, 2003.
43. van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423: 773-777, 2003.
44. Buhrman G, Parker B, Sohn J, Rudolph J, Mattos C. Structural mechanism of oxidative regulation of the phosphatase Cdc25B via an intramolecular disulfide bond. Biochemistry 44: 5307-5316, 2005.
45. Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem 273: 15366-15372, 1998. 46. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible
inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 277: 20336-20342, 2002.
47. Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J 2: 5501-5510, 2003.
48. Savitsky PA, Finkel T. Redox regulation of Cdc25C. J Biol Chem 77: 20535-20540, 2002.
49. van der Wijk T, Blanchetot C, Overvoorde J, den Hertog J. Re-dox-regulated rotational coupling of receptor protein-tyrosine phosphatase alpha dimers. J Biol Chem 278: 13968-13974, 2003. 50. Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated
hydrogen peroxide reversibly inhibits protein-tyrosine phospha-tase 1B in vivo and enhances the early insulin action cascade. J Biol Chem 276: 21938-21942, 2001.
51. Meng TC, Hsu SF, Tonks NK. Development of a modified in-gel assay to identify protein tyrosine phosphatases that are oxi-dized and inactivated in vivo. Methods 35: 28-36, 2005.
52. Meng TC, Tonks NK. Analysis of the regulation of protein ty-rosine phosphatases in vivo by reversible oxidation. Methods Enzymol 366: 304-318, 2003.
53. Meng TC, Fukada T, Tonks NK. Reversible oxidation and inacti-vation of protein tyrosine phosphatases in vivo. Mol Cell 9: 387-399, 2002.
54. Meng TC, Buckley DA, Galic S, Tiganis T, Tonks NK. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem 279: 37716-37725, 2004.
55. Singh DK, Kumar D, Siddiqui Z, Basu SK, Kumar V, Rao KV. The strength of receptor signaling is centrally controlled through a cooperative loop between Ca2+ and an oxidant signal. Cell 121: 281-293, 2005.
56. Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reac-tive oxygen species promote TNFalpha-induced death and sus-tained JNK activation by inhibiting MAP kinase phosphatases. Cell 120: 649-661, 2005.
57. Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique charac-teristics compared to other NADPH oxidases. Cell Signal 18: 69-82, 2006.
58. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biol-ogy of ageing. Nature 408: 239-247, 2000.
59. Willcox JK, Ash SL, Catignani GL. Antioxidants and prevention of chronic disease. Crit Rev Food Sci Nutr 44: 275-295, 2004. 60. Szatrowski TP, Nathan CF. Production of large amounts of
hy-drogen peroxide by human tumor cells. Cancer Res 51: 794-798, 1991.
61. Feig DI, Reid TM, Loeb LA. Reactive oxygen species in tumori-genesis. Cancer Res 54: 1890s-1894s, 1994.
62. Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275: 1649-1652, 1997.
63. Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the su-peroxide-generating oxidase Mox1. Nature 401: 79-82, 1999. 64. Arnold RS, Shi J, Murad E, Whalen AM, Sun CQ, Polavarapu R,
Parthasarathy S, Petros JA, Lambeth JD. Hydrogen peroxide mediates the cell growth and transformation caused by the mi-togenic oxidase Nox1. Proc Natl Acad Sci USA 98: 5550-5555, 2001.
65. Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy S, Arnold RS, Lambeth JD. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc Natl Acad Sci USA 99: 715-720, 2002.
66. Mitsushita J, Lambeth JD, Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene trans-formation. Cancer Res 4: 3580-3585, 2004.
67. Palka HL, Park M, Tonks NK. Hepatocyte growth factor receptor tyrosine kinase Met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J Biol Chem 278: 5728-5735, 2003. 68. Myers MP, Tonks NK. PTEN. sometimes taking it off can be
better than putting it on. Am J Hum Genet 61: 1234-1238, 1997. 69. Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP,
Hem-mings BA, Wigler MH, Downes CP, Tonks NK. The lipid phos-phatase activity of PTEN is critical for its tumor supressor func-tion. Proc Natl Acad Sci USA 95: 13513-13518, 1998.