_Journal
otOrgang.
metalliC
Chemistry
E L S E V I E R Journal of Organometallic Chemistry 512 (I 996) 101 - 110Reactions of ether-phosphine ruthenium hydride complexes with carbon
disulfide and phenylacetylene: crystal structures
of RuCl(P ~ O)3(
n2-$2 CH)
and Ru(CO) CI(P ~ O)2( r/2-$2 CH)
1Ekkehard Lindner a,*, Ying-Chih Lin b,,, Michael Gepr~igs a, Kuang-Hway Yih b,
Riad F a w z i a, Manfred Steimann b
a lnstitutfiir Anorganische Chemie der Universitfit, Aufder Morgenstelle 18, D-72076 T'fibingen, Germany Department of Chemistry, National Taiwan University, Taiwan 106, Taiwan
Received 6 June 1995; in revised form 11 September 1995
Abstract
The dithioformato complexes RuCI(P ~ O ) 3 ( ' r / 2 - S 2 C H ) (2a,b) and RuCI(P n O)(P ~ O X ' r / 2 - S 2 C H ) (3a,b) are accessible by insertion of CS 2 into the R u - H bond of the (ether-phosphine)(hydrido)ruthenium complexes RuCIH(P O O)(P ~ 0 ) 2 (la,b) [P ~ O ffi r/l(P) - coordinated, P O O = r/2(O,P)-chelated; O,P ffi diphenyl(2-methoxyethyl)phosphine (a), (1,3-dioxan-2-ylmethyl) diphenylphosphine (b)]. Treatment of 2a, 3a and 2b, 3b with carbon monoxide in CH2CI 2 results in the formation of the carbonyl complexes Ru(CO)CI(P ~ O)2(rl2-S2CH) (4a,b). The structures of 2a and 4a were determined by single crystal X-ray diffraction analyses. Crystal data for 2a: space group Pca2j with a = 17.578(4) ,~, b = 14.215(3) ,~, c = 17.934(4) .~, V = 4481(2) ,~3, Z = 4. The smacture was refined to R = 0.037, wR = 0.082. Crystal data for 4a: space group P 2 1 / n with a = 12.009(2) ,~, b ffi 17.143(4) ,~, c = 16.510(4) ,~,, /3 = 107.92(2) °, V ffi 3234(1) ,~3, Z = 4. The structure was refined to R = 0.035, wR ffi 0.087. la,b react with phenylacetylene to give the acetylide complexes RuCI(P N O)2(C~CPh) (5a,b) with evolution of dihydrogen. Carbonylation of 5a yields the cis-Tll(P)-coordinated complex RuCI(CO)2(P ~ O)2(C~-CPh) (6). If chloride is abstracted from Ru(CO)CI(P ~ O)2(T/2-S2CH) (4a) with AgBF 4 in THF the P O O-chelated complex [Ru(CO)(P n O)(P ~ OX-02-S2CH)][BF4] (7) is obtained. Upon reaction of 7 with PhC=-CH the R u - O bond is cleaved and instead of an r/Lvinylidene species the acetylide complex Ru(COXP ~ O)2(T/2-S2CH)(C~-CPh) (8) is formed.
Keywords: Ruthenium; Hydride; Ether-phosphine; Carbon disulfide; Phenylacetylene; Hydrogen bonding; Crystal structure; Group 8
1. I n t r o d u c t i o n
R e c e n t l y we reported on the synthesis, reactivity and d y n a m i c b e h a v i o r o f s o m e transition metal c o m p l e x e s containing hemilabile ether-phosphines (O,P) [1]. These systems, in w h i c h the ether o x y g e n atom is incorporated in o p e n - c h a i n or c y c l i c ether moieties, act as m o n o d e n - tate (P ~ O = r/l(P)-coordinated) or bidentate (P n O = r/2(O,P)-coordinated) ligands. T h e ether o x y g e n atom is able to stabilize coordinatively unsaturated metal c o m - plexes by f o r m a t i o n o f w e a k m e t a l - o x y g e n bonds, and the ether part o f this type o f ligand is regarded as an intramolecular solvent [2]. The m e t a l - o x y g e n b o n d strength depends on the ring size, number, position and
* Corresponding authors.
Dedicated to Professor Marvin Rausch on the occasion of his 65th birthday.
0022-328X/96/$15.00 © 1996 Elsevier Science S.A. All rights reserved SSDI 0 0 2 2 - 3 2 8 X ( 9 5 ) 0 5 9 5 0 - 4
nucleophilicity o f the ether o x y g e n atoms, and the basicity o f the metal center [3]. In reactions o f chelated (ether-phosphine)metal c o m p l e x e s with some small m o l e c u l e s such as sulfur dioxide, ethene, carbon m o n o x i d e , c a r b o n disulfide, and phenylacetylene the m e t a l - o x y g e n b o n d is easily cleaved to form the substi- tuted c o m p l e x e s that have been reported by us recently
[4].
The reactions o f c a r b o n disulfide and phenylacety- lene with transition metal c o m p l e x e s have attracted a great deal o f attention in recent years, o w i n g mainly to their potential as sources o f C1 chemistry in organic synthesis [5], and as catalysts for polymerization and h y d r o g e n a t i o n o f alkynes [6].
T h e cleavage o f the w e a k m e t a l - o x y g e n bond in (ether-phosphine)metal c o m p l e x e s enables the addition o f small molecules under very mild conditions. Herein we describe the reactions o f an (ether-phosphine)(hy-
102 E. Lindner et al./ Journal of Organometallic Chemistry 512 (1996) 101-110
drido)ruthenium complex with carbon disulfide, carbon monoxide and phenylacetylene to form ruthenium com- plexes containing dithioformato, carbonyl and phenyl- acetylide groups.
2. Experimental section
2.1. Materials
All manipulations were performed under argon using vacuum-line and standard Schlenk techniques. Solvents were dried and deoxygenated by refluxing over the appropriate reagents prior to use. n-Hexane, diethyl ether, tetrahydrofuran and benzene were distilled from sodium/benzophenone. Acetonitrile and dichloro- methane were distilled from calcium hydride, and methanol from magnesium. IR spectra were measured on a Bruker IFS 48 instrument and were referenced to a polystyrene standard, using cells equipped with sodium chloride windows. FAB mass spectra were recorded on a Finnigan MAT TSQ 70 (10 kV, 50 °C) spectrometer. Elemental analyses were performed with a Carlo Erba 1106 analyzer; C1 and S analyses were carried out according to Schbniger [7] and analyzed as described by Dirschel and Erne [8] and Wagner [9]. Ru was deter- mined according to the literature [10]. 31p{IH} NMR spectra were obtained on a Bruker WP 80 and a Bruker DRX 250 spectrometer operating at 32.39 and 101.25 MHz respectively, external standard (coaxial insert) 1% H3PO 4 in acetone-d 6 for T < 273 K. IH and t3C{LH} NMR spectra were measured with Bruker DRX 250 and Bruker AMX 400 spectrometers at 250.13 and 62.90, 400.13 and 100.62 MHz respectively, tH and ~3C chem- ical shifts were measured relative to partially deuterated solvent peaks which are reported relative to TMS. All other solvents and reagents were of reagent grade and were used as-received. RuC13, PhC---CH and CS 2 were purchased from Heraeus, Merck and Riedel-de Hafin respectively. The starting complexes RuC1H(P N O)(P ~ O) z (la,b) were synthesized as previously described [111.
2.2. Chloro-tris[diphenyl(2-methoxyethyl)phosphine- P](~2-dithioformato)ruthenium(II) (2a) and chloro- bis[ diphenyl( 2-methoxyethyl)phosphine-P ;O' ,P' h7 2 dithioformato)ruthenium(H) (3a)
A solution of RuC1H(P n O)(P ~ 0 ) 2 (la) (400 rag, 0.46 mmol) in 20 ml of n-hexane was treated with carbon disulfide (126 mg, 1.65 mmol) at ambient tem- perature. Instantaneously the reaction mixture turned to orange-red. After 10 min of stirring an orange-red precipitate was formed. The precipitate was collected by filtration (G4) and dried in vacuo. Further purification was accomplished by recrystallization from a mixture of
CH 2C12/n-hexane (1:10). Analysis of the spectral data showed a mixture of the two complexes 2a and 3a. The ratio 2 a / 3 a of ca. 4:3 was obtained by integration of the absorptions in the 3tp{IH} NMR spectrum. Overall yield 329 mg (85%). When n-hexane was slowly dif- fused into a CHzC12 solution of a mixture of 2a and 3a different shaped crystals of these compounds were ob- tained, which could be separated mechanically for ana- lytic characterization. Spectroscopic data of 2a: m.p. 141-143 °C (dec.). Anal. Found: C, 58.38; H, 5.38; C1, 3.80; Ru, 10.62; S, 6.76. C46H52C103P3RuS 2. Calc.: C, 58.37; H, 5.54; C1, 3.75; Ru, 10.68; S, 6.78%. IR (KBr): 6(HCS) 1225 (m), Ua~(CS 2) 907 (m) cm -1. 31p{JH} NMR (101.25 MHz, CDC13, 298 K): 3 22.6 (t, 2j(pp) 30.5 Hz, pt ~O), 20.1 (d, 2j(pp) 30.5 Hz, p2,3 ~ O). tH NMR (400.13 MHz, CDC13, 298 K): 6 10.29 (dt, 4j(PH) 5.1, 4j(PH) 3.6 Hz, 1 H, HCSe), 7.65-6.70 (m, 30 H, Ph), 3.68-3.01 (m, 12 H, PCH2CH2), 2.82 (s, 9 H, OCH3). 13C{I H} NMR (100.62 MHz, CDC13, 298 K): 6 230.3 (s, CS2), 138.3-126.4 (m, C-Ph), 69.2, 67.9 (s, PCH2CH2), 57.9, 57.5 (s, OCH3), 26.3 (m, PCH2). MS [FAB, 3-nitrobenzyl- alcohol (NOBA)] m / e 945.6 [M+], 910.4 [M+-C1]. Spectroscopic data of 3a: m.p. 138-140 °C (dec.). Anal. Found: C, 53.24; H, 4.72; C1, 5.09; Ru, 14.74; S, 9.33. C3~H35C102P2RuS 2. Calc.: C, 53.02; H, 5.02; CI, 5.05; Ru, 14.39; S, 9.13%. IR (KBr): 6(HCS) 1226 (m), t,as(CS 2) 907 (m) cm - t . 31p{IH} NMR (101.25 MHz, CDC13, 298 K): 6 49.6 (d, 2j(pp) 30.4 Hz, P A O), 46.2 (d, 2j(pp) 30.4 Hz, P ~ O ) . ~H NMR (400.13 MHz, CDC13, 298 K): 6 11.62 (dd, 4j(PH) 3.0, 4j(PH) 2.0 Hz, 1 H, HCS2), 8.07-6.58 (m, 20 H, Ph), 3.53-2.62 (m, 8H, PCHzCH2). 13C{1H} NMR (100.62 MHz, CDC13, 298 K): 6 239.9 (s, CS2). MS (FAB, NOBA) m / e 701.4 [M+], 667.3 [M+-CI]. 2.3. Chloro-tris[(1,3-dioxan-2-ylmethyl)diphenylphos- phine-P](Tle-dithioformato)ruthenium(ll) (2b) and chloro-bis[ (1,3-dioxan-2-ylmethyl)diphenylphosphine- P ;O',P'](Tle-dithioformato)ruthenium(II) (3b)
A solution of RuC1H(P n O)(P ~ 0 ) 2 (lb) (400 mg, 0.401 mmol) in 20 ml of diethyl ether was treated with carbon disulfide (126 mg, 1.65 mmol) at room tempera- ture. Instantaneously the reaction mixture turned to orange-red. The solution was concentrated under vac- uum to give an orange-red precipitate. The precipitate was collected by filtration (G4) and dried in vacuo. Analysis of the spectral data showed a mixture of the complexes 2b and 3b. The ratio 2 b / 3 b of ca. 1 : 18 was obtained by integration of the absorptions in the s~ p{¿ H} NMR spectrum. Overall yield 295 mg (92%). Spectro- scopic data of 2b: because of the low yield, 2b could not be separated from 3b, therefore an elemental analy- sis of 2b was not available. 3tp{JH} NMR (101.25 MHz, CDC13, 298 K): 6 24.6 (t, 2j(pp) 26.3 Hz,
E. Lindner et al./Journal of Organometallic Chemistry 512 (1996) 101-110 103 pl ~ O), 21.5 (d, 2j(pp) 26.3 Hz, p2,3 ~ O). IH (400.13 MHz, CDC13, 298 K): ~ 10.17 (dt, 4j(PH) 4.8, 4j(PH) 3.6 Hz, 1 H, HCS2), 8.22-6.81 (m, 30 H, Ph), 4.81-1.00 (m, 27 H, alkanes). 13C{IH} NMR (100.62 MHz, CDC13, 298 K): t5 137.2-125.1 (m, C-Ph), 104.4 (d, 2J(PC) 14.1 Hz, PCH2CH), 66.9 (s, OCH2CH2), 35.1 (m [12], J(PC) 17.1 Hz, PCH2), 25.4 (s, OCH2CH2). Spectro- scopic data of 3b: m.p. 110-112 °C (dec.). Anal. Found: C, 53.55; H, 5.19; C1, 5.13; Ru, 12.97; S, 7.81 ~}. C35H39C104P2RuS 2. Calc.: C, 53.46; H, 5.00; CI, 4.51; Ru, 12.85; S, 8.16%. IR (KBr) 3(HCS) 1243 (m), Va~(CS 2) 928 (m) cm -l. 31P{JH} NMR (101.25 MHz, CDC13, 298 K): 6 44.5 (d, 2j(pp) 31.5 Hz, P NO), 22.6 (d, 2j(pp) 31.5 Hz, P ~ O). IH NMR (400.13 MHz, CDC13, 298 K): 3 11.68 (dd, 4j(PH) 5.1, 4j(PH) 3.6 Hz, 1 H, HCS2), 7.89-6.22 (m, 20 H, Ph), 4.47-0.59 (m, 18 H, alkanes). 13C{IH} NMR (100.62 MHz, CDC13, 298 K): 6 239.8 (s, CS2), 138.7-126.5 (m, C-Ph), 100.6 (d, 2j(PC) 20.1 Hz, PCH2CH, P N O), 100.3 (d, 2J(PC) 5.0 Hz, PCH2CH, P ~ O), 67.4 (m, OCH2CH 2, P N O), 66.4 (m, OCH2CH 2, P ~ O), 37.8 (m [12], J(PC) 26.2 Hz, PCH 2, P f~ O), 35.1 (m [12], J(PC) 17.1 Hz, PCH 2, P ~ O), 26.3 (s, OCHzCH z, P A O), 25.1 (s, OCH2CH 2, P ~ O). MS (FAB, NOBA)
m / e
786.6 [M÷], 751.6 [M+-C1], 673.7 [M+-C1-HCS~].
2.4. Carbonyl(chloro)-bis[diphenyl(2-methoxyeth-
yl )phosphine-P ] ( 71e-dithioformato )ruthenium( I I ) ( 4a )
Carbon monoxide was bubbled for 10 min into a stirred solution of a 4:3 mixture of 2a (171 mg, 0.181 mmol) and 3a (129 mg, 0.184 mmol) in 20 ml of dichloromethane at ambient temperature. A color change from red to yellow occurred within 5 min. Subsequently the solvent was removed under vacuum. The residue was redissolved in 5 ml of CH2C12. n-Hexane (15 ml) was added to the solution and a yellow precipitate was formed. The precipitate was collected by filtration (G4) washed with n-hexane (2 × 10 ml) and then dried in vacuo yielding 253 mg (95%) of 4a; m.p. 157-159 °C (dec.). Anal. Found: C, 52.64; H, 5.18; C1, 4.90; Ru, 13.92; S, 8.73. C 3 2 H 3 5 C 1 0 3 P 2 R u S 2. Calc.: C, 52.63; H, 4.83; C1, 4.85; Ru, 13.84; S, 8.78%. IR (KBr): ~,(CO) 1946 (vs) cm -I, 8(HCS) 1226 (m), Uas(CS 2) 924 (m) c m - 1 . 31p{IH} NMR (101.25 MHz, CDC13, 298 K): 6 24.7. IH NMR (400.13 MHz, CDCl 3, 298 K): 6 10.40 (t, 4j(PH) 3.5 Hz, 1 H, HCS2), 7.75-7.04 (m, 20 H, Ph), 3.72-3.54 (m, 8 H, PCHzCH2), 3.40 (s, 6 H, OCH3). 13C{IH} NMR (100.62 MHz, CDC13, 298 K): 232.1 (t, 3j(PC) 5.0 Hz, CS2), 202.5 (t, 2J(PC) 12.1 Hz, CO), 133.9-127.6 (m, C-Ph), 68.2 (s, PCH2CH2),
~The elemental analysis was obtained from a 1:18 mixture of 2 b / 3 b . 58.2 (s, OCH3), 27.0 (m [12], J(PC) 13.8 Hz, PCH2). MS (FAB, NOBA)
m / e
730.5 [M+], 695.5 [M+-C1], 667.5 [M+-C1-CO].2.5. Carbonyl(chloro)-bis[(1,3-dioxan-2-ylmethyl)di-
phenylphosphine-P] )(~ Cdithioformato)ruthenium(H)
(4b)
Carbon monoxide was bubbled for 5 min into a stirred solution of a 1:18 mixture of 2b (16 mg, 0.015 mmol) and 3b (284 mg, 0.361 mmol) in 20 ml of dichloromethane at ambient temperature. A spontaneous color change from red to yellow occurred. Subsequently the solvent was removed under vacuum. The residue was washed with 10 ml of n-hexane to give a yellow precipitate, which was collected by filtration (G4) and dried in vacuo to yield 300 mg (98%) of 4b; m.p. 151-153 °C (dec.). Anal. Found: C, 53.17; H, 5.09; C1, 4.56; Ru, 12.72; S, 7.01. C36H39CIOsP2RuS 2. Calc.: C, 53.10; H, 4.83; C1, 4.35; Ru, 12.41; S, 7.88%. IR (KBr): v(CO) 1947 (vs) cm -I, 8(HCS) 1227 (m), Va~(CS 2) 927 (m) cm -I. 31p{IH} NMR (101.25 MHz, CDCI 3, 298 K): 6 25.1. IH NMR (400.13 MHz, CDC13, 298 K): 3 10.50 (t, 4j(PH) 3.5 Hz, 1 H, HCS2), 7.82-7.03 (m, 20 H, Ph), 4.34-0.81 (m, 18 H, alkanes). 13C{IH} NMR (100.62 MHz, CDC13, 298 K): 3 231.8 (t, 3j(PC) 5.2 Hz, CS2), 202.7 (t, 2j(PC) 12.4 Hz, CO), 134.2-127.2 (m, C-Ph), 99.9 (s, PCH2CH), 66.7, 66.4 (s, OCH2CH2), 32.5 (m [12], J(PC) 15.1 Hz, PCH2), 25.2 (s, OCH2CH2). MS (FAB, NOBA)
m / e
737.6 [M+-HCSz], 637.2 [M+-HCS2-C1-CO].2.6. Chloro-bisldiphenyl(2-methoxyethyl)phosphine-
O,P](phenylacetylido)ruthenium(ll) (5a)
Addition of phenylacetylene (46 mg, 0.45 mmol) to a solution of la (400 mg, 0.46 mmol) in 20 ml of diethyl ether, followed by a short period of stirring at room temperature, gave a yellow solution which was concen- trated to 5 ml. Upon cooling the solution below 0 °C, a yellow precipitate was obtained which was isolated by filtration (G4) and washed with n-hexane (2 × 10 ml) and subsequently dried under vacuum to yield 240 mg (72%) of 5a; m.p. 100-102 °C (dec.); Anal. Found: C, 62.54; H, 5.76; CI, 4.72; Ru, 13.68. C38H39C102P2Ru. Calc.: C, 62.84; H, 5.41; CI, 4.88; Ru, 13.92%. IR (KBr): v(C--C) 2064 (vs) cm-i. 31p{iH} NMR (32.39 MHz, CHzC12, 248 K): 6 23.5. IH NMR (400.13 MHz, CDC13, 298 K): 8 7.50-7.10 (m, 25 H, Ph), 3.87 (s, 4 H, PCHzCH2), 3.30 (s, 6 H, OCH3), 2.71 (m, 4 H, PCH2). '~C{IH} NMR (100.62 MHz, 298 K): 6 134.2- 127.3 (m, C-Ph), 110.4 (br s, PhC), 108.6 (br s, RuC), 70.0 (m, PCH2CHz), 59.2 (s, OCH3), 30.7 (m, PCH2). MS (FAB, NOBA)
m / e
726.1 [M+], 691.1 [M+-C1], 589.1 [M+-C1-PhC2 ].104 E. Lindner et al. / Journal of Organometallic Chemistry 512 (1996) 101-110
2.7. Chloro-bis[(1,3-dioxan-2-ylmethyl)diphenylphos- phine-O,P](phenylacetylido)ruthenium(ll) (5b )
To a yellow solution of l b (400 mg, 0.401 mmol) in 10 ml of diethyl ether was added phenylacetylene (93 mg, 0.91 mmol) at room temperature. After 5 min of stirring the solvent was removed under vacuum and was washed with 10 ml of n-hexane to give a yellow precipitate. The precipitate was collected by filtration (G4) and dried in vacuo to yield 293 mg (90%) of 5b; m.p. 105-107 °C (dec.). Anal. Found: C, 62.26; H, 5.85; C1, 4.40; Ru, 12.84. C42H43C104P2Ru. Calc.: C, 62.25; H, 5.35; CI, 4.38; Ru, 12.47%. IR (KBr) v ( C - C ) 2065 (vs) cm -I. 31P{IH} NMR (32.39 MHz, CH2C12, 248 K): 6 44.3 (d, 2j(pp) 47.0 Hz), 32.6 (d, 2j(pp) 47.0 Hz). 13C{IH} NMR (100.62 MHz, CDCI 3, 298 K): 137.1-122.1 (m, C-Ph), 104.7 (br s, PhC), 104.2 (br s, RuC), 100.6, 100.4 (s, PCH2CH), 66.9, 66.7, 66.5, 65.8 (s, OCH2CH2), 38.0, 36.4 (m, PCH2), 25.9, 24.7 (each s, OCH2CH2). MS (FAB, NOBA) m / e 809.7 [M+], 775.7 [M+-C1], 673.7 [M+-CI-PhC2].
2.8. Chloro(dicarbonyl)-bis[diphenyl(2-methoxy-
ethyl )phosphine-P] (phe nylacetylido )ruthenium( l I ) (6)
Carbon monoxide was bubbled for 15 min into a stirred solution of 5a (200 mg, 0.275 mmol) in 20 ml of diethyl ether at room temperature. Subsequently the solvent was removed under vacuum. The residue was washed with 10 ml of n-hexane to give a yellow precipitate, which was collected by filtration (G4) and dried in vacuo to yield 200 mg (93%) of 6; m.p. 140-142 °C (dec.). Anal. Found: C, 60.97; H, 5.52; C1, 4.97; Ru, 12.51. C40H39C1OaP2Ru. Calc.: C, 61.41; H, 5.02; CI, 4.53; Ru, 12.92%. IR (KBr): v(C--C) 2047 (m) cm -1, v(CO) 1989 (vs), 1942 (vs) cm -I. 31p{tH} NMR (101.25 MHz, CDCI 3, 298 K): 6 16.7. IH NMR (400.13 MHz, CDC13, 298 K): 6 8.17-7.04 (m, 25 H, Ph), 3.75-3.41 (m, 8 H, PCH2CH2), 3.14 (s, 6 H, OCH3). 13C{IH} NMR (100.62 MHz, CDC! 3, 298 K): 6 113.4 (br s, PhC), 110.2 (br s, RuC), 67.8 (s, PCHECH2), 58.1 (s, OCH3), 26.7 (m, PCH2). MS (FAB, NOBA) m / e 753.4 [M+-CO], 726.6 [M +-
2co1.
2.9. Carbonyl-bis[diphenyl(2-methoxyethyl)phosphine- P ;O' ,P' ] ( rl 2-dithioformato )ruthenium( H ) tetr afluor obo- rate (7)
A mixture of 4a (197 mg, 0.27 mmol) and AgBF 4 (53 mg, 0.27 mmol) dissolved in 15 ml of tetrahydrofu- ran was stirred for 5 h. The solution was stirred and monitored by 31 p{iH} NMR. After complete disappear-
31 I
ance of the P{ H} resonance of complex 4a the reac- tion was finished and the solution was filtered through Celite. The filtrate was dried in vacuo to yield 172 mg
(82%) of 7 as a yellow oil. IR (CH2C12): v(CO) 1987 (vs) cm -l. 31p{IH} NMR (32.39 MHz, THF, 248 K): 8 46.0 (d, 2j(pp) 240.0 Hz, P N O), 18.3 (d, 2j(pp) 240.0 Hz, P ~ O). 1H NMR (400.13 MHz, CDC13, 298 K): 12.17 (pseudo t, 4j(PH) 6.4 Hz, 1 H, HCS2), 7.54-7.13 (m, 20 H, Ph), 3.74-3.36 (m, 8 H, PCH2CH2). |3C{IH} NMR (100.62 MHz, CD3COCD 3, 298 K): 8 233.2 (s, CS2), 203.5 (br s, CO), 134.9-128.2 (m, C-Ph), 69.3 (s, PCH2CH2), 58.9 (s, OCH3), 28.9 (m [12], J(PC) 5.0 Hz, PCH2). MS (FAB, NOBA) m / e 694.7 [M ÷- BEn]. 2.10. Carbonyl-bis[ diphenyl(2-methoxyethyl)phosphine- P ] ( ~7 2-dithiof ormato )( phenylacetylido )ruthenium( ll ) (8)
Phenylacetylene (0.46 mg, 0.45 mmol) was added to a solution of 7 (200 mg, 0.257 mmol) in 5 ml of tetrahydrofuran and 10 ml of methanol. The solution was stirred for 3 days and a yellow precipitate was formed. The precipitate was collected by filtration (G4) and dried in vacuo. Yield 159 mg (78%) of 8; m.p. 152-154 °C (dec.); Anal. Found: C, 59.91; H, 5.22; Ru, 12.78; S, 8.04. C40H4103P2RuS 2. Calc.: C, 60.28; H, 5.19; Ru, 12.68; S, 8.05%. IR (KBr): v(C-=C)2120 (m) cm - t , u(CO) 1940 (vs) cm -1. 31p{1H} NMR (101.25 MHz, CDC13, 298 K): 8 18.3 (s). IH NMR (400.13 MHz, CDC13, 298 K): 8 10.39 (t, 4j(PH) 3.5 Hz, 1 H, HCS2), 7.93-7.09 (m, 25 H, Ph), 3.62-3.41 (m, 8 H, PCH2CH2), 3.19 (s, 6 H, OCH3). 13C{aH} NMR (62.90 MHz, CDCI 3, 298 K): 8 236.3 (t, 3j(PC) 5.2 Hz, CS2), 202.2 (t, 2J(PC) 13.1 Hz, CO), 133.4-124.2 (m, C-Ph), 113.0 (br s, PhC), 110.0 (t, 2j(PC) 17.7 Hz, RuC), 68.1 (s, PCH2CH2), 57.9 (s, OCH3), 29.1 (m [12], J(PC) 15.0 Hz, PCH2). MS (FAB, NOBA) m / e 795.5 [M+], 695.2 [M+-PhC2 ].
2.11. Structure determination of 2a and 4a
Crystal data and details of data collection are sum- marized in Table 1. The atomic coordinates and equiva- lent isotropic displacement parameters for 2a and 4a are given in Tables 2 and 3. Single crystals were obtained by slow diffusion of n-hexane into concentrated CH2CI 2 solutions of 2a and 4a respectively. All crystals were mounted on a glass fiber and transferred to a P4 Siemens diffractometer taking rotation photographs and perform- ing a photo search to find a suitable reduced cell (graphite-monochromated Mo K ct radiation).
The lattice constants were determined with 25 pre- cisely centered high-angle reflections and refined by least-squares methods. All structures were solved by Patterson methods [ 13] and refined by least-squares with anisotropic thermal parameters for all nonhydrogen atoms. Hydrogen atoms were included in calculated positions (riding model). For complex 2a the Flack parameter was determined [-0.04(3)]. An absorption
E. Lindner et al./ Journal of Organometallic Chemistry 512 (1996) 101-110 105 Table 1
Crystal data and collection parameters for 2a and 4a
2a 4 a
Formula FW Crystal size
(mm 3)
Crystal system Orthorhombic Space group Pca2
a (,~) 17.578(4) b (,~) 14.215(3) c (,~.) 17.934(4) (°) v (~3) 4481(2) Z 4 Calc. density 1.403 (g cm -3) h, k, 1 range T (°C) F(000) (e) /x(Mo K a) (mm- ~ ) Scan range (20) 4-50 Reflections 14927 collected No. of unique 7891 reflections Obs. data 6358 [1 > 2o'(I)] No. of parameters 507 Goodness of fit 1.432 RI a 0.037 wR 2 b 0.082 C 46 H 52CIO3P3 RuS2 946.43 0.10 ×0.30× 0.40 C32 H 35CIO3P2RuS2 730.18 0.30 × 0.30 × 0.35 Monoclinic P 2 j / n 12.009(2) 17.143(4) 16.510(4) 107.92(2) 3234(1) 4 1.500 20, - 1 6 ~ 1 3 , ±21 +14,20, +19 - 100 - 100 1960 1496 0.649 0.827 4-50 11390 5700 4480 371 1.573 0.035 0.087 a R t = E I t F o l - l e e II/EI Fo 1.
b wR2 = {E[wfFd - F~)2]}/(E[wfFd)2]} oS.
correction @scan was applied to all structures. Maxi- m u m and m i n i m u m peaks in the final difference synthe- sis were 0.94 and - 0.58 (2a), and 1.25 and - 0.74 (4a) e,~ -3 respectively. Because of disorder the displace- ment parameters of C(30) are very high. Further details of the crystal structure investigations are available on request from the Fachinformationszentrum Karlsruhe, Gesellschafl f'fir wissenschafllich-technische Informa- tion m b H , D-76344 Eggenstein-Leopoldshafen, on quot- ing the despository number CSD-58961, the names of the authors and the journal citation.
3 . R e s u l t s a n d d i s c u s s i o n
3.1. Insertion o f CS 2 into the R u - H bond o f the com- plexes RuCIH(P N O)(P ~ O) e (la,b)
Treatment of the starting compounds RuCIH(P n O)(P ~ 0 ) 2 ( l a , b ) [P ~ O = d i p h e n y l ( 2 - m e t h o x y - ethyl)phosphine (a), (1,3-dioxan-2-ylmethyl)diphenyl- phosphine (b)] with CS 2 afforded mixtures of the air- stable and o r a n g e - r e d complexes R u C I ( P ~ O ) 3 ( r / 2 -
S2CH) (2a,b) and RuCI(P n O)(P ~ O)(r/2-S2CH) (3a,b) with ratios of 4:3 and 1:18 respectively (Scheme 1). From this ratio, it can be derived that the steric
Table 2
Atomic coordinates (× 104) and equivalent isotropic displacement parameters (~2 × 103) for 2a Atom x y z Ueq Ru(1) 9489(1) 8201(1) 2321(1) 17(1) CI(I) 8962(1) 8117(1) 1038(1) 25(1) S(I) 9747(1) 8507(1) 3569(1) 25(I) S(2) 8545(1) 9 3 2 6 ( 1 ) 2799(1) 26(1) P(1) 1 0 5 3 1 ( 1 ) 7 1 6 7 ( 1 ) 2254(1) 21(1) P(2) 1 0 1 1 8 ( 1 ) 9 6 1 1 ( 1 ) 1933(1) 20(1) P(3) 8511(1) 7085(1) 2646(1) 20(1) 0(1) 10797(3) 5061(5) 3829(4) 82(2) 0(2) 9353(3) 11980(4) 976(3) 82(2) 0(3) 6281(2) 7 1 7 3 ( 3 ) 2000(2) 39(1) C(I) 1 1 5 3 6 ( 3 ) 7 7 1 9 ( 4 ) 3396(3) 27(1) C ( 2 ) 12205(3) 8072(4) 3695(3) 32(1 ) C ( 3 ) 12769(3) 8413(4) 3232(3) 37(1) C ( 4 ) 12659(3) 8388(4) 2465(3) 34(2) C ( 5 ) 11998(3) 8019(3) 2165(2) 26(1) C ( 6 ) 1 1 4 2 4 ( 2 ) 7 6 6 3 ( 3 ) 2628(3) 22(1) C ( 7 ) 1 1 5 7 6 ( 3 ) 6 0 8 1 ( 5 ) 1398(4) 42(2) C ( 8 ) 11840(4) 5681(6) 754(4) 59(2) C ( 9 ) 11434(4) 5734(6) 106(4) 57(2) C(10) 1 0 7 3 3 ( 4 ) 6206(5) 93(4) 48(2) C(11) 1 0 4 6 7 ( 3 ) 6630(4) 745(3) 32(1) C(12) 10884(3) 6573(4) 1403(3) 28(I) C(13) 10400(3) 6178(4) 2919(3) 31(1) C(14) 11057(3) 5546(4) 3167(4) 39(1) C(15) 11339(4) 4613(7) 4235(4) 76(3) C(16) 1 1 5 9 5 ( 3 ) 1 0 1 8 7 ( 4 ) 1431(3) 33(1) C(17) 1 2 1 9 2 ( 3 ) 10159(4) 922(3) 38(1) C(18) 12160(3) 9550(5) 318(4) 45(2) C(19) 11533(3) 8983(4) 207(3) 36( I ) C(20) 10935(3) 9005(4) 714(3) 27(1) C(21) 1 0 9 6 5 ( 3 ) 9 5 9 7 ( 4 ) 1328(3) 27(1) C(22) 9915(3) 1 1 1 7 5 ( 4 ) 2899(3) 32(1) C(23) 1 0 0 7 3 ( 3 ) 1 1 6 7 5 ( 5 ) 3547(4) 45(2) C(24) 1 0 6 7 7 ( 4 ) 1 1 4 5 0 ( 5 ) 3991(3) 47(2) C(25) 1 1 1 4 3 ( 4 ) 1 0 7 0 0 ( 5 ) 3789(3) 45(2) C(26) 1 1 0 0 0 ( 3 ) 1 0 1 5 9 ( 4 ) 3153(3) 34(1) C(27) 1 0 3 7 7 ( 3 ) 1 0 3 9 2 ( 4 ) 2701(3) 27(1) C ( 2 8 ) 9 4 9 6 ( 3 ) 1 0 3 3 5 ( 4 ) 1339(3) 28(1) C(29) 9870(4) 1 1 2 3 2 ( 4 ) 1035(4) 39( 1 ) C(30) 8979(8) 12113(12) 323(5) 200(11) C(31) 8765(3) 6 5 6 5 ( 5 ) 4136(3) 38(1) C(32) 8686(4) 6660(6) 4905(3) 53(2) C(33) 8122(5) 7228(6) 5178(4) 64(2) C ( 3 4 ) 7616(4) 7676(5) 4703(4) 51(2) C ( 3 5 ) 7707(3) 7588(4) 3942(3) 35(1) C(36) 8284(3) 7042(4) 3645(3) 26(1) C(37) 8855(3) 561 4 ( 4 ) 1676(3) 32(1) C(38) 8903(3) 4671 (5) 1446(4) 42(1 ) C(39) 8650(3) 3959(4) 1911(4) 41(1) C(40) 8344(3) 4173(4) 2603(4) 40(1) C(41) 8297(3) 5097(4) 2837(3) 33(1) C(42) 8559(2) 5828(3) 2377(3) 26(1 ) C(43) 7604(2) 7415(4) 2196(3) 26( 1 ) C(44) 6939(2) 6747(3) 2288(4) 29( 1 ) C(45) 5651(3) 6 6 2 2 ( 4 ) 2037(4) 49(2) C(46) 9005(3) 9 2 3 4 ( 4 ) 3624(3) 28( 1 )
106 E. Lindner et al. / Journal of Organometallic Chemistry 512 (1996) 101-110
effect of the cyclic ether-phosphine ligand controls the formation of complex 3b. 2a,b and 3a,b are moderately soluble in CH2C12 and CHC13, slightly soluble in ace- tone and tetrahydrofuran, and insoluble in diethyl ether and n-hexane. In the FAB mass spectrum, two parent peaks with the typical Ru isotope distribution are in agreement with the molecular masses of 2a and 3a respectively. Because of the low yield of complex 2b, in the FAB mass spectrum of a mixture of 2b and 3b only the parent peak corresponding to 3b appears. The 1H NMR spectra of mixtures of 2a, 3a and 2b, 3b respec- tively display the proton resonances of the dithiofor- mato groups as a doublet of triplets and a doublet of doublets respectively. The former resonance is traced back to two chemically equivalent r/t(P)-coordinated phosphine molecules p 2 ~ O , p3 ~ 0 and another r/l(p)-coordinated phosphine ligand P ¿ ~ O and stems from compounds 2a,b. The latter resonance results from two inequivalent phosphorus atoms in the spectrum of 3a,b. In the 3¿p{IH} NMR spectrum of the mixtures of 2a, 3a and 2b, 3b respectively, two sets of resonances are clearly distinguishable. The triplet and the doublet with a ratio of 1:2 are assigned to the pl ~ O ligand and to the two chemically equivalent p 2 ~ O and p 3 ~ O ligands of complexes 2a,b. The AB pattern results from two inequivalent phosphorus atoms in 3a,b and the downfield resonance is in the typical range of "r/e(o,P) - coordinated ether-phosphine ligands [3]. The medium intensity absorptions between 907-928 and 1225-1243 cm-~ in the IR spectra of 2a, 3a and 2b, 3b are attributed to Vas(CS 2) and 6(HCS) of the bidentate dithioformato ligand [14]. In the ~3C{tH} NMR spec-
trum of a mixture of 2a and 3a two singlets at lowest field are attributed to the carbon atoms of the dithiofor- mato groups [14]. The relatively upfield resonance of the two singlets is assigned to the carbon atom of the dithioformato group of 2a and it is consistent with a higher electron density at ruthenium in 2a. Because of the low yield of complex 2b, a mixture of 2b and 3b shows only one singlet at lowest field in the 13C{IH} NMR spectrum, attributed to the carbon atom of the dithioformato group of 3b.
Supposedly the formation of 2a,b and 3a,b proceeds via the intermediates A and B, in which carbon disul- fide is ~--bonded to the ruthenium center [15]. This addition either requires an R u - O bond cleavage or an O,P ligand dissociation in la,b to give A and B respec- tively. Subsequently, an insertion of CS 2 into the R u - H bond follows to give the dithioformato complexes men- tioned above. S S
II
II
c H H Co~p4" I
~ P ~ O u " ~ S ~ 0 C1 C1 A BIn order to know how the complexes 2a,b and 3a,b are related to each other, the following experiments have been carried out in the case of 2a and 3a. Mixtures of the compounds were reacted with a hundred-fold excess of carbon disulfide in CH2C1 z for two days, and
a ph2PcOH2PcH2OCHs b Ph2PCH2-~O0 ~ H /~/~S 2 0 S..,..., I f P ~ C~ 2a, b CS21 -O,P H ~ 0 ~ . ..~ P~O 'R6 C~ la, b H H ~0,..
[
,S .IP~O .... R6 ... COk..p~l~p~
o- o,p " o ~ / [ X c °
Cl CI .30, b 4a, b Ph Ph PhC~ CH ~0o. If . . o ~ / o . , . 1 7.o cI o..~j 5a 5b/
CO P~O = r/LP-coordinated 1 P^O T/2-O,P-coordinoted PhO,P = Ether-phosphine ligand I~
oc.. 'l' ,.co " Ru ....
o~FJt \p o
CI B S c h e m e 1.E. Lindner et al./Journal of Organometallic Chemistry 512 (1996) 101-110 107 the ratio of the complexes did not change. Within 2 h
even in refluxing THF the mentioned ratio was formed again. Upon treatment of the obtained 4:3 mixture of 2a and 3a with a two-fold excess of PhzPCHzCH2OCH 3 in refluxing toluene for 1 h, the ratio changed to 5:1. To sum up these findings, it can be said that 2a is formed from 3a and Ph2PCHzCHzOCH3; but an ether-phos- phine dissociation from 2a to give 3a does not occur.
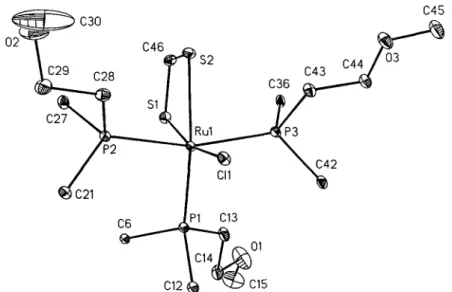
To confirm the insertion of CS 2 into the R u - H bond, complex 2a was characterized by X-ray diffraction anal- ysis. The ORrEP diagram with atom labeling is shown in Fig. 1. Both sulfur atoms of the incoming CS 2 molecule are coordinated to the ruthenium metal.
The distorted octahedral geometry around the metal atom is due to a small S ( 1 ) - R u - S ( 2 ) angle of 70.98(4) ° in 2a (Table 4). This angle is approximately equal to the corresponding value of 69.83(5) ° found in Os(r/2- SzCH)(P/Pr3)3(CO)C1 [15]. The two R u - S bond lengths are significantly different, which is caused by the differ- ent trans-directing influence of the trans-positioned phosphine and chlorine in 2a.
Interestingly, the action of carbon disulfide on the corresponding tris(triphenylphosphine)ruthenium com- ~llex RuCIH(PPh3) 3, according to the 3]p{1H} and
I
C{ H} NMR spectra, affords more than ten products. Compared with the above-mentioned system this reac- tion proceeds completely unselectively.
3.2. Carbonylation o f complexes 2 and 3 to form the complexes Ru(CO)CI(P ~ 0 ) 2 (77 2_S 2 CH) (4a, b)
Mixtures of 2a, 3a and 2b, 3b respectively were allowed to react with carbon monoxide in CH2C12 to give yellow air-stable products which were identified as
the complexes Ru(CO)CI(P~O)2(r/2-S2CH) (4a,b). Upon this reaction, not only was one of the ether-phos- phine ligands in 2a,b substituted by carbon monoxide, but also the R u - O bond of 3a,b was cleaved to form the carbonylation products 4a,b. Because of the weaker R u - O bond of the cyclic ether-phosphine ligand [3a], the rate of formation of 4b was faster than that of 4a. 4a and 4b are easily soluble in polar, but insoluble in nonpolar, solvents. In the mass spectra of 4a,b, the molecular ion along with the [M+-CO] peak are de- tected with highest intensity. The IR spectra show one strong carbonyl stretching band at 1946 c m - t for 4a and 1947 c m - ~ for 4b. At lowest field one triplet with a small 4j(PH) coupling constant was found in the ~H NMR spectra for 4a,b, which is characteristic of two
trans-phosphine ligands [16]; it is attributed to the pro-
ton of the dithioformato group. Owing to the coupling with two equivalent P atoms, the ~3C{IH} NMR spectra of 4a and 4b reveal two triplets at lowest field, which are assigned to the carbon atoms of the dithioformato ligand and the terminal carbonyl group respectively.
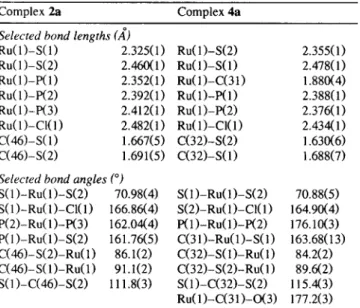
The structure of complex 4a was determined by X-ray crystal structure analysis to ensure the geometry and nature of the carbonylation reaction. An ORTEP plot of 4a is shown in Fig. 2. The ruthenium-carbon bond length (Ru(1)-C(31) 1.880(4) ,~) in 4a is shorter than the corresponding R u - C O distance (2.064(17) ,~) in
cis-Ru(CO)CI(PPh3)2(rf-S2CH) [16], which can be ex-
plained by the replacement of the chloride with the better o-donating dithioformato group in 4a. The R u - P distances and that of R u - C I (Table 4) in 4a are identical with those in cis-Ru(CO)CI(PPhO2(7)2-S2CH) ( R u - P 2.396(2) and 2.398(2) .~; Ru-C1- 2.421(3) A). These bond lengths are in the expected range [17].
~ C 3 0
02 T
C46
C21
C12
C45
$2
C43 C 4 4 F
''~'
) ~1013
108 E. Lindner et al./ Journal of Organometallic Chemistry 512 (1996) 101-110
Table 3
Atomic coordinates (× 10 4) and equivalent isotropic displacement parameters (/~2 × 103) for 4a Atom x y Z Ueq Ru(1) 519(1) 2 1 9 2 ( 1 ) 3975(1) 21(1) CI(1) -61(1) 833(1) 3707(1) 43(1) P(2) 2457(1) 1 8 5 3 ( 1 ) 4024(1) 22(1) P(1) - 1458(1) 2 4 4 1 ( 1 ) 3924(1) 24(1) S(1) - 11(1) 2 5 3 2 ( 1 ) 2444(1) 60(1) S(2) 890(1) 3 5 2 7 ( 1 ) 3841(1) 46(1) 0(1) -4537(2) 1 3 5 5 ( 2 ) 2656(2) 48(1) 0(2) 2776(3) 125(2) 2400(2) 53(1) 0(3) 1397(2) 2 2 7 3 ( 2 ) 5870(2) 48(1) C(1) - 2596(3) 3 7 1 2 ( 2 ) 2936(3) 39(1) C ( 2 ) -2937(4) 4 4 9 2 ( 3 ) 2812(3) 54(1) C(3) - 2630(5) 5 0 0 9 ( 3 ) 3470(4) 62(1 ) C(4) - 1947(5) 4 7 7 1 ( 3 ) 4267(3) 60(1) C(5) - 1599(4) 3 9 9 5 ( 2 ) 4405(3) 43(1 ) C(6) - 1943(3) 3 4 5 7 ( 2 ) 3747(2) 29(1 ) C ( 7 ) -2613(3) 2 5 3 7 ( 2 ) 5192(2) 32(1) C(8) - 2863(3) 2 2 7 0 ( 2 ) 5920(2) 36(1 ) C ( 9 ) -2285(3) 1 6 2 1 ( 2 ) 6354(2) 40(1) C(10) - 1471(3) 1 2 3 3 ( 2 ) 6065(3) 39(1) C(11) - 1228(3) 1 4 9 0 ( 2 ) 5335(2) 33(1) C(12) - 1795(3) 2 1 5 1 ( 2 ) 4900(2) 27(1) C(13) - 2541(3) 1 8 8 8 ( 2 ) 3092(2) 34(1) C(14) -3813(3) 1 9 6 1 ( 3 ) 3093(3) 52(1) C(15) - 4822(5) 1 3 9 5 ( 4 ) 1773(3) 72(2) C(16) 3760(3) 3 0 3 9 ( 2 ) 5026(2) 34(1) C(17) 4507(4) 3 6 7 9 ( 2 ) 5211(3) 42(1) C(18) 4949(3) 3 9 7 7 ( 2 ) 4590(3) 44(1) C(19) 4647(4) 3 6 3 8 ( 2 ) 3796(3) 41(1) C(20) 3910(3) 2 9 9 4 ( 2 ) 3609(2) 32(1) C(21) 3462(3) 2 6 8 8 ( 2 ) 4230(2) 25(1) C(22) 2613(3) 687(2) 5261(2) 34(1) C(23) 3209(4) 172(2) 5890(3) 40(1) C(24) 4422(4) 134(2) 6138(2) 37(1 ) C(25) 5033(3) 621(2) 5756(2) 36(1) C(26) 4442(3) 1 1 4 0 ( 2 ) 5133(2) 31(1 ) C(27) 3220(3) 1 1 8 2 ( 2 ) 4876(2) 25(1) C(28) 2706(3) 1 3 9 3 ( 2 ) 3080(2) 31(1) C(29) 2230(4) 558(3) 2922(3) 47(1 ) C(30) 2358(5) 324(3) 1555(3) 56(1 ) C(31) 1048(3) 2 2 3 4 ( 2 ) 5171(2) 33(1) C(32) 429(4) 3 4 3 7 ( 3 ) 2809(4) 64(2) Table 4
Selected interatomic distances (,~) and angles (°) for 2a and 4a
Complex 2a Complex 4a
Selected bond lengths (A)
Ru(1)-S(1) 2.325(1) Ru(1)-S(2) 2.355(1) Ru(1)-S(2) 2.460(1) Ru(1)-S(1) 2.478(1) Ru(1)-P(1) 2.352(1) Ru(1)-C(31) 1.880(4) Ru(1)-P(2) 2.392(1) Ru(1)-P(1) 2.388(1) Ru(1)-P(3) 2.412(1) Ru(1)-P(2) 2.376(1) Ru(1)-Cl(l) 2.482(1) Ru(1)-CI(1) 2.434(1) C(46)-S(1) 1.667(5) C(32)-S(2) 1.630(6) C(46)-S(2) 1.691(5) C(32)-S(1) 1.688(7)
Selected bond angles (°)
S(I)-Ru(1)-S(2) 70.98(4) S(1)-Ru(1)-S(2) 70.88(5) S(1)-Ru(I)-CI(1) 166.86(4) S(2)-Ru(1)-CI(1) 164.90(4) P(2)-Ru(1)-P(3) 162.04(4) P(1)-Ru(I)-P(2) 176.10(3) P(1)-Ru(1)-S(2) 161.76(5) C(31)-Ru(1)-S(I) 163.68(13) C(46)-S(2)-Ru(1) 86.1(2) C(32)-S(1)-Ru(I) 84.2(2) C(46)-S(1)-Ru(1) 91.1(2) C(32)-S(2)-Ru(1) 89.6(2) S(I )-C(46)-S(2) 111.8(3) S(1)-C(32)-S(2) 115.4(3) Ru(1)-C(31)-O(3) 177.2(3)
3.3. Preparation o f the acetylide-ruthenium complexes
RuCt(P n 0)2(C =-- CPh) (Sa,b)
C o m p a r e d with the bis(chelate)ruthenium complexes ClzRu(P n 0 ) 2 [18], the starting compounds RuC1H(P n O)(P ~ O) z ( l a , b ) show a different behavior toward phenylacetylene. Instead of the formation of r/Lvinyli- dene complexes, hydrogen is evolved and the acetylide complexes RuCI(P n O ) 2 ( C = C P h ) (5a,b) are formed.
5a and 5b are obtained in high yields as yellow air-stable products which are soluble in dichloromethane but insoluble in nonpolar solvents. The FAB mass spectra show parent peaks corresponding to the molecu- lar masses of 51 and 5b. The IR spectra include a strong band at 2064 c m - I for 5a and 2065 cm-~ for 5b, which can be assigned to the C - - C stretching mode of the acetylide moiety [19]. In the 31 p{i H} N M R spectrum of 5a only one singlet appears, which indicates the
cis-position of the two chemically equivalent r/2(O,P) -
P1
12
C14~,~ "~ ' ~ C13
C 3 2 ~
$2
03
sly.,.. R c31
7"- ""-'-"--4
C28
CI1C30
C21
2C27
)C29
E. Lindner et al. / Journal of Organometallic Chemistry 512 (1996) 101-110 109 chelated ether-phosphines. Interestingly, the 31p{IH}
NMR spectrum of 5b shows an AB pattern in the typical range of r/2(O,P)-coordinated O,P ligands [3a]. It is attributed to the unsymmetric geometry of 5b caused by the steric hindrance of the cyclic ether-phos- phines. The 13C{tH} NMR spectra of 5a and 5b reveal two broad singlets at 6 110.4 and 108.6 for 5a and
104.7 and 104.2 for 5b, which are assigned to the carbon atoms of the acetylide moieties [20]. According to the literature [20], the formation of 5a and 5b may be due to the oxidative addition of P h C - C H to the ruthe- nium metal, or an acid-base reaction between the hy- dride and PhC---CH followed by a reductive elimination of H z.
3.4. Carbonylation of complex 5a to form complex RuCI(CO)z(P ~ 0)2(C--CPh) (6)
In previous work [la,21] we investigated the car- bonylation of RuC12(P D 0 ) 2 leading to the cleavage of two R u - O bonds to give the all-trans-RuC12(CO)2(P ~
0 ) 2 product. Although the geometry of RuCI(P D O)z(C=CPh) (5a) is comparable with that of RuC12(P
A 0 ) 2, the carbonylation of 5a results in the complex RuCI(CO)2(P ~ O)2(C=--CPh) (6) with a cis-arrange-
ment of the carbonyl and phosphine ligands (Scheme 1). Compound 6 is a pale yellow air-stable complex which is soluble in dichloromethane but insoluble in nonpolar solvents. The FAB mass spectrum of 6 shows a base peak corresponding to [M-CO] + . Two strong terminal carbonyl stretching bands in the IR spectrum of 6 display the cis-position of the two molecules. One
medium IR absorption at 2047 cm -l and two broad singlets at 6 113.4 and 110.2 in the 13C{IH} NMR spectrum confirm the acetylide moiety in complex 6. The 31p{IH} NMR spectrum of 6 exhibits one reso- nance, indicating the cis-position of the two equivalent
r/l(P)-coordinated ether-phosphines [22].
3.5. Chloride abstraction from complex Ru(CO)CI(P ~ O)2(rle-SeCH) (4a) to form the complex [Ru(CO)(P ¢q O)(P ~ O)(n 2-$2 CH)1IBF4 ] (7)
r/Z(O,P)-chelated complexes of the type [Cp* Ru(L)- (P N O)][BPh 4] (Cp* = @-CsM%; L = CO, P(OEt) 3) [22] have been obtained by chloride abstraction from
Cp* Ru(L)(P ~ O)C1 with NaBPh 4 in CHaC12. The rate of this reaction depends on the basicity of the metal [23]. The employment of NaBPh 4 to remove C1- from 4a was not successful. Obviously, the strong electron withdrawing properties of the carbonyl ligand and the weak electron donating function of the dithioformato group in 4a decrease the electron density at the central metal.
However, the intramolecular coordination of the ether moiety in 4a, which leads to the r/2(O,P)-chelated complex [Ru(CO)(P ('10)(P ~ O)(r/2-S2CH)][BF4] (7), succeeded with AgBF 4 in tetrahydrofuran at room tem- perature (Scheme 2). Compound 7 was obtained as a yellow oil which decomposed upon exposure to air. The FAB mass spectrum of 7 shows a base peak correspond- ing to [Ru(CO)(P D O)(P ~ O)('r/2-S2CH)] +, formed by loss of the BF 4- group. The 31p{IH} NMR spectrum of 7 exhibits an AB pattern resulting from two different coordination modes of the ether-phosphine ligands. The high field doublet occurs in the typical range of an r/l(P)-coordinated ether-phosphine ligand [22], and the downfield doublet reveals an r/2(O,P)-coordinated ether-phosphine. The 13C{IH} NMR spectrum of 7 re- veals two broad singlets at lowest field, which are assigned to the carbon atoms of the dithioformato ligand and the terminal carbonyl group respectively. The shift of the absorption of the carbonyl stretching vibration to higher wavelengths in the IR spectrum of 7 is attributed to the cationic character of the metal center.
3.6. Synthesis o f complex Ru(CO)(P ~ O)e(~l z- SzCH)(C=- CPh) (8)
Methods for the synthesis of a series of r/Lvinyli- dene complexes using CpRuCI(PR3) 2 [24] or [Cp * RuCl{r/Z(P,O) - ipr 2 PCH2CO2CH 3}] [25] and 1-al- kynes have been reported. Furthermore, the reaction of the cationic r/2(O,P)-chelated ruthenium complex [Cp* Ru(L)(P D O)][BPh 4] (Cp* = r/5-CsM%; L = CO, P(OEt) 3, P ~ O) with phenylacetylene yielded the corre- sponding cationic r/l-vinylidene complexes [4,23]. To explore the reactivity of complex 7, we studied its behavior towards phenylacetylene.
The unexpected formation of the acetylide complex Ru(CO)(P ~ O)2(r/2-S2CH)(C~CPh) (8) was achieved by the reaction of 7 with phenylacetylene. It is a yellow
H \ H \ -l+
c~,
S.... ... RU'I "P'"O Ag~IF4 S ... Rlu..i>,p~ 0 O ~ p / ' ] ' ~ . C O - A g C I p / l ~ C 0 o k...o 4a 7 PhC~CH BF4- _ HBF 4 H \ C~. //'~S S.,,.,,. I .... P~O RtS O~P/" t ~'CO Ph Scheme 2.
110 E. Lindner et al./Journal of Organometallic Chemistry 512 (1996) 101-110
air-stable crystalline solid and is soluble in polar sol- vents like CH2C12 and CHC13. The F A B mass spec- trum o f 8 shows a parent p e a k which corresponds to the fragment [ R u ( C O ) ( P ~ O ) 2 ( r / 2 - S 2 C H ) ( C ~ C P h ) ] +. The 13C{tH} N M R spectrum o f 8 indicates one broad singlet and one triplet at 6 1 13.0 and 1 10.0 for the acetylide carbon atoms, and the c o r r e s p o n d i n g IR spectrum shows a m e d i u m band at 2 1 2 0 c m - t which is assigned to the C - C bond. C o m p o u n d 8 reveals a singlet in the 3~ p{j H} N M R spectrum, attributable to the t w o chemical equiva- lent r/l(P)-coordinated ether-phosphines. One triplet with a small 4 j ( P H ) c o u p l i n g constant at lowest field in the ~ H N M R spectrum o f 8 corresponds to the proton o f the dithioformato g r o u p and is characteristic for the
t r a n s - p o s i t i o n o f the two ether-phosphines [16].
3.7. C o n c l u s i o n
This study describes the chemical behavior o f the tris(ether-phosphine)(hydrido)ruthenium(II) c o m p l e x e s RuC1H(P N O)(P ~ 0 ) 2 ( l a , b ) provided with different steric-demanding ether-phosphine ligands towards some small molecules. T o establish the influence o f the steric e n c u m b r a n c e around the ruthenium center, and o f the R u - O b o n d strength towards i n c o m i n g substrates, the t w o ligands P h 2 P C H 2 C H z O C H 3 and P h 2 P C H 2- C 4 H 7 0 2 have been e m p l o y e d . O n e h y d r o g e n and one chlorine ligand and three ether-phosphines with m e r -
position in l a , b f o r m the octahedral 18-electron precur- sors only w e a k l y protected by intramolecular chelation o f the ether moiety. H e n c e , the facile cleavage o f one R u - O bond o f the chelated ether-phosphine is used to activate c a r b o n disulfide, phenylacetylene, and carbon m o n o x i d e . T h e reaction o f l b , which contains the steri- cally more d e m a n d i n g and w e a k e r R u - O b o n d e d ligand P h 2 P C H 2 C 4 H 7 0 2 , with c a r b o n disulfide prefers the formation o f the less e n c u m b e r e d dithioformato c o m - p o u n d 3b. C o m p a r e d with the c o r r e s p o n d i n g triph- e n y l p h o s p h i n e c o m p l e x RuC1H(PPh3) 3, the ether-phos- phine c o m p l e x e s l a , b react with CS 2 to proceed c o m - pletely selectively. To sum up the results, it can be said that the hindrance o f the ether-phosphine ligand in the dithioformato and acetylido products and the weakness o f the R u - O bond respectively are in the order P h 2 P C H 2 C 4 H 7 0 2 > P h z P C H z C H z O C H 3.
Acknowledgements
The support o f this research by the Fonds der C h e m i s c h e n Industrie is gratefully a c k n o w l e d g e d . W e thank the B A S F Aktiengesellschaft for valuable starting materials. W e also a c k n o w l e d g e a scholarship from the D A A D / N S C ( G e r m a n A c a d e m i c E x c h a n g e S e r v i c e / National Science Council, Taipei, Taiwan) P r o g r a m o f Research Visits to G e r m a n y by N S C - s p o n s o r e d Ph.D. candidates.
References and notes
[1] (a) E. Lindner, U. Schober, R. Fawzi, W. Hiller, U. Englert and P. Wegner, Chem. Ber., 120 (1987) 1621; (b) E. Lindner and U. Schober, Inorg. Chem., 27 (1988) 212; (c) E. Lindner and B. Karle, Chem. Ber., 123 (1990) 1469; (d) B. de Klerk-Engels, J.H. Groen, K. Vrieze, A. M~ckel, E. Lindner and K. Goubitz,
lnorg. Chim. Acta., 195 (1992) 237; (e) E. Lindner, M. Haustein, H.A. Mayer, H. Ktlhbauch, K. Vrieze and B. de Klerk-Engels,
Inorg. Chim. Acta., 215 (1994) 165.
[2] A. Bader and E. Lindner, Coord. Chem. Rev., 108 (1991) 27. [3] (a) E. Lindner, A. M~kel, H.A. Mayer, H. Kiihbauch, R. Fawzi
and M. Steimann, lnorg. Chem., 32 (1993) 1266; (b) E. Lind- ner, Q. Wang, H.A. Mayer, R. Fawzi and M. Steimann,
Organometallics, 12 (1993) 1865.
[4] E. Lindner, M. Haustein, R. Fawzi, M. Steimann and P. Weg- ner, Organometallics, 13 (1994) 5021.
[5] (a) K.H. Yih, Y.C. Lin, M.C. Cheng and Y. Wang,
Organometallics, 13 (1994) 1561; (b) K.H. Yih, Y.C. Lin, G.H. Lee and Y. Wang, J. Chem. Soc., Chem. Commun., (1995) 223. [6] C. Bianchini, C. Bohanna, M.A. Esteruelas, P. Frediani, A. Meli, L.A. Oro and M. Peruzzini, Organometallics, 11 (1992) 3837.
[7] (a) W. Sch~niger, Microehim. Acta., (1955) 123; (b) W. Sch~niger, Microchim. Acta., (1956) 869.
[8] A. Dirschel and F. Erne, Microchim. Acta., (1961) 866. [9] S. Wagner, Microchim. Acta., (1957) 19.
[10] E. Lindner, A. Bader and H.A. Mayer, Z. Anorg. Allg. Chem., 5 9 8 / 5 9 9 (1991) 235.
[11] E. Lindner, M. Kemmler, T. Schneller and H.A. Mayer, lnorg. Chem., 34 (1995) 5489.
[12] Part of an AXX' spectrum, J = I J J(PC) + 3j(PC) 1.
[13] G.M. Sheldrick, SHELXL-93 program, University of G~ttingen. [14] (a) K.K. Pandey, Coord. Chem. Rev., 140 (1995) 37; (b) S.D.
Robinson and A. Sahajpal, lnorg. Chem., 16 (1977) 2718; (c) P.B. Critchlow and S.D. Robinson, lnorg. Chem., 17 (1978) 1902.
[15] H. Werner, M.A. Tena, N. Mahr, K. Peters and H.G. von Schnering, Chem. Ber., 128 (1995) 41.
[16] S. Gopinathan, I.R. Unni and C. Gopinathan, Polyhedron, 6
(1987) 1859.
[17] (a) E. Lindner, U. Schober, R. Fawzi, W. Hiller, U. Englert and P. Wegner, Chem. Ber., 120 (1987) 1621; (b) H. Werner, A. Stark, M. Schulz and J. Wolf, Organometallics, 11 (1992) 1126. [18] E. Lindner, M. Gepr~igs, K. Gierling, R. Fawzi and M. Steimann,
lnorg. Chem., in press.
[19] (a) C. Bianchini, C. Bohanna, M.A. Esteruelas, P. Frediani, A. Meli, L.A. Oro and M. Peruzzini, Organometallics, 11 (1992) 3837; (b) G. AIbertin, S. Antoniutti, E.D. Ministro and E. Bordignon, J. Chem. Soc., Dalton Trans., (1992) 3203. [20] (a) M. Helliwell, K.M. Stell and R.J. Mawby, J. Organomet.
Chem., 365 (1988) C32; (b) J.M. Bray and R.J. Mawby, J.
Chem. Soc., Dahon Trans., (1989) 589; (c) M.J. Tenorio, M.C. Puerta and P. Valerga, J. Chem. Soc., Chem. Commun., (1993) 1750; (d) A. Salvini, P. Frediani, M. Bianchi and F. Piacenti,
lnorg. Chim. Acta., 227 (1994) 247.
[21] E. Lindner, A. M6ckel, H.A. Mayer and R. Fawzi, Chem. Ber., 125 (1992) 1363.
[22] E. Lindner, M. Haustein, H.A. Mayer, K. Gierling, R. Fawzi and M. Steimann, Organometallics, 14 (1995) 2246.
[23] E. Lindner, S. Pautz and M. Haustein, J. Organomet. Chem., in press.
[24] M.I. Bruce and R.C. Wallis, Aust. J. Chem., 32 (1979) 1471. [25] T. Braun, P. Steinert and H. Werner, J. Organomet. Chem., 488