Hemeoxygenase-1 expression in response to arecoline-induced oxidative stress in
human umbilical vein endothelial cells
Thu-Ching Hung
a, Li-Wen Huang
b, Shu-Jem Su
c, Bau-Shan Hsieh
a, Hsiao-Ling Cheng
a, Yu-Chen Hu
a,
Yen-Hui Chen
d, Chi-Ching Hwang
e, Kee-Lung Chang
e,⁎
a
Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
b
Department of Medical Laboratory Science and Biotechnology, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
c
Bachelor Degree Program of Health Beauty, Department of Medical Technology, School of Medicine and Health Sciences, FooYin University, Kaohsiung 83101, Taiwan
d
Wu Kun-Che Gynecology, Obstetrics and Pediatrics Hospital, Kaohsiung 80654, Taiwan
eDepartment of Biochemistry, Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
a b s t r a c t
a r t i c l e i n f o
Article history:
Received 15 September 2009
Received in revised form 14 February 2010 Accepted 15 May 2010
Available online xxxx
Keywords: Arecoline
Reactive oxygen species Hemeoxygenase-1 MAPK
HUVEC
Background: Arecoline, the most abundant areca alkaloid, has been reported to stimulate reactive oxygen species (ROS) production in several cell types. Overproduction of ROS has been implicated in atherogenesis. Hemeoxygenase-1 (HO-1) has cytoprotective activities in vascular tissues. This study investigated the effect of arecoline on adhesion molecule expression and explored the role of HO-1 in this process.
Methods: Human umbilical vein endothelial cells (HUVECs) were treated with arecoline, then ROS levels and the expression of adhesion molecules and HO-1 were analyzed and potential signaling pathways investigated.
Results: After 2 h of arecoline treatment, ROS production was stimulated and reached a maximum at 12 h. Expression of the adhesion molecules ICAM and VCAM was also induced. Glutathione pretreatment completely blocked arecoline-stimulated ROS production and VCAM expression, but not ICAM expression. Arecoline also induced HO-1 expression and this effect was partly due by ROS stimulation. Inhibition of c-jun N-terminal kinase (JNK) by SP600125, p38 by SB 203580, or tyrosine kinase by genistein reduced arecoline-induced HO-1 expression. In contrast, inhibition of ERK (extracellular signal-related MAP kinase) by PD98059 had no effect. Transfection of HUVECs with the GFP/HO-1 gene, which resulted in a 5-fold increase in HO-1 activity, markedly, but not completely, inhibited the decrease in cell viability caused by arecoline. Conclusions: This study demonstrates that, in HUVECs, arecoline stimulates ROS production and ICAM and VCAM expression. HO-1 expression is also upregulated through the ROS, tyrosine kinase, and MAPK (JNK and p38) signaling pathways.
Crown Copyright © 2010 Published by Elsevier Ireland Ltd. All rights reserved.
1. Introduction
It has been estimated that there are 200 to 600 million betel quid
chewers in the world. Betel quid chewing is thought to cause oral
submucous
fibrosis and oral cancer. Betel quid usually comprises a
piece of areca nut and lime with or without Piper betle leaves. Areca
nut contains many polyphenols and several alkaloids arecoline being
the major alkaloid. Recent evidence suggests that arecoline is
cytotoxic and genotoxic for various kinds of cells
[1,2]
. It also
produces reactive oxygen species (ROS) and depletes intracellular
thiols in human peripheral blood lymphocytes
[3]
. Overproduction of
intracellular ROS has been implicated in a variety of pathological
conditions, including cancer, diabetes, and cardiovascular diseases,
such as atherosclerosis. Atherosclerosis is a complex disease with a
chronic in
flammatory pathogenesis
[4]
. ROS contribute to the
pathogenesis of atherosclerosis by altering endothelial functions,
including causing injury to endothelial cells and increasing cell
adhesion molecule-mediated leukocyte adhesion to endothelial cells
[5]
.
In response to ROS, several stress proteins are regulated as a
cytoprotective response to diminish cellular damage. Hemeoxygenase-1
(HO-1), a 32-kDa enzyme, is the rate-limiting enzyme in the conversion of
heme into biliverdin, carbon monoxide, and free iron. HO-1 can be
induced in both endothelial and vascular smooth muscle cells by several
stimuli, including oxidized low-density lipoprotein
[6,7]
, heavy metals
[8]
,
in
flammatory cytokines
[9,10]
, and oxidative stress
[11]
. There is evidence
that HO-1 plays a key role in protection against oxidative stress
[12]
and
that mitogen-activated protein kinases (MAPKs) as well as other kinases
[13]
, including tyrosine kinases
[14]
, are involved in HO-1 activation.
International Journal of Cardiology xxx (2010) xxx–xxx
⁎ Corresponding author. Department of Biochemistry, Faculty of Medicine, College of Medicine, Kaohsiung Medical University, No. 100, Shih-Chuan 1st Rd. Kaohsiung 80708, Taiwan. Tel.: + 886 7 3121101x2138; fax: + 886 7 322 3075.
E-mail addresses:[email protected],[email protected] (K.-L. Chang).
0167-5273/$– see front matter. Crown Copyright © 2010 Published by Elsevier Ireland Ltd. All rights reserved. doi:10.1016/j.ijcard.2010.05.015
Contents lists available at
ScienceDirect
International Journal of Cardiology
j o u r n a l h o m e p a g e : w w w. e l s e v i e r. c o m / l o c a t e / i j c a r d
Prospective cohort studies have indicated that the habit of
chewing betel nut is an independent risk factor of cardiovascular
disease in humans
[15,16]
; however, it is still unclear whether there is
any direct relationship between betel quid chewing and the
development of cardiovascular disease. Moreover, there have not
been any studies on a possible association between arecoline and
atherosclerosis. The aim of this study was to determine whether
arecoline affected the process of atherosclerosis. We measured ROS
production and cell adhesion molecule expression in
arecoline-treated human umbilical vein endothelial cells (HUVECs) and
investigated whether HO-1 played a protective role against
areco-line-stimulated effects. The contribution of the activation of MAPKs
and/or other kinases to arecoline-stimulated HO-1 induction was also
examined.
2. Materials and methods
2.1. Cell culture and viability
HUVECs were isolated from human umbilical vein by digestion with 0.1% collagenase type IV (Sigma, St. Louis, MO, USA) as reported previously[17]. Cells at passages 3–5 were used for experiments and were cultured in 1.5% gelatin-coated dishes in Medium 199 (Gibco, Grand Island, NY, USA) containing 2 mML-glutamine, penicillin (50 U/ml), streptomycin (50μg/ml), and 2% low serum growth supplement (50× LSGS, Cascade Biologics, Portland, OR, USA) at 37 °C in an atmosphere of 5% CO2/
95% air. To evaluate viability, XTT labeling mixture (Boehringer Mannheim, Germany) was added to each well (final concentration of 0.3 mg/ml) and the cells incubated for 4 h, then the absorbance of the sample at 450 nm was measured using a microplater reader (EL312e; Bio-Tek Instruments, Winooski, VT, USA) and the viability calculated. Experiments were repeated for three times.
2.2. PBMC adhesion assay
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy human whole blood using Ficoll-Paque (Invitrogen, Carlsbad, CA, USA) as reported previously [18]. The isolated PBMCs (3 × 106
cells/well) were added to cultured HUVECs that were untreated or had been treated with arecoline for 72 h, then, 2 h later, the non-adhesive PBMCs were removed and the adherent PBMCs and HUVECs washed gently washing with medium, then observed on an inverted microscope (Olympus CHX41; Olympus, Tokyo, Japan).
2.3. Intracellular ROS detection
2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA; Molecular Probes, Eugene,
OR, USA) was used to measure intercellular ROS production. In brief, 10μM H2DCF-DA
was added to the culture medium 30 min before the end of treatment. At the end of treatment, the cells were harvested by trypsinization and resuspended in Hanks' balanced salt solution (HBSS; Gibco, Grand Island, NY, USA), then thefluorescence of the dichlorofluorescein formed from the oxidation of H2DCF-DA by cellular oxidants
was measured using a FACScanflow cytometry (Beckman Coulter-Epics XL; Beckman Coulter Inc, Fullerton, CA, USA) with an excitation wavelength of 488 nm and an emission wavelength of 525 nm. Data were analyzed using WinMDI 2.8 software. As a positive control, 100μM H2O2was added to HUVECs 2 h before cell harvesting.
2.4. ICAM-1 and VCAM-1 expression
After treatment, cells were harvested and washed twice with HBSS, then incubated for 30 min at 4 °C with monoclonal anti-human ICAM-1 or anti-human VCAM-1 antibodies (Santa Cruz Biotechnology, CA, USA). After washing, the cells were incubated withfluorescein isothiocyanate-labeled secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), then werefixed in 4% paraformaldehyde and washed. ICAM-1 and VCAM-1 expression was detected by FACS cytometry (Becton Dickinson, San Jose, CA) and analyzed using WinMDI Software with a minimum of 1 × 104
cells/sample being evaluated in each case.
2.5. RT-PCR
RT-PCR was performed as described previously[18]. Total RNA was extracted using REzol C&T reagent (PROtech Technology, Taipei, Taiwan) and complementary DNA (cDNA) synthesized. Three micrograms of cDNA was used for PCR amplification (Promega, Madison, WI, USA) in a reaction volume of 50μl containing 25 μl of 2× PCR Master Mix and 1μl of each specific primer. The reaction mixture was heated to 94 °C for 5 min, then amplification was performed for 30 cycles of 94 °C for 45 s, 55 °C for 45 s, and 72 °C for 1 min on a thermal cycler, then the reaction mixture was heated at 72 °C for 7 min. The primers used for PCR were 5′-CCAGCGGGCCAGCAACAAAGTGC-3′ and 5′-AAGCCTTCAGTGCCCACGGTAAGG-3′ for HO-1 and 5′-GGTCGGAGTCAACG-GATTTG-3′ and 5′-ATGAG CCCCAGCCTTCTCCAT-3′ for β-actin. The amplified PCR
products were separated by electrophoresis in a 2% agarose gel and the intensity of the HO-1 band calculated by densitometry and the results expressed as a percentage of the optical density of the correspondingβ-actin band.
2.6. Western blot analysis
Western blotting was performed as described previously[19]. Cytosolic extracts were prepared using ice-cold lysis buffer and incubation on ice for 20 min. After centrifugation, the protein in the supernatants was quantified using a protein assay kit from Bio-Rad Laboratories (Hercules, CA, USA). Forty micrograms of protein per lane was electrophoresed on 10% or 12% SDS-polyacrylamide gels. After transfer of the protein from the gel to nitrocellulose membranes, the membranes were blocked at room temperature for 1 h in phosphate-buffered saline (PBS) plus 0.1% Tween 20 (PBS-T) containing 5% fat-free powdered milk, then incubated for 2 h at room temperature with monoclonal mouse anti-human ICAM-1 or VCAM-1 antibody or rabbit polyclonal anti-human HO-1 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing, the membranes were incubated at 25 °C for 60 min with the appropriate horseradish peroxidase-labeled secondary antibody (PharMingen, San Diego, CA) and the proteins visualized by chemiluminescence detection (PerkinElmer Life Sciences, Inc. Madison, WI, USA).β-actin was used as the internal control.
Fig. 1. Arecoline induces ROS production. ROS production was determined using H2DCF-DA andflow cytometry. The gray peak represents the vehicle control. Cells were
treated with different concentrations of arecoline for 2 h (A) or with 50μg/ml of arecoline for different periods (B). The results are expressed as the mean ± S.D. for three separate experiments.
2.7. Cloning of full length HO-1 cDNA
Based on the sequence of human HO-1 (NM_002133), we designed 5′ and 3′ gene-specific primers (5′-AAGCTTATGGAGCGTCCGCAACCC-3′; 5′-GGATCCTCACATGGCA-TAAAGCCCTAC-3′) to amplify a segment containing HindIII and BamHI restriction enzyme sites. Three micrograms of cDNA was used for PCR amplification (Promega,
Madison, WI, USA) in a reaction volume of 50μl containing 5 μl of 10×Pfu DNA Polymerase Buffer, 1μl of dNTP (10 μM), 1 μl of each specific primer, and 0.6 μl of Pfu DNA Polymerase (2 units/μl) (Promega, Madison, WI, USA). The reaction mixture was heated to 94 °C for 5 min, then amplification was performed for 30 cycles of 94 °C for 30 s, 55 °C for 1 min, and 72 °C for 3 min with a thermal cycler, then the mixture was heated at 72 °C for 7 min. The HO-1 PCR segment was then digested with HindIII and
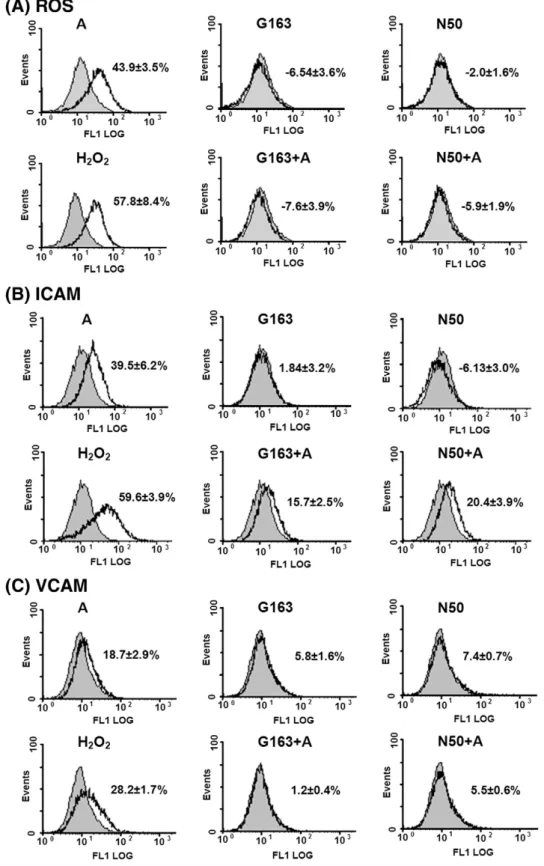
Fig. 2. Glutathione pretreatment reduced arecoline-induced ROS production and ICAM and VCAM expression. Cells were pretreated with reduced glutathione (163μM; G163) or N-acetylcysteine (50μM; N50) for 2 h, then incubated for 2 h or 72 h in the presence of the same agent with or without arecoline (50 μg/ml; A). Intracellular ROS production (A) or expression of ICAM (B) and VCAM (C) was measured byflow cytometry as described in Section 2. The gray peak represents the vehicle control; H2O2-treated cells were used as the
positive control. The results are expressed as the mean ± S.D. for three separate experiments.
BamHI at 37 °C for 3 h (Promega, Madison, WI, USA) and the target sequence recovered from an agarose gel. The pEGFP-C1 vector (Clontech, Mountain View, CA, USA) was cleaved by the same enzymes and recovered from the agarose gel as the vector segment. These two recovered segments were ligated with T4 DNA ligase (Promega, Madison, WI, USA) at 16 °C for 16 h and the pEGFP-C1/HO-1 nucleotide sequence analyzed by gel electrophoresis.
2.8. Plasmid construction
The pEGFP-C1/HO-1 sequence was digested with HindIII and BamHI and the 867 bp segment recovered from an agarose gel. The segments were inserted into the HindIII
and BamHI sites of the pEGFP-C1 vector to generate the pEGRP-C1/HO-1 vector. DH5α competent bacteria were transformed with the recombinant plasmid, which carries a kanamycin resistance gene. A number of independently transformed bacterial colonies were selected and grown in small-scale cultures. Recombinant plasmid pEGFP-C1/HO-1 was identified by restriction enzyme (HindIII/BamHI) analysis.
2.9. Cell transfection
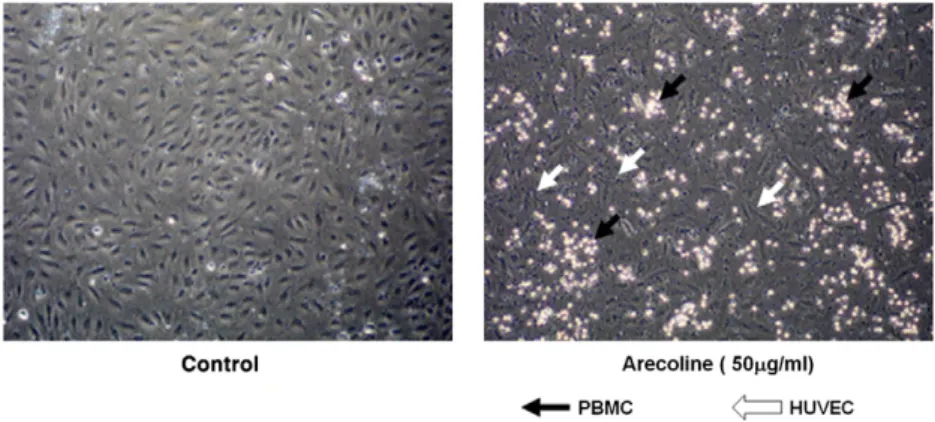
Transfection was performed using lipofectamine2000 (Invitrogen, Life technolo-gies, Carlsbad, CA, USA) and following the manufacturer's protocol and the transfected Fig. 3. Arecoline induces PBMC adhesion to HUVECs. Human PBMCs cells isolated using Ficoll-Paque (PBMC; 3 × 106
cells/well) were added to HUVECs that had been left untreated or treated with arecoline 50μg/ml for 72 h, then, 2 h later, the non-adherent cells were removed and the adherent cells washed gently with medium and PBMC adhering to HUVECs observed by inverted microscopy.
Fig. 4. A HO-1 inhibitor increases arecoline-induced ROS production and ICAM and VCAM expression. Cells were pretreated for 30 min with the HO-1 inhibitor ZnPP (5μM), then incubated for 2 h or 72 h in the presence of the inhibitor with arecoline (50μg/ml; A50). Intracellular ROS production or ICAM and VCAM expression was measured by flow cytometry as described in Section 2. The gray peak represents the vehicle control. The results are expressed as the mean ± S.D. for three separate experiments.
cells incubated in fresh HUVEC growth medium. Cells transfected with empty vector served as controls.
2.10. Statistical analysis
All data are the mean ± S.D. The significance of differences was determined by one-way ANOVA followed by Fisher's test. Statistical analyses were performed using SAS (version 6.011; SAS Institute Inc, Cary, NC. USA). P valueb0.05 was considered statistically significant.
3. Results
3.1. Arecoline induces ROS production
To determine whether arecoline caused ROS generation, HUVECs were treated with different concentrations of arecoline for 2 h or with 50μg/ml (212 μM) of arecoline for different times. Using 2 h of treatment, ROS production was induced by arecoline in a dose-dependent manner (Fig. 1A), while, using 50μg/ml of arecoline, the amount of ROS increased up to 12 h of treatment, then decreased slightly to a plateau from 24 to 72 h (Fig. 1B). Treatment of HUVECs for 72 h, the IC50value of arecoline was 50μg/ml
(data not shown) which was chosen for the evaluation of adhesion molecule expression and cell viability.
3.2. Arecoline induces ICAM and VCAM expression
To examine whether the arecoline-induced ROS production had an effect on adhesion molecule expression, HUVECs were treated with 50μg/ml (212 μM) of arecoline for 2 h or 72 h, then ROS or ICAM and VCAM levels were determined by FACS cytometry with H2O2-treated HUVECs as the positive control. As shown inFig. 2, after
arecoline treatment, ROS production was induced (Fig. 2A, top left panel) or expression of ICAM (Fig. 2B, top left panel) and VCAM (Fig. 2C, top left panel) was increased. In addition, the adherence of PBMC to HUVECs was increased (Fig. 3).
3.3. Glutathione reduces arecoline-induced ICAM and VCAM expression
To examine whether glutathione could reverse these changes, HUVECs were pretreated with 163μM of GSH (reduced glutathione) or 50 μM N-acetylcysteine (NAC), a GSH producer, for 2 h before and during arecoline treatment. GSH or NAC totally inhibited arecoline-induced ROS production (Fig. 2A). In addition, induction of VCAM expression was almost completely inhibited (Fig. 2C), while induction of ICAM expression was decreased, but not totally inhibited (Fig. 2B). These results suggest that ROS are partially responsible for the induction of VCAM and ICAM expression.
3.4. Arecoline induces HO-1 expression
HO-1 is known to be a cytoprotective protein involved in defense against oxidative stress-induced cellular damage, especially in the vascular system. To investigate the role of HO-1 in HUVECs during arecoline treatment, the cells were treated with 5μM ZnPP, a HO-1 inhibitor, for 30 min, then were incubated for 2 h or 72 h with 50μg/ml (212μM) of arecoline in the continued presence of the inhibitor, and ROS production or ICAM and VCAM expression was measured. As shown inFig. 4, ZnPP pretreatment induced ROS production and ICAM and VCAM expression even in the absence of arecoline treatment, while co-treatment with arecoline and ZnPP resulted in even higher levels of ROS, ICAM, and VCAM. These results suggest that HO-1 protects endothelial cells against injury caused by ROS and PBMC adherence induced by adhesion molecules.
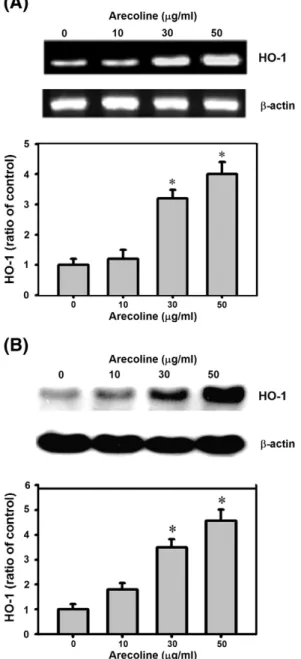
HO-1 is reported to be expressed in response to ROS[11]. Since arecoline induced ROS production, we investigated whether HO-1 expression in HUVECs was changed by arecoline treatment. After 12 h of arecoline treatment, when ROS production is maximal (Fig. 1), HO-1 mRNA and protein levels were examined. Levels of HO-1 mRNA (Fig. 5A) and protein (Fig. 5B) were increased in a concentration-dependent manner by arecoline treatment. Using 50μg/ml of arecoline, an increase was observed after 2 h treatment and was maximal at 24 h (data not shown).
Fig. 5. Arecoline induces HO-1 expression. Cells were treated with different concentrations of arecoline for 12 h, then HO-1 mRNA levels were measured by RT-PCR (A) and protein levels measured by Western blotting (B). *: Pb0.05 compared to untreated controls.
Fig. 6. Glutathione decreases, but does not completely block, arecoline-induced HO-1 expression. Cells were pretreated for 2 h with reduced glutathione (163μM, GSH) or N-acetylcysteine (50μM, NAC), then incubated for 24 h in the presence of the same agent with arecoline (50μg/ml; A50) and HO-1 expression was measured by Western blotting withβ-actin as the internal control. After densitometric analysis, the density of the band was expressed as the relative density compared to that in untreated cells (control), taken as 100%. The results are expressed as the mean ± S.D. for three separate experiments. #: Pb0.05 compared to the arecoline-treated cells.
3.5. HO-1 expression is not only due to ROS
To examine whether arecoline-induced HO-1 expression was due solely to ROS production, HUVECs were pretreated for 2 h with GSH 163μM or NAC 50 μM at the same concentration as inFig. 2which completely inhibited arecoline-induced ROS production, then were treated with 50μg/ml of arecoline for 24 h, when HO-1 expression was induced.Fig. 6shows that either GSH or NAC inhibited almost all of the arecoline-induced HO-1 expression, but not completely, indicating that arecoline still induced some HO-1 expression even when ROS production was completely blocked. This shows that arecoline-induced HO-1 expression was not only due to ROS and that other factors were involved.
3.6. MAP kinase pathways in the regulation of HO-1 expression by arecoline
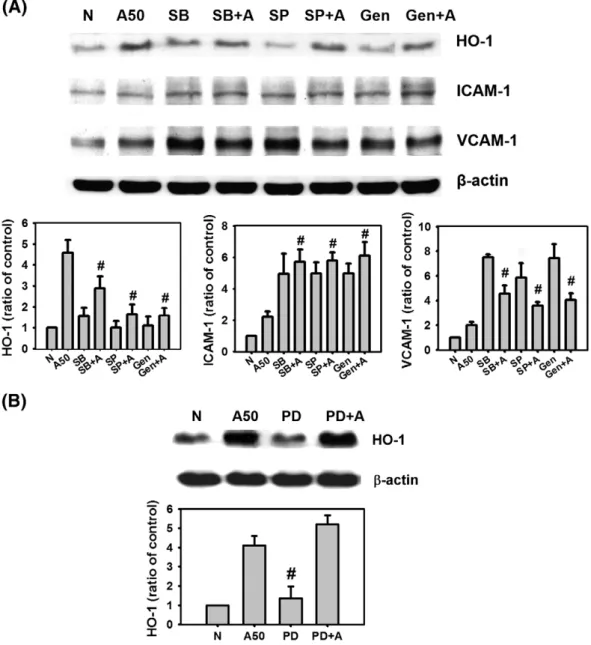
MAP kinases contribute to the regulation of HO-1 expression in many cell types [20,21], but the relative contribution of different pathways to HO-1 upregulation by arecoline in HUVECs is not known. Since arecoline treatment (50μg/ml) strongly increased HO-1 expression at 24 h, these conditions were used to investigate the signaling pathways involved in HO-1 induction using selective pharmacological agents. HUVECs were pretreated with selective pharmacological agents for 2 h, then with arecoline (50μg/ml) in the presence of the same agent for 24 h.Fig. 7A shows that inhibition of p38 with SB 203580 (SB) or of c-jun N-terminal kinase (JNK) with SP600125 (SP) reduced arecoline-induced HO-1 expression, indicating involvement of
these pathways. Addition of genistein (Gen), a tyrosine kinase inhibitor, also reduced arecoline-induced HO-1 expression, indicating tyrosine kinase activation was involved. In contrast, inactivation of ERK (extracellular signal-related MAP kinase) with PD98059 (PD), an inhibitor of the ERK upstream activators MAPK kinase MKK1 and MKK2, had no effect (Fig. 7B). In accordance with the cytoprotective role of HO-1 and these findings, ICAM and VCAM expression was increased by SB 203580, SP600125, or genistein pretreatment of arecoline-treated HUVECs, while HO-1 expression was reduced (Fig. 7A) and PD98059 again had no effect (data not shown). These data show that HO-1 upregulation by arecoline occurs via the JNK and p38 MAPK pathways, but not the ERK pathway, and also via tyrosine kinase activation.
3.7. Effect of HO-1 overexpression on arecoline cytotoxicity
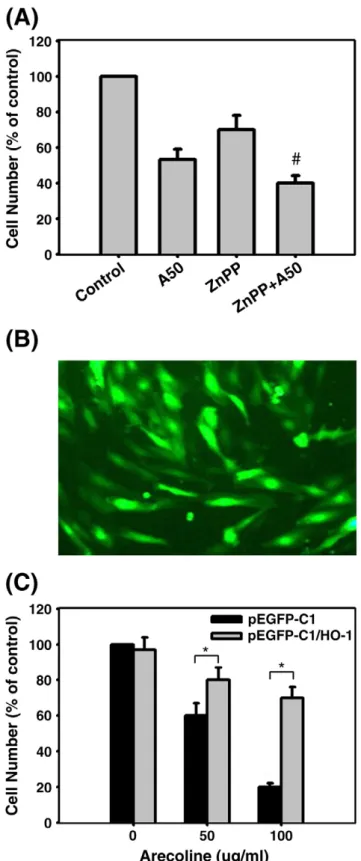
To investigate the role of HO-1 in the arecoline-induced cytotoxicity for HUVECs, cells were incubated with or without 5μM ZnPP for 30 min, then were treated with 50μg/ml (212 μM) arecoline for 72 h, when cell viability was examined. As shown in Fig. 8A, ZnPP pretreatment alone decreased cell viability by 30% and arecoline alone decreased viability by 47% compared to the control group, while co-treatment resulted in a decrease of 62%, indicating that HO-1 acted as a cytoprotective agent against arecoline-induced toxicity. Furthermore, we performed a gene transfer experiment in which HO-1 protein fused in-frame to the C-terminal portion of GFP (GFP/HO-1) or GFP protein alone was expressed under the regulation of the CMV promoter in HUVECs. Transfected cells could be detected by GFPfluorescence (Fig. 8B). Cells transfected with
Fig. 7. Contribution of MAP kinases to arecoline-induced HO-1 expression. Cells were pretreated for 2 h with the p38 inhibitor SB 203580 (10μM; SB), the JNK inhibitor SP600125 (10μM; SP), or the tyrosine kinase inhibitor genistein (5 μM; G) (A) or with an inhibitor of the ERK upstream activators MAPK kinase MKK1 and MKK PD98059 (20 μM; PD) (B), then incubated for 24 h in the presence of the same inhibitor with arecoline (50μg/ml; A50) when HO-1, ICAM and VCAM expression was measured by Western blotting with β-actin as the internal control. After densitometric analysis, the density of the band was expressed as the relative density compared to that in untreated cells (control), taken as 100%. The results are expressed as the mean ± S.D. for three separate experiments. #: Pb0.05 compared to the arecoline-treated cells.
GFP/HO-1 showed a 5-fold increase in HO-1 levels compared to GFP-transfected cells (data not shown). As shown inFig. 8C, there was no significant difference in cell viability between arecoline-treated GFP/HO-1 and GFP-transfected cells, whereas GFP/
HO-1 transfection, but not GFP transfection, inhibited the decrease in cell viability caused by arecoline. However, the damage caused by arecoline was not completely prevented even when HO-1 was overexpressed 5-fold, suggesting that multiple cytoprotective factors are needed to completely prevent arecoline-induced damage of HUVECs.
4. Discussion
In this study, we found that arecoline stimulated ROS production
in HUVECs and that this was related to ICAM and VCAM expression
and resulted in PBMC adhesion. Glutathione pretreatment completely
inhibited arecoline-stimulated ROS production and VCAM expression,
but not ICAM expression. Simultaneously, arecoline treatment
increased HO-1 expression. Inhibition of c-jun N-terminal kinase
(JNK), p38, or tyrosine kinase inhibited the induction of HO-1
expression, suggesting that arecoline induced the upregulation of
HO-1 via these signaling pathways. Overexpression of HO-1 as a result
of gene transfer attenuated arecoline-induced cell damage, but not
completely, indicating that other cytoprotective mechanisms were
needed to overcome arecoline cytotoxicity for HUVECs.
There are a variety of intracellular sources of ROS in vascular cells,
including mitochondrial respiration, NADPH oxidase, lipooxygenase,
cyclooxygenases, xanthine oxidase, and the uncoupling of nitric oxide
synthesis
[22]
. Arecoline is reported to markedly increase
mitochon-drial membrane potential hyperpolarization in KB epithelial cells
[2]
,
to induce cyclooxygenase-2 expression in human sperm cell, buccal
mucosal
fibroblasts and primary oral keratinocytes
[23,24]
, and to
increase eNOS expression in HUVECs
[25]
, showing that arecoline can
induce ROS generation in several ways. ROS plays both deleterious
and bene
ficial roles. Ironically, various ROS-mediated actions protect
cells against ROS-induced oxidative stress and re-establish or
maintain the
“redox balance”, also termed “redox homeostasis”. This
“two-faced” character of ROS is clearly substantiated
[26,27]
. Studies
have also revealed roles for hemeoxygenase in addition to heme
metabolism. The HO-1 isoform is involved in healing, psoriasis,
keratinocyte proliferation, and, in its role as a heat shock protein,
protection against cellular oxidative stress
[28
–30]
.
This study showed that ROS induced HO-1 expression, which
protected against ROS-induced oxidative stress, such as the increase
in ICAM and VCAM expression in arecoline-treated HUVEC. ROS
production was induced by arecoline after 2 h of treatment and was
maximal at 12 h, a time at which increased HO-1 expression was seen,
followed by attenuation of the enhancement of ICAM and VCAM
expression. However, HO-1 expression alone was not suf
ficient to
completely prevent arecoline-induced damage. Given that HUVECs
transfected with the HO-1 gene showing 5-fold higher HO-1 activity
were not completely protected from the damage caused by arecoline,
it is clear that other cytoprotective pathways are needed to block the
effect of arecoline in HUVECs. In addition, arecoline probably has
other effects on HUVECs. For example, arecoline induces cell cycle
arrest at the M/G2 phase and changes apoptotic-related protein
expression in HUVECs (our unpublished data).
The MAP kinases, ERK, p38, and JNK, and ROS are upstream
activators of transcription factors, such as AP-1, NF-
κB, and Nrf2,
which are implicated in HO-1 expression
[21,31]
. MAP kinases
regulate HO-1 expression in many cell types
[20,21]
, but the roles of
ROS and the MAP kinase cascade in arecoline-stimulated HO-1
induction in HUVECs have not been previously reported. Our results
showed that glutathione completely blocked arecoline-stimulated
ROS production, but only partially inhibited HO-1 expression,
indicating that factors other than ROS were also involved. Inhibition
of JNK, p38, or tyrosine kinase activity impaired arecoline-induced
HO-1 expression, indicating that these proteins were involved in this
process. However, it is not known whether HO-1 induction is an
adaptive and/or persistent response to arecoline and further studies,
such as the use of si-RNA or knockout mice, are required to fully clarify
Fig. 8. HO-1 attenuates arecoline-induced cytotoxicity in HUVECs. (A) Cells were pretreated for 30 min with the HO-1 inhibitor ZnPP (5μM), then incubated for 72 h with arecoline (50μg/ml; A50) and cell viability measured. (B) Expression of the transfected GFP/HO-1 gene shown byfluorescence microscopy. (C) Cells transfected with GFP or GFP/HO-1 were treated with arecoline for 72 h, then cell viability was measured. The results are expressed as the mean ± S.D. for three separate experiments. *: Pb0.05 compared to the corresponding untreated controls; #: Pb0.05 compared to the arecoline-treated cells.
the role of HO-1 in HUVECs and the signi
ficance of HO-1 induction in
arecoline-associated atherosclerosis.
In conclusion, this study shows that arecoline stimulates ROS
production in HUVECs and that this is associated with ICAM and
VCAM expression. In addition, HO-1 expression is upregulated by
arecoline-induced oxidative stress through ROS and the tyrosine
kinase and MAPK (JNK and p38) signaling pathways.
Con
flict of interest
None.
Acknowledgements
This study was supported by grants from the National Science
Council, Taiwan (NSC 94-2320-B-037-039; NSC 95-2320-B-037-011;
and NSC 96-2320-B-037-003). The authors of this manuscript certify
that they have complied with the Principles of Ethical Publishing in
the International Journal of Cardiology
[32]
.
References
[1] Chang YC, Tai KW, Chou MY, Tseng TH. Synergistic effects of peroxynitrite on arecoline-induced cytotoxicity in human buccal mucosalfibroblasts. Toxicol Lett 2000;118:61–8.
[2] Chang MC, Ho YS, Lee PH, et al. Areca nut extract and arecoline induced the cell cycle arrest but not apoptosis of cultured oral KB epithelial cells: association of glutathione, reactive oxygen species and mitochondrial membrane potential. Carcinogenesis 2001;22:1527–35.
[3] Kumpawat K, Deb S, Ray S, Chatterjee A. Genotoxic effect of raw betel-nut extract in relation to endogenous glutathione levels and its mechanism of action in mammalian cells. Mutat Res 2003;538:1–12.
[4] Lusis AJ. Atherosclerosis. Nature 2000;407:233–41.
[5] Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol 2001;280:C719–41.
[6] Jeney V, Balla J, Yachie A, et al. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002;100:879–87.
[7] Shi W, Haberland ME, Jien ML, Shih DM, Lusis AJ. Endothelial responses to oxidized lipoproteins determine genetic susceptibility to atherosclerosis in mice. Circula-tion 2000;102:75–81.
[8] Stewart D, Killeen E, Naquin R, Alam S, Alam J. Degradation of transcription factor Nrf2 via the ubiquitin–proteasome pathway and stabilization by cadmium. J Biol Chem 2003;278:2396–402.
[9] Terry CM, Clikeman JA, Hoidal JR, Callahan KS. Effect of tumor necrosis factor-alpha and interleukin-1 alpha on heme oxygenase-1 expression in human endothelial cells. Am J Physiol 1998;274:H883–91.
[10] Terry CM, Clikeman JA, Hoidal JR, Callahan KS. TNF-alpha and IL-1alpha induce heme oxygenase-1 via protein kinase C, Ca2+, and phospholipase A2 in endothelial cells. Am J Physiol 1999;276:H1493–501.
[11] Keyse SM, Tyrrel RM. Heme oxygenase is the major 32-kDa stress protein induced in human skinfibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA 1989;86:99–103.
[12] Yachie A, Niida Y, Wada T, et al. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest 1999;103:129–35. [13] Kiemer AK, Bildner N, Weber NC, Vollmar AM. Characterization of heme
oxygenase 1 (heat shock protein 32) induction by atrial natriuretic peptide in human endothelial cells. Endocrinology 2003;144:802–12.
[14] Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 2006;86:583–650.
[15] Guh JY, Chuang LY, Chen HC. Betel-quid use is associated with the risk of the metabolic syndrome in adults. Am J Clin Nutr 2006;83:1313–20.
[16] Yen AM, Chen LS, Chiu YH, Boucher BJ, Chen TH. A prospective community-population-registry based cohort study of the association between betel-quid chewing and cardiovascular disease in men in Taiwan (KCIS no. 19). Am J Clin Nutr 2008;87:70–8.
[17] Su SJ, Huang LW, Pai LS, Liu HW, Chang KL. Homocysteine at pathophysiologic concentrations activates human monocyte and induces cytokine expression and inhibits macrophage migration inhibitory factor expression. Nutrition 2005;21: 994–1002.
[18] Chang KL, Hung TC, Hsieh BS, Chen YH, Chen TF, Cheng HL. Zinc at pharmacologic concentrations affects cytokine expression and induces apoptosis of human peripheral blood mononuclear cells. Nutrition 2006;22:465–74.
[19] Chang KL, Kung ML, Chow NH, Su SJ. Genistein arrests hepatoma cells at G2/M phase: involvement of ATM activation and upregulation of p21waf1/cip1 and Wee1. Biochem Pharmacol 2004;67:717–26.
[20] Maines MD, Gibbs PE. 30 some years of heme oxygenase: from a“molecular wrecking ball” to a “mesmerizing” trigger of cellular events. Biochem Biophys Res Commun 2005;338:568–77.
[21] Prawan A, Kundu JK, Surh YJ. Molecular basis of heme oxygenase-1 induction: implications for chemoprevention and chemoprotection. Antioxid Redox Signal 2005;7:1688–703.
[22] Dröge W. Free radicals in the physiological control of cell function. Physiol Rev 2002;82:47–95.
[23] Chang MC, Wu HL, Lee JJ, et al. The induction of prostaglandin E2 production, interleukin-6 production, cell cycle arrest, and cytotoxicity in primary oral keratinocytes and KB cancer cells by areca nut ingredients is differentially regulated by MEK/ERK activation. J Biol Chem 2004;279:50676–83.
[24] Tsai CH, Chou MY, Chang YC. The up-regulation of cyclooxygenase-2 expression in human buccal mucosalfibroblasts by arecoline: a possible role in the pathogenesis of oral submucousfibrosis. J Oral Pathol Med 2003;32:146–53.
[25] Kuo FC, Wu DC, Yuan SS, et al. Effects of arecoline in relaxing human umbilical vessels and inhibiting endothelial cell growth. J Perinat Med 2005;33:399–405. [26] Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease.
Arterioscler Thromb Vasc Biol 2005;25:29–38.
[27] Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007;39:44–84.
[28] Choi AM, Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol 1996;15:9–19.
[29] Clark JE, Green CJ, Motterlini R. Involvement of the heme oxygenase–carbon monoxide pathway in keratinocyte proliferation. Biochem Biophys Res Commun 1997;241:215–20.
[30] Hanselmann C, Mauch C, Werner S. Haem oxygenase-1: a novel player in cutaneous wound repair and psoriasis? Biochem J 2001;353:459–66.
[31] Ryter SW, Choi AM. Heme oxygenase-1: redox regulation of a stress protein in lung and cell culture models. Antioxid Redox Signal 2005;7:80–91.