Synthesis, Molecular and Photovoltaic Properties of Donor
−Acceptor
Conjugated Polymers Incorporating a New Heptacylic
Indacenodithieno[3,2
‑b]thiophene Arene
Huan-Hsuan Chang, Che-En Tsai, Yu-Ying Lai, De-Yang Chiou, So-Lin Hsu, Chain-Shu Hsu,
and Yen-Ju Cheng*
Department of Applied Chemistry, National Chiao Tung University, 1001 Ta Hsueh Road, Hsin-Chu, 30010 Taiwan
*
S Supporting InformationABSTRACT:
We have developed a new multifused
indacenodithieno[3,2-b]thiophene arene (IDTT) unit where
the central phenylene is covalently fastened with the two outer
thieno[3,2-b]thiophene (TT) rings, forming two
cyclo-pentadiene rings embedded in a heptacyclic structure. This
rigid and coplanar IDTT building block was copolymerized
with electron-de
ficient acceptors,
4,7-dibromo-2,1,3-benzothia-diazole (BT), 4,7-dibromo-5,6-di
fluoro-2,1,3-benzothiadiazole
(FBT) and 1,3-dibromo-thieno[3,4-c]pyrrole-4,6-dione
(TPD) via Stille polymerization, respectively. Because the
higher content of the thienothiophene moieties in the fully coplanar IDTT structure facilitates
π-electron delocalization, these
new polymers show much improved light-harvesting abilities and enhanced charge mobilities compared to PDITTBT copolymer
using hexacyclic diindenothieno[3,2-b]thiophene (DITT) as the donor moieties. The device using PIDTTBT:PC
71BM (1:4, w/
w) exhibited a decent power conversion e
fficiency (PCE) of 3.8%. Meanwhile, the solar cell using PIDTTFBT:PC
71BM (1:4 in
wt %) blend exhibited a greater V
ocvalue of 0.9 V and a larger J
scof 10.08 mA/cm
2, improving the PCE to 4.2%. The enhanced
V
ocis attributed to the lower-lying of HOMO energy level of PIDTTFBT as a result of
fluorine withdrawing effect on the BT
unit. A highest PCE of 4.3% was achieved for the device incorporating PIDTTTPD:PC
71BM (1:4 in wt %) blend.
■
INTRODUCTION
Polymer solar cells (PSCs) using organic p-type (donor) and
n-type (acceptor) semiconductors have attracted tremendous
scienti
fic and industrial interest.
1The most critical challenge at
molecular level is to develop p-type conjugated polymers that
can simultaneously possess su
fficient solubility for
process-ability and miscibility with an n-type material, low band gap
(LBG) for strong and broad absorption spectrum to capture
more solar photons, and high hole mobility for e
fficient charge
transport.
2The most useful approach to make a LBG polymer
is to connect electron-rich donor and electron-de
ficient
acceptor segments along the conjugated polymer backbone.
Thiophene and benzene aromatic rings are the most important
ingredients to comprise p-type conjugated polymers.
Benzene-based derivatives such as tricyclic 2,7-
fluorene or 2,7-carbazole
units have shown to serve as useful building blocks to construct
donor
−acceptor polymers having deep-lying HOMO energy
levels that contribute to achieve high open-circuit voltage (V
oc)
(>0.8 V) for PSCs.
3However, the intrinsic drawback is that
these polymers usually possess relatively large optical band gaps
(>2 eV) that limit their ability to harvest sunlight and thus
result in moderate short-circuit current (J
sc). On the other
hand, because of the lower aromaticity to adapt more quinoidal
resonance structure, thiophene-based D
−A polymers have
better light absorption ability to permit greater J
sc. However,
their V
ocvalues are generally limited to ca. 0.6 V as a result of
the high-lying HOMO levels.
4Thieno[3,2-b]thiophene (TT)
unit emerges as a superior thiophene-based building block to
achieve high mobility p-type semiconductors.
5This compact
structure actually possesses higher aromatic stabilization energy
than a thiophene, which can potentially lower the HOMO level
for higher V
oc.
6
Moreover, the C
2hsymmetry and coplanar
geometry may promote more ordered packing and stronger
interchain interactions to obtain exceptional hole mobility,
which is bene
ficial for J
sc.
7Introducing the alkyl chains into the
two
β-positions of thieno[3,2-b]thiophene unit is usually
necessary to improve the solubility of the resulting polymers.
Unfortunately, these side chains inevitably impose a negative
e
ffect on the effective conjugation due to severe steric
hindrance-induced twisting between the neighboring aryls.
8By implementing forced planarization via covalently fastening
adjacent aromatic units in the polymer backbone, advantageous
intrinsic properties such as reduced band gap and enhanced
charge mobility can be retained.
9Therefore, it is of interest to
integrate benzene and thieno[3,2-b]thiophene units into a
molecular entity with forced rigidi
fication to simultaneously
Received: September 18, 2012
Revised: November 4, 2012
Published: November 27, 2012
extend the conjugation while maintaining the coplanarity.
However, development of ladder-type architectures requires
elegant molecular design and synthesis.
10Recently, we
first
reported a multifused hexacyclic
diindenothieno[3,2-b]-thiophene (DITT) unit, where the central TT ring is connected
with two outer phenyl rings through two embedded
cyclo-pentadienyl (CP) rings (Scheme 1).
11This electron-rich unit
was copolymerized with electron-de
ficient benzothiadiazole
acceptor to obtain a donor
−acceptor copolymer PDITTBT
(Scheme 1). Nevertheless, this polymer is short of absorption
coverage at the visible and near IR region due to the fact that
DITT
possesses high content of high-aromaticity benzene rings
(two benzene rings and one thieno[3,2-b]thiophene), leading
to relatively large optical band gap and thus limited
photocurrent. By reversing the arrangement of TT and benzene
units in the DITT framework, we present here a new
multifused heptacyclic structure,
indacenodithieno[3,2-b]-thiophene (IDTT), where the central phenylene ring is fused
with two outer TT rings by two carbon bridges. Compared to
hexacyclic DITT unit, this heptacyclic IDTT has extended
conjugation length with greatly increasing the content of the
thiophene moieties (one benzene and two
thieno[3,2-b]-thiophene units). Furthermore, the placement of the
thienothiophene units at the two terminal sides of IDTT is
advantageous for facile
α-bromination or stannylation for
subsequent polymerization. Four side chains introduced at the
bridging carbons in IDTT guarantee solubility without twisting
the coplanarity. Meanwhile, exploitation of suitable
electron-de
ficient acceptors in combination with IDTT donor in the
polymeric backbone is required. Benzothiadiazole (BT)
12and
thieno[3,4-c]pyrrole-4,6-dione (TPD)
13are the widely used
electron-de
ficient acceptors due to their suitable electron
affinity and easy availability. 5,6-Difluorobenzothiadiazole
(FBT) unit with two
fluorine atoms on BT unit also emerges
as a superior derivative for adjusting the molecular properties.
14In this research, we report the detailed synthesis of the
distannyl-IDTT monomer which was copolymerized with BT,
FBT, TPD acceptor moieties to prepare a new class of D
−A
alternating IDTT-based copolymers (Scheme 1). Their
thermal, optical and electrochemical properties have been
carefully characterized. Preliminary results showed that the
IDTT-based polymers are promising for photovoltaic solar cell
applications.
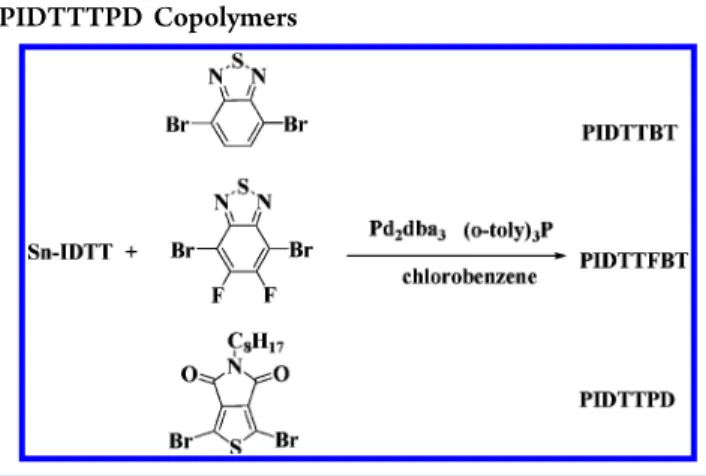
Synthesis. The synthetic route for Sn-IDTT monomer is
depicted in Scheme 2. Stille coupling of diethyl
2,5-dibromoterephthate with
2-(tributylstannyl)thieno[3,2-b]-thiophene yielded compound 1. Double nucleophilic addition
of the ester groups in 1 by (4-octyloxy)phenyl magnesium
bromide led to the formation of benzylic alcohols in 2 which
was subjected to intramolecular Friedel
−Crafts cyclization
under acidic condition to furnish the heptacyclic IDTT arene in
a good yield of 81%. Bromination of IDTT by
N-bromosuccinimide generated Br-IDTT in a high yield of
87%. Finally, Br-IDTT was lithiated by n-butyllithium followed
by quenching with trimethyltin chloride to a
fford the distannyl
Scheme 1. Chemical Structures of Hexacyclic DITT and
Heptacyclic IDTT Arenes and Their Corresponding
Donor
−Acceptor Copolymers
Sn-IDTT
in a moderate yield of 54%. Sn-IDTT monomer was
copolymerized with 4,7-dibromo-2,1,3-benzothiadiazole (BT),
4,7-dibromo-5,6-di
fluoro-2,1,3-benzothiadiazole (FBT) and
1,3-dibromo-thieno[3,4-c]pyrrole-4,6-dione (TPD) acceptor
monomers via Stille coupling to a
fford three donor−acceptor
copolymers,
poly(indacenodithieno[3,2-b]thiophene-alt-benzo-thiadiazole) (PIDTTBT, M
n= 16.6 kDa, PDI = 1.7),
poly(indacenodithieno[3,2-b]thiophene-alt-di
fluorobenzothia-diazole) (PIDTTFBT, M
n= 24.0 kDa, PDI = 1.2) and
poly(indacenodithieno[3,2-b]thiophene-alt-thieno[3,4-c]-pyrrole-4,6-dione) (PIDTTTPD, M
n= 31.3 kDa, PDI = 2.0),
respectively (Scheme 3). These copolymers puri
fied by
successive Soxhlet extraction and precipitation showed
narrower molecular weight. The resulting copolymers
flanked
with four side chains on IDTT unit possess excellent
solubilities in common organic solvents, such as chloroform,
toluene, and THF.
Thermal and Optical Properties. The thermal stability of
the polymers was analyzed by thermogravimetric analysis
(TGA) (Figure S1, Supporting Information). PIDTTBT,
PIDTTFBT, and PIDTTTPD exhibited su
fficiently high
decomposition temperatures (T
d) of 414, 391, and 384
°C,
respectively.
UV
−vis absorption spectra of the three polymers in THF
solutions and in thin
films are shown in Figure 1 and the
correlated optical parameters are summarized in Table 1.
PIDTTBT
and PIDTTFBT showed two obvious absorption
bands in the spectra. The longer wavelength absorbance is
attributed to the intramolecular charge transfer (ICT) between
the electron-rich and the electron-de
ficient segments. However,
the localized transition bands and ICT bands of PIDTTTPD
overlap into a broad band covering the whole visible region
from 400 to 700 nm, indicating that the accepting strength of
TPD
is weaker than that of BT unit. The absorption spectra of
the three polymers shift toward longer wavelengths from the
solution state to the solid state, indicating that the coplanar
structure of IDTT is capable of inducing strong interchain
interactions. The optical band gaps (E
gopt) deduced from the
onset of absorption in the solid state are determined to be 1.69
eV for PIDTTBT, 1.77 eV for PIDTTFBT and 1.95 eV for
PIDTTTPD. Note that the optical band gap of PDITTBT is
2.15 eV, which is signi
ficantly larger than that of PIDTTBT,
suggesting that the heptacyclic IDTT unit with higher content
of thienothiophene moieties indeed facilitates the
π-electron
delocalization compared to the hexacyclic DITT unit.
Electrochemical Properties. Cyclic voltammetry (CV)
was employed to examine the electrochemical properties and
determine the highest occupied molecular orbital (HOMO)
and lowest unoccupied molecular orbital (LUMO) energies of
the polymers (Figure 2, Table 1). The three polymers showed
stable and reversible p-doping and n-doping processes, which
are important prerequisites for p-type semiconductor materials.
The LUMO energy levels of PIDTTBT, PIDTTFBT, and
PIDTTTPD
are determined to be
−3.40, −3.50, and −3.18 eV,
respectively. The LUMO energy levels are higher than that of
the PC
71BM acceptor (
−3.8 eV) to ensure energetically
favorable electron transfer. It should be noted that the
HOMO energy of PIDTTFBT (
−5.30 eV) is lower than that
of PIDTTBT (
−5.43 eV) due to the two electron-withdrawing
fluorine atoms on the BT units.
14Furthermore, PIDTTTPD
shows the lowest HOMO energy level of
−5.45 eV, indicating
that TPD acceptor unit is also capable of lowering the HOMO
energy level of the resulting polymer. The HOMO energy
levels are within the ideal range to ensure better air-stability and
greater attainable V
oc.
Theoretical Calculations. In order to gain more insight
into the molecular orbital properties of the polyaromatic
π-electron systems, quantum
−chemical calculations were
per-formed with the Gaussian09 suite employing the B3LYP and
TD-B3LYP density functionals in combination with the
6-311G(d,p) basis set. Considering an insigni
ficant effect on
Scheme 3. Synthesis of PIDTTBT, PIDTTFBT, and
PIDTTTPD Copolymers
electronic properties, all the side-chain substituents were
replaced with methyl groups for simplicity. Repeating units,
denoted as IDTTBT, IDTTFBT, and IDTTTPD, in their most
stable conformations were used as simplified model compounds
for PIDTTBT, PIDTTFBT, and PIDTTTPD, respectively.
The calculated HOMO/LUMO energy, excitation energy,
oscillator strength, and con
figurations of the excited states are
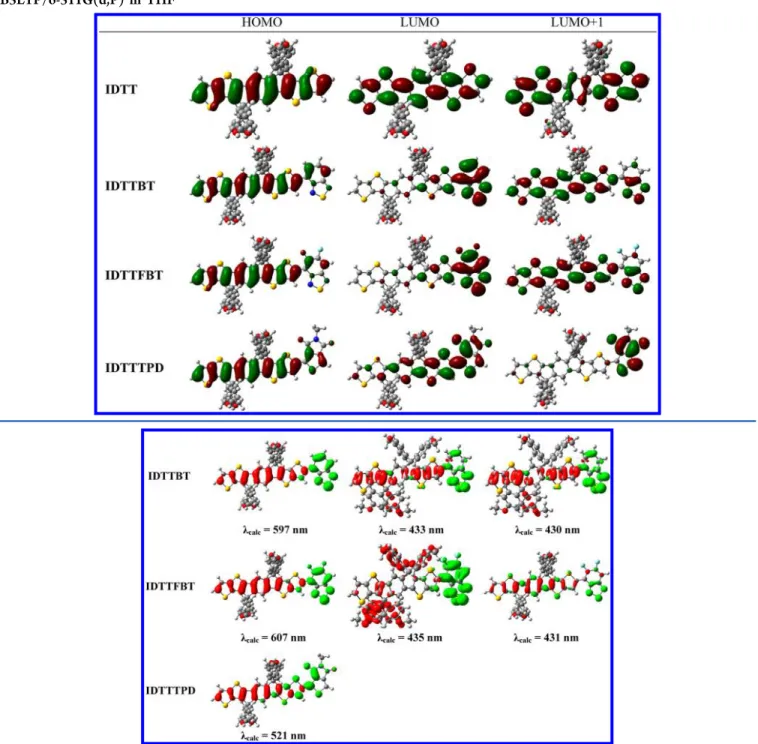
summarized in Table 2 and the frontier orbitals, HOMO (H),
LUMO (L) and the closeby LUMO+1 (L+1) are illustrated in
Table 3. The HOMO electron density distribution of
IDTTTPD
is analogous to that of IDTTFBT and IDTTBT,
where the electron density is not only distributed
homoge-neously along the molecular backbone of the IDTT moiety but
also on parts of the acceptor. Given that the contribution of the
IDTT
moiety to the HOMO energy level should be similar
among the three compounds, the di
fference in the HOMO
energy level therefore mainly depends on the nature of the
acceptors. Consistent with the electrochemical experiments, the
calculated HOMO energy levels of the three model compounds
follow the order: IDTTTPD (
−5.25 eV) < IDTTFBT (−5.23
eV) < IDTTBT (
−5.18 eV). On the contrary, the LUMO of
IDTTTPD
is higher in energy than that of IDTTFBT and
IDTTBT. IDTTFBT and IDTTBT have similar LUMO
electron density patterns of which the electron density is
mainly located on the acceptors (BT and FBT). Instead, the
electron density of LUMO in IDTTTPD is not only localized
on TPD unit but span from TPD to IDTT moieties.
Involvement of the IDTT orbitals in the LUMO of IDTTTPD
may result in a high-lying energy level of LUMO.
Table 1. Summary of the Intrinsic Properties of the Polymers
aλmax(nm)
polymer Mn(kDa) PDI Td(°C) Egopt(eV) (Film) THF film HOMO (eV) LUMO(eV)
PIDTTBT 16.6 1.7 414 1.69 420 430 −5.30 −3.40 622 645 PIDTTFBT 24.0 1.2 391 1.77 415 424 −5.43 −3.50 616 635 PIDTTTPD 31.3 2.0 384 1.95 588 591 −5.45 −3.18 aE
goptfrom the onset of UV spectra in thinfilm.
Figure 2. Cyclic voltammograms of PIDTTBT, PIDTTFBT, and PIDTTTPDin the thinfilms at a scan rate of 50 mV/s.
Table 2. Calculated
aHOMO/LUMO Energy, Excitation Energy, Oscillator Strength, and Configurations (with Large CI
Coe
fficients) of the Excited States
excitation energy
compound HOMO (eV) LUMO (eV) λmax,exp(nm)b λcalcd(nm) oscillator strength symmetry configurationc
IDTT −5.22 −1.87 393, 417 420 1.1262 singlet-A H→L 321 0.2295 singlet-A H−4→L H→L+2 IDTTBT −5.18 −2.78 622 597 0.6822 singlet-A H→L 420 433 0.7493 singlet-A H−1→L 430 0.315 singlet-A H→L+1 H−1→L H→L+1 H−2→L IDTTFBT −5.23 −2.89 616 607 0.6499 singlet-A H→L 415 435 0.2193 singlet-A H−3→L H−2→L H−1→L H→L+1 431 0.8399 singlet-A H→L+1 H−3→L H−2→L H−1→L IDTTTPD −5.25 −2.53 588 521 1.4519 singlet-A H→L
aTD−B3LYP/6-311G(d,p), PCM=THF.bExperimental values were measured for nonsimplified IDTT, IDTTBT, IDTTFBT, IDTTTPD in THF
As listed in Table 2, although there are variations in the
absolute values, the calculated absorptions are still in good
agreement with the experimental values. In order to have
further understanding of these electronic transitions, electron
density di
fference maps (EDDMs) were conducted (Figure
3).
15The electronic transitions can therefore be visualized
through EDDMs. Red indicates a decrease in charge density,
while green indicates an increase. For IDTTBT, the lowest
energy singlet electronic transition (
λ
calc= 597 nm;
λ
max,exp=
622 nm) indicates a pronounced intramolecular charge transfer
(ICT) from IDTT to BT. The transitions at
λ
calc= 433 and 430
nm are close in energy and may not be separable. In fact, in the
UV absorption spectrum, only one absorption peak (
λ
max,exp=
420 nm) was observed. On the basis of the EDDMs, both
transitions belong to charge separations from the molecular
backbone of IDTT and one 4-methoxyphenyl side chain to the
BT
unit. The lowest energy singlet electronic transition (
λ
calc=
607 nm;
λ
max,exp= 616 nm) of IDTTFBT is a ICT band, which
is basically caused by the electron transfer from IDTT to FBT.
The transitions at
λ
calc= 435 and 431 nm are also inseparable in
the experimental spectrum (
λ
max,exp= 415 nm). One at
λ
calc=
435 nm is assigned to the ICT from the
flanking
4-methoxyphenyl groups of IDTT to BT, and the other one at
λ
max= 431 nm only occurs at localized regions. Lastly, the most
Table 3. Plots (Isovalue = 0.02 au) of Frontier Orbitals of IDTT, IDTTBT, IDTTFBT, and IDTTPD Calculated at the Level of
B3LYP/6-311G(d,P) in THF
Figure 3.Electron density difference maps (EDDMs) of selected singlet electronic transitions of IDTTBT (at 597, 433, and 430 nm), IDTTFBT (at 607, 435, and 431 nm), and IDTTTPD (at 521 nm). Red indicates a decrease in charge density, while green indicates an increase. All EDDMS were plotted with isovalue 0.0012 au.
probable vertical excitation of IDTTTPD is calculated to be
521 nm, which deviates slightly from the
λ
max,expof 589 nm. It
mainly results from the ICT from IDTT to TDP and the
π−π*
transition within the IDTT unit has a minor contribution.
Organic Field E
ffect Transistors. To investigate the
mobilities of the polymers, organic
field-effect transistors
(OFETs) were fabricated in the bottom-contact/top-contact
geometry as described in the Experimental Section (Table 4
and Figure 4). The hole mobilities were deduced from the
transfer characteristics of the devices in the saturation regime.
The polymers-based OFETs using SiO
2as gate dielectric was
treated with octadecyltrichlorosilane (ODTS) to form a
self-assembly monolayer (SAM). With the modi
fication of a
ODTS-SAM layer along with annealing temperature at 150
Table 4. FET Characteristics of the Polymer Thin Films
polymer SAM layer annealing temperature (°C) mobility (cm2V−1s−1) on/off V
t(V)
PIDTTBT ODTS 200 7× 10−3 1.9× 103 −13.8
PIDTTFBT ODTS 150 1× 10−2 1.4× 107 −21.7
PIDTTTPD ODTS 200 1× 10−3 3.2× 104 −25.4
Figure 4.. Typical output curves (a, c, e) and transfer plots (b, d, f) of the OFET devices based on PIDTTBT, PIDTTFBT, and PIDTTTPD, respectively, with ODTS-SAM layer.
and 200
°C, the hole mobilities of the three polymers were
determined to be 7
× 10
−3cm
2V
−1s
−1, 1
× 10
−2cm
2V
−1s
−1,
and 1
× 10
−3cm
2V
−1s
−1with the on
−off ratios of 1.9 × 10
3,
1.4
× 10
7, and 3.2
× 10
4for PIDTTBT, PIDTTFBT, and
PIDTTTPD
devices, respectively. It should be mentioned that
PDITTBT
using hexacyclic DITT as the donor showed a much
lower mobility of 7
× 10
−5cm
2V
−1s
−1.
11This result again
indicates that higher content of thienothiophene unit can
improve the charge transporting properties.
Photovoltaic Characteristics. Bulk heterojunction
photvoltaic cells were fabricated by spin-coating the blends from
o-dichlorobenzene solutions at various polymer-to-PC
71BM ratios
on the basis of ITO/PEDOT:PSS/polymer:PC
71BM/Ca/Al
con
figuration and their performances were measured under a
simulated AM 1.5 G illumination of 100 mW/cm
2. The
asymmetric PC
71BM was used due to its stronger light
absorption in the visible region than that of PC
61BM. The
characterization data are summarized in Table 5 and the J
−V
curves of these polymers are shown in Figure 5. The blend ratio
of the active layers shown in the Table 5 is the result of the
optimized conditions for the devices. The device based on the
PIDTTBT:PC
71BM (1:4 in wt %) blend exhibited a V
ocof 0.84
V, a J
scof 8.32 mA/cm
2, a FF of 55%, delivering a decent PCE
of 3.8%. It is noteworthy that the device based on PDITTBT
polymer using the hexacyclic DITT unit as the donor only
exhibited a lower V
ocof 0.88 V, a J
scof 7.46 mA/cm
2, resulting
in a lower PCE of 2.7%.
11Meanwhile, the device using
PIDTTFBT:PC
71BM (1:4 in wt %) blend exhibited a greater
V
ocvalue of 0.9 V and a larger J
scof 10.08 mA/cm
2with an
improved PCE to 4.2%. The enhanced V
ocis attributed to the
lower-lying of HOMO energy level of PIDTTFBT as a result of
fluorine withdrawing effect on the FBT unit. More
encourag-ingly, the device based on the PIDTTTPD:PC
71BM (1:4 in wt
%) blend exhibited a high V
ocof 0.90 V, a J
scof 7.99 mA/cm
2, a
FF of 60%, leading to a highest PCE of 4.3%. Even though the
device derived from PIDTTFBT has the highest V
ocand J
sc, it
does not show a better PCE than the PIDTTTPD-based
device, which is due to the fact that the device of PIDTTTPD
has a much larger FF than that of PIDTTBT. To con
firm the
accuracy of the measurements of the devices, the corresponding
external quantum e
fficiency (EQE) spectra were measured
under illumination of monochromatic light (Figure 4). The J
scvalues calculated from integration of the EQE spectra agree well
with the J
scobtained from the J
−V measurements.
■
CONCLUSIONS
Compared to thiophene unit, C
2h-symmetry
thieno[3,2-b]-thiophene (TT) unit with higher aromatic stabilization energy
and coplanar geometry is a promising building block for
donor
−acceptor conjugated polymers to obtain higher V
ocand
J
sc. However, introducing aliphatic side chains on the
β-positions of TT units leads to severe steric hindrance-induced
twisting between the neighboring aryls in the polymer
backbone. A straightforward approach to circumvent this
deficiency is to embed TT units in a multifused ladder-type
structure. A hexacyclic diindenothieno[3,2-b]thiophene
(DITT) unit has been
first developed. Nevertheless, DITT
possesses high content of high-aromaticity benzene rings
resulting in relatively large optical band gap and thus limited
J
sc. By reversing the arrangement of TT and benzene units in
the DITT framework, we have successfully developed a new
multifused heptacyclic structure,
indacenodithieno[3,2-b]-thiophene (IDTT), where the central phenylene ring is fused
with two outer TT rings. The ladder-type IDTT framework can
be easily constructed by Friedel
−Crafts annulation. The optical
and electrochemical properties of the resulting polymers have
been characterized experimentally and theoretically. Compared
to the hexacyclic DITT unit, this heptacyclic IDTT has
extended conjugation length with signi
ficantly increasing the
content of the thiophene moieties. Because of the more
favorable
π-electron delocalization, IDTT-based polymers show
much improved light-harvesting abilities and enhanced charge
mobilities. The device using PIDTTBT:PC
71BM (1:4, w/w)
blend exhibited an improved e
fficiency of 3.8%. Meanwhile, the
device using PIDTTFBT:PC
71BM (1:4, w/w) blend exhibited
a greater V
ocvalue of 0.9 V as a result of the
fluorine
withdrawing e
ffect on the BT unit, and a larger J
scof 10.08 mA/
cm
2with a higher PCE of 4.2%. The device based on the
Table 5. PSCs Characteristics
polymer blend ratio polymer:PC71BM Voc (V) Jsc (mA/cm2) (%)FF PCE(%) PIDTTBT 1: 4 0.84 8.32 55 3.8 PIDTTFBT 1: 4 0.90 10.08 46 4.2 PIDTTTPD 1: 4 0.90 7.99 60 4.3Figure 5.J−V characteristics of ITO/PEDOT:PSS/polymer:PC71BM/Ca/Al under illumination of AM1.5, 100 mW/cm2, and corresponding EQE spectra.
PIDTTTPD:PC
71BM (1:4 in wt %) blend exhibited a high V
ocof 0.90 V and a highest PCE of 4.3%. We envisage that further
improvement of device performance is highly achievable by
optimizing the processing conditions which are underway in
our laboratories. This research demonstrated that the new
heptacyclic indacenodithieno[3,2-b]thiophene is one of the
most promising building blocks for constructing
high-perform-ance conjugated polymers.
■
EXPERIMENTAL SECTION
General Measurement and Characterization. All chemicals are purchased from Aldrich or Acros and used as received unless otherwise specified.1H and13C NMR spectra were measured using Varian 300 and 400 MHz instrument spectrometers. Thermogravimetric analysis (TGA) was recorded on a Perkin-Elmer Pyris under nitrogen atmosphere at a heating rate of 10°C/min. Absorption spectra were recorded on a HP8453 UV−vis spectrophotometer. The molecular weight of polymers was measured on a Viscotek VE2001GPC, and polystyrene was used as the standard (THF as the eluent). The electrochemical cyclic voltammetry (CV) was conducted on a CH Instruments Model 611D. A carbon glass coated with a thin polymer film was used as the working electrode and Ag/Ag+electrode as the reference electrode, while 0.1 M tetrabutylammonium hexafluoro-phosphate (Bu4NPF6) in acetonitrile was the electrolyte. CV curves were calibrated using ferrocence as the standard, whose oxidation potential is set at −4.8 eV with respect to zero vacuum level. The HOMO energy levels were obtained from the equation HOMO = −(Eoxonset − E(ferrocene)onset + 4.8) eV. The LUMO levels of polymer were obtained from the equation LUMO =−(Eredonset− E(ferrocene)onset + 4.8) eV.
OFET Fabrication. An n-type heavily doped Si wafer with a SiO2 layer of 300 nm and a capacitance of 11 nF/cm2was used as the gate electrode and dielectric layer. Thinfilms (40−60 nm in thickness) of polymers were deposited on octadecyltrichlorosilane (ODTS)-treated SiO2/Si substrates by spin-coating their o-dichlorobenzene solutions (5 mg/mL). Then, the thin films were annealed at different temperatures (150 or 250 °C) for 30 min. Gold source and drain contacts (40 nm in thickness) were deposited by vacuum evaporation on the organic layer through a shadow mask, affording a bottom-gate, top-contact device configuration. Electrical measurements of OTFT devices were carried out at room temperature in air using a 4156C, Agilent Technologies. Thefield-effect mobility was calculated in the saturation regime by using the equation Ids= (μWCi/2L)(Vg− Vt)2, where Idsis the drain-source current,μ is the field-effect mobility, W is the channel width (1 mm), L is the channel length (100μm), Ciis the capacitance per unit area of the gate dielectric layer, Vg is the gate voltage, and Vtis threshold voltage.
PSCs Fabrication. ITO/Glass substrates were ultrasonically cleaned sequentially in detergent, water, acetone and iso-propanol (IPA). The cleaned substrates were covered by a 30 nm thick layer of PEDOT:PSS (Clevios P provided by Stark) by spin coating . After annealing in a glovebox at 150°C for 30 min, the samples were cooled to room temperature. Polymers were dissolved in o-dichlorobenzene (ODCB), and then PC71BM (purchased from Nano-C) was added. The solution was then heated at 80°C and stirred overnight at the same temperature. Prior to deposition, the solution werefiltered (0.45 μm filters). The solution of polymer:PC71BM was then spin coated to form the active layer. The cathode made of calcium (35 nm thick) and aluminum (100 nm thick) was sequentially evaporated through a shadow mask under high vacuum (<10−6Torr). Each sample consists of 4 independent pixels defined by an active area of 0.04 cm2. Finally, the devices were encapsulated and characterized in air.
Synthesis of Compound 1. A mixture of diethyl 2,5-dibromoterephthalate (6.05 g, 15.9 mmol), 2-(tributylstannyl)thieno-[3,2-b]thiophene (15.71 g, 36.6 mmol), Pd(PPh3)4(0.74 g, 0.6 mmol), and degassed toluene (80 mL) was heated to 130°C under nitrogen atmosphere for 16 h. The reaction mixture was poured into water (150 mL) and extracted with ethyl acetate (300 mL× 3). The combined organic layer was dried over MgSO4and the solvent was removed in
vacuo. The residue was purified by column chromatography on silica gel (hexane/ethyl acetate, v/v, 20/1) and then recrystallized from hexane to give a light yellow solid 1 (6.9 g, 87%):1H NMR (CDCl3, 300 MHz, ppm)δ 1.13 (t, J = 7.1 Hz, 6 H), 4.21−4.28 (q, J = 7.1 Hz, 4 H), 7.26−7.29 (m, 4 H), 7.40 (d, J = 5.1 Hz, 2 H), 7.89 (s, 2 H);13C NMR (CDCl3, 75 MHz, ppm)δ 13.8, 61.8, 119.3, 119.4, 127.4, 132.0, 133.8, 134.1, 139.3, 139.9, 142.0, 167.4; MS (EI, M+•, C24H18O4S4) calcd 498.01, found 498.
Synthesis of Compound 2. A Grignard reagent was prepared by the following procedure. To a suspension of magnesium turnings (3.2 g, 132.0 mmol) and 3−4 drops of 1,2-dibromoethane in dry THF (120 mL) was slowly added 1-bromo-4-(octyloxy)benzene (34.23 g, 120.0 mmol) dropwise and the mixture was then stirred for 1 h. To a THF (50 mL) solution of 1 (4.80 g, 9.6 mmol) under nitrogen atmosphere was added dropwise the freshly prepared 4-(octyloxy)phenyl-1-magnesium bromide (1 M, 76.8 mL, 76.8 mmol) at room temperature. The resulting mixture was heated at the refluxing temperature for 16 h, cooled to room temperature, poured into water (100 mL), and extracted with ethyl acetate (150 mL× 3). The collected organic layer was dried over MgSO4. After removal of the solvent under reduced pressure, the residue was purified by column chromatography on silica gel (hexane/ethyl acetate, v/v, 30/1) to furnish a yellow solid 2 (5.17 g, 43.7%):1H NMR (CDCl 3, 300 MHz, ppm)δ 0.88, (t, J = 6.8 Hz, 12 H), 1.28−1.45 (m, 40 H), 1.75−1.80 (m, 8 H), 3.42 (s, 2 H), 3.94 (t, J = 6.6 Hz, 8 H), 6.27 (s, 2 H), 6.80 (d, J = 9 Hz, 8 H), 6.90 (s, 2 H), 7.08 (d, J = 9 Hz, 8 H), 7.13 (d, J = 5.1 Hz, 2 H), 7.30 (d, J = 5.1 Hz, 2 H);13C NMR (CDCl3, 75 MHz, ppm)δ 14.1, 22.7, 26.0, 29.2, 29.3, 29.4, 31.8, 68.0, 82.4, 113.8, 119.3, 120.3, 127.0, 129.1, 132.3, 136.0, 138.7, 139.5, 139.9, 143.9. 145.4. 158.3; MS (FAB, M+•, C76H94O6S4) calcd 1230.59, found 1230.
Synthesis of IDTT. To a solution of 2 (1.00 g, 0.81 mmol) in THF (100 mL) was added concentrated sulfuric acid (0.5 mL). The mixture was stirred for 2 h at 90°C, cooled to room temperature, poured into water (250 mL), and extracted with ethyl acetate (500 mL× 3). The combined organic layer was dried over MgSO4and the solvent was removed under reduced pressure. The residue was then purified by column chromatography on silica gel (hexane/ethyl acetate, v/v, 80/1) to afford an yellow solid IDTT (1.58 g, 81%):1H NMR (CDCl
3, 400 MHz, ppm)δ 0.87 (t, J = 6.8 Hz, 12 H), 1.26−1.43 (m, 40 H), 1.70− 1.77 (m, 8 H), 3.89 (t, J = 6.6 Hz, 8 H), 6.79 (d, J = 8.8 Hz, 8 H), 7.19 (d, J = 8.8 Hz, 8 H), 7.25−7.29 (m, 4 H), 7.45 (s, 2 H) ;13C NMR (CDCl3, 75 MHz, ppm)δ 14.1, 22.6, 26.1, 29.2, 29.3, 29.3, 31.8, 62.2, 67.9, 114.3, 116.7, 120.4, 126.3, 129.2, 133.6, 134.9, 136.0, 141.7, 143.0. 146.3. 153.5. 158.2; MS (FAB, M+•, C76H90O4S4) calcd 1195.79, found 1195.
Synthesis of Monomer Br-IDTT. To a solution of IDTT (1.1 g, 0.92 mmol) in THF (30 mL) was added N-bromosuccinimide (0.38 g, 2.12 mmol) at room temperature. The reaction mixture, kept away from light, was stirred for 12 h at room temperature, quenched by water (50 mL + 150 mL), and extracted with ethyl acetate (150 mL× 3). The collected organic layer was dried over MgSO4. After removal of the solvent under vacuum, the residue was purified by column chromatography on silica gel (hexane/ethyl acetate, v/v, 100/1) and then recrystallized from hexane to give a brown solid Br-IDTT(1.08 g, 87%):1H NMR (CDCl 3, 300 MHz, ppm)δ 0.88 (t, J = 6.3 Hz, 12 H), 1.27−1.42 (m, 40 H), 1.72−1.77 (m, 8 H), 3.90 (t, J = 6.5 Hz, 8 H), 6.80 (d, J = 8.6 Hz, 8 H), 7.14 (d, J = 8.6 Hz, 8 H), 7.27 (s, 2 H), 7.44 (s, 2 H);13C NMR (CDCl 3, 75 MHz, ppm)δ 14.1, 22.6, 26.0, 29.2, 29.3, 29.3, 31.8, 62.1, 67.9, 112.5, 114.4, 116.8, 123.1, 129.0, 133.9, 134.5, 135.8, 139.9, 142.1. 146.6. 153.6. 158.3; MS (FAB, M+·, C76H88 Br2O4S4) calcd 1353.58, found 1353.
Synthesis of Sn-IDTT. To a THF (30 mL) solution of Br-IDTT (0.85 g, 0.63 mmol) was added a hexane solution of n-BuLi (2.5M, 1.58 mmol) dropwise at −78 °C. The mixture was stirred at this temperature for 1 h and a THF solution of chlorotrimethylstannane (1.0 M, 1.89 mmol) was then introduced dropwise. It was quenched with water (50 mL) and extracted with ether (50 mL × 3). The collected organic layer was dried over MgSO4 and the solvent was removed in vacuo. The residue was recrystallized from hexane to give a brown solid Sn-IDTT(0.52 g, 54%):1H NMR (CDCl
ppm)δ 0.37 (s, 18 H), 0.87 (t, J = 6.6 Hz, 12 H), 1.27−1.41 (m, 40 H), 1.69−1.78 (m, 8 H), 3.89(t, J = 6.6 Hz, 8 H), 6.79 (d, J = 8.7 Hz, 8 H), 7.19 (d, J = 8.7 Hz, 8 H), 7.30 (s, 2 H), 7.42 (s, 2 H);13C NMR (CDCl3, 100 MHz, ppm) δ −8.1, 14.1, 22.6, 26.1, 29.2, 29.3, 29.3, 31.8, 62.1, 67.9, 114.2, 116.6, 127.4, 129.3, 135.2, 136.1, 139.1, 140.3, 143.0, 143.8. 145.8. 153.3. 158.1.
Synthesis of PIDTTBT. To a 50 mL round-bottom flask were introduced Sn-IDTT (200 mg, 0.131 mmol), 4,7-dibromo-2,1,3-benzothiadiazole (38.5 mg, 0.131 mmol), Pd2(dba)3(4.8 mg, 0.005 mmol), tri(o-tolyl)phosphine (12.8 mg, 0.042 mmol), and dry chlorobenzene (7 mL). The mixture was bubbled with nitrogen for 10 min at room temperature. The reaction was then carried out in a microwave reactor under 270 W for 50 min. In order to end-cap the resultant polymer, tributyl(thiophen-2-yl)stannane (24.4 mg, 0.059 mmol) was added to the mixture, and the microwave reaction was continued for 10 min under 270 W. Subsequent to tributyl(thiophen-2-yl)stannane, another end-capping reagent, 2-bromothiophene (11.6 mg, 0.063 mmol), was added and the reaction was continued for another 10 min under otherwise identical conditions. The mixture was then added into methanol dropwise. The precipitate was collected by filtration and washed by Soxhlet extraction with acetone and hexane sequentially for 3 days. The crude polymer was dissolved in hot THF and the residual Pd catalyst and Sn metal in the THF solution was removed by Pd−thiol gel and Pd-TAAcOH (Silicycle Inc.). After filtration and removal of the solvent, the polymer was redissolved in THF and reprecipitated by methanol. The resultant polymer was collected byfiltration and dried under vacuum for 1 day to afford a dark-purplefiber-like solid (150 mg, 84%, Mn= 16600, PDI = 1.70): 1H NMR (CDCl
3, 300 MHz)δ 0.87 (br, 12 H), 1.26−1.42 (br, 40 H), 1.74 (br, 8 H), 3.91 (br, 8 H), 6.84 (br, 8 H), 7.19 (br, 8 H), 7.35− 7.55 (m, 2 H), 7.75 (br, 2 H), 8.57 (br, 2 H).
Synthesis of PIDTTFBT. To a 50 mL round-bottom flask were introduced Sn-IDTT (200 mg, 0.131 mmol), 4,7-dibromo-5,6-difluoro-2,1,3-benzothiadiazole (43.2 mg, 0.131 mmol), Pd2(dba)3 (4.8 mg, 0.005 mmol), tri(o-tolyl)phosphine (12.8 mg, 0.042 mmol), and dry chlorobenzene (7 mL). The mixture was bubbled with nitrogen for 10 min at room temperature. The reaction was then carried out in a microwave reactor under 270 W for 50 min. In order to end-cap the resultant polymer, tributyl(thiophen-2-yl)stannane (24.4 mg, 0.059 mmol) was added to the mixture, and the microwave reaction was continued for 10 min under 270 W. Subsequent to tributyl(thiophen-yl)stannane, another end-capping reagent, 2-bromothiophene (11.6 mg, 0.063 mmol) was added and the reaction was continued for another 10 min under otherwise identical conditions. The mixture was then added into methanol dropwise. The precipitate was collected by filtration and washed by Soxhlet extraction with acetone and hexane sequentially for three days. The crude polymer was dissolved in hot THF and the residual Pd catalyst and Sn metal in the THF solution was removed by Pd−thiol gel and Pd−TAAcOH (Silicycle Inc.). After filtration and removal of the solvent, the polymer was redissolved in THF and reprecipitated by methanol. The resultant polymer was collected byfiltration and dried under vacuum for 1 day to afford a give a dark-purple fiber-like solid (160 mg, 86%, Mn= 24000, PDI = 1.18):δ1H NMR (CDCl3, 300 MHz)δ 0.86 (br, 12 H), 1.26−1.29 (br, 40 H), 1.74 (br, 8 H), 3.91 (br, 8 H), 6.84 (br, 8 H), 7.27 (br, 8 H), 7.54 (br, 2 H), 8.66 (br, 2 H). Synthesis of PIDTTTPD. To a 50 mL round-bottom flask were introduced Sn-IDTT (200 mg, 0.131 mmol), 1,3-dibromo-thieno[3,4-c]pyrrole-4,6-dione (55.6 mg, 0.131 mmol), Pd2(dba)3(4.8 mg, 0.005 mmol), tri(o-tolyl)phosphine (12.8 mg, 0.042 mmol), and dry chlorobenzene (7 mL). The mixture was bubbled with nitrogen for 10 min at room temperature. In order to end-cap the resultant polymer, tributyl(thiophen-2-yl)stannane (24.4 mg, 0.059 mmol) was added to the mixture, and the microwave reaction was continued for 10 min under 270 W. Subsequent to tributyl(thiophen-2-yl)stannane, another end-capping reagent, 2-bromothiophene (11.6 mg, 0.063 mmol) was added, and the reaction was continued for another 10 min under otherwise identical conditions. The mixture was then added into methanol dropwise. The precipitate was collected by filtration and washed by Soxhlet extraction with acetone and hexane sequentially for
three days. The crude polymer was dissolved in hot THF and the residual Pd catalyst and Sn metal in the THF solution was removed by Pd−thiol gel and Pd−TAAcOH (Silicycle Inc.). After filtration and removal of the solvent, the polymer was redissolved in THF and reprecipitated by methanol. The resultant polymer was collected by filtration and dried under vacuum for 1 day to give a dark-purple fiber-like solid (170 mg, 89%, Mn = 31300, PDI = 2.04): δ 1H NMR (CDCl3, 300 MHz)δ 0.87 (br, 15 H), 1.27−1.43 (m, 50 H), 1.75 (br, 10 H), 3.92 (br, 10 H), 6.84 (br, 8 H), 7.19 (br, 8 H), 7.50 (br, 2 H), 8.52 (br, 2 H).
■
ASSOCIATED CONTENT
*
S Supporting InformationComputational details, thermogravimetric analysis, and
1H and
13
C NMR spectra of the new compounds and copolymers. This
material is available free of charge via the Internet at http://
pubs.acs.org/.
■
AUTHOR INFORMATION
Corresponding Author
*E-mail: [email protected].
NotesThe authors declare no competing
financial interest.
■
ACKNOWLEDGMENTS
We thank the National Science Council and the
“ATU
Program
” of the Ministry of Education, Taiwan, for financial
support. We are also grateful to the National Center for
High-performance Computing (NCHC) in Taiwan for computer
time and facilities.
■
REFERENCES
(1) Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J. Science 1995, 270, 1789.
(2) (a) Thompson, B. C.; Fréchet, J. M. J. Angew. Chem., Int. Ed. 2008, 47, 58. (b) Cheng, Y.-J.; Yang, S.-H.; Hsu, C.-S. Chem. Rev. 2009, 109, 5868. (c) Chen, J.; Cao, Y. Acc. Chem. Res. 2009, 42, 1709. (d) Li, Y.; Zou, Y. Adv. Mater. 2008, 20, 2952. (e) Li, Y. Acc. Chem. Res. 2012, 45, 723. (f) Huo, L.; Hou, J. Polym. Chem. 2011, 2, 2453. (g) Duan, C.; Huang, F.; Cao, Y. J. Mater. Chem. 2012, 22, 10416. (h) Zhou, H.; Yang, L.; You, W. Macromolecules 2012, 45, 607.
(3) (a) Svensson, M.; Zhang, F.; Veenstra, S. C.; Verhees, W. J. H.; Hummelen, J. C.; Kroon, J. M.; Inganäs, O.; Andersson, M. R. Adv. Mater. 2003, 15, 988. (b) Zhou, Q.; Hou, Q.; Zheng, L.; Deng, X.; Yu, G.; Cao, Y. Appl. Phys. Lett. 2004, 84, 1653. (c) Zhang, F.; Mammo, W.; Andersson, L. M.; Admassie, S.; Andersson, M. R.; Inganäs, O. Adv. Mater. 2006, 18, 2169. (d) Zhang, F.; Bijleveld, J.; Perzon, E.; Tvingstedt, K.; Barrau, S.; Inganäs, O.; Andersson, M. R. J. Mater. Chem. 2008, 18, 5468. (e) Schulz, G. L.; Chen, X.; Holdcroft, S. Appl. Phys. Lett. 2009, 94, 023302. (f) Huang, F.; Chen, K.-S.; Yip, H.-L.; Hau, S. K.; Acton, O.; Zhang, Y.; Luo, J.; Jen, A. K.-Y. J. Am. Chem. Soc. 2009, 131, 13886.
(4) (a) Zhu, Z.; Waller, D.; Gaudiana, R.; Morana, M.; Mühlbacher, D.; Scharber, M.; Brabec, C. Macromolecules 2007, 40, 1981. (b) Mühlbacher, D.; Scharber, M.; Zhengguo, M. M.; Zhu, M. M. Z.; Waller, D.; Gaudiana, R.; Brabec, C. Adv. Mater. 2006, 18, 2884. (c) Kim, J. Y.; Lee, K.; Coates, N. E.; Moses, D.; Nguyen, T.-Q.; Dante, M.; Heeger, A. J. Science 2007, 317, 222. (d) Peet, J.; Kim, J. Y.; Coates, N. E.; Ma, W. L.; Moses, D.; Heeger, A. J.; Bazan, G. C. Nat. Mater. 2007, 6, 497. (e) Chen, C.-H.; Hsieh, C.-H.; Dubosc, M.; Cheng, Y.-J.; Hsu, C.-S. Macromolecules 2010, 43, 697.
(5) (a) Li, Y.; Wu, Y.; Liu, P.; Birau, M.; Pan, H.; Ong, B. S. Adv. Mater. 2006, 18, 3029. (b) McCulloch, I.; Heeney, M.; Bailey, C.; Genevicius, K.; MacDonald, I.; Shkunov, M.; Sparrowe, D.; Tierney, S.; Wagner, R.; Zhang, W.; Chabinyc, M. L.; Kline, R. J.; Mcgehee, M. D.; Toney, M. F. Nat. Mater. 2006, 5, 328.
(6) (a) Subramanian, G.; Schleyer, P. R.; Jiao, H. Angew. Chem., Int. Ed. Engl. 1996, 35, 2638. (b) Hess, B. A., Jr.; Schaad, L. J. J. Am. Chem. Soc. 1973, 95, 3907.
(7) (a) Hayashi, N.; Mazaki, Y.; Kobayashi, K. J. Chem. Soc., Chem. Commun. 1994, 2351. (b) DeLongchamp, D. M.; Kline, R. J.; Lin, E. K.; Fischer, D. A.; Richter, L. J.; Lucas, L. A.; Heeney, M.; McCulloch, I.; Northrup, J. E. Adv. Mater. 2007, 19, 833. (c) Chabinyc, M. L.; Toney, M. F.; Kline, R. J.; McCulloch, I.; Heeney, M. J. Am. Chem. Soc. 2007, 129, 3226.
(8) (a) Zhang, X.; Kohler, M.; Matzger, A. J. Macromolecules 2004, 37, 6306. (b) Miguel, L. S.; Matzger, A. J. Macromolecules 2007, 40, 9233. (c) Miguel, S, L.; Matzger, A, J. J. Org. Chem. 2007, 72, 442. (d) Henssler, J, T.; Zhang, X.; Matzger, A, J. J. Org. Chem. 2009, 74, 9112.
(9) (a) Liang, Y.; Wu, Y.; Feng, D.; Tsai, S.-T.; Son, H.-J.; Li, G.; Yu, L. J. Am. Chem. Soc. 2009, 131, 56. (b) He, F.; Wang, W.; Chen, W.; Xu, T.; Darling, S. B.; Strzalka, J.; Liu, Y.; Yu, L. J. Am. Chem. Soc. 2011, 133, 3284. (c) Zhang, Y.; Zou, J.; Yip, H.-L.; Chen, K.-S.; Zeigler, D. F.; Sun, Y.; Jen, A. K.-Y. Chem. Mater. 2011, 23, 2289. (d) Cheng, Y.-J.; Wu, J.-S.; Shih, P.-I.; Chang, C.-Y.; Jwo, P.-C.; Kao, W.-S.; Hsu, C.-S. Chem. Mater. 2011, 23, 2361. (e) Wu, J.-S.; Cheng, Y.-J.; Dubosc, M.; Hsieh, C.-H.; Chang, C.-Y.; Hsu, C.-S. Chem. Commun. 2010, 46, 3259. (f) Wu, J.-S.; Cheng, Y.-J.; Lin, T.-Y.; Chang, C.-Y.; Shih, P.-I.; Hsu, C.-S. Adv. Funct. Mater. 2012, 22, 1711. (g) Wang, J.-Y.; Hau, S. K.; Yip, H.-L.; Davies, J. A.; Chen, K.-S.; Zhang, Y.; Sun, Y.; Jen, A. K.-Y. Chem. Mater. 2011, 23, 765. (h) Ashraf, R. S.; Chen, Z.; Leem, D. S.; Bronstein, H.; Zhang, W.; Schroeder, B.; Geerts, Y.; Smith, J.; Watkins, S.; Anthopoulos, T. D.; Sirringhaus, H.; Mello, J. C.; de; Heeney, M.; McCulloch, I. Chem. Mater. 2011, 23, 768. (i) Chen, C.-H.; Cheng, Y.-J.; Dubosc, M.; Hsieh, C.-H.; Chu, C.-C.; Hsu, C.-S. Chem. Asian. J. 2010, 5, 2480. (j) Zhang, M.; Guo, X.; Wang, X.; Wang, H.; Li, Y. Chem. Mater. 2011, 23, 4264. (k) Cheng, Y.-J.; Chen, C.-H.; Lin, Y.-S.; Chang, C.-Y.; Hsu, C.-S. Chem. Mater. 2011, 23, 5068. (l) Chen, C.-H.; Cheng, Y.-J.; Chang, C.-Y.; Hsu, C.-S. Macromolecules 2011, 44, 8415. (m) Cheng, Y.-J.; Ho, Y.-J.; Chen, C.-H.; Kao, W.-S.; Wu, C.-E.; Hsu, S.-L.; Hsu, C.-S. Macromolecules 2012, 45, 2690. (n) Chen, Y. -C.; Yu, C. -Y.; Fan, Y.-L.; Huang, L.-I.; Chen, C.-P.; Ting, C. Chem. Commun. 2010, 46, 6503. (o) Zhang, Y.; Zou, J.; Yip, H.-L.; Chen, K.-S.; Davies, J. A.; Sun, Y.; Jen, A. K. Y. Macromolecules 2011, 44, 4752.
(10) (a) Goldfinger, M. B.; Crawford, K. B.; Swager, T. M. J. Am. Chem. Soc. 1997, 119, 4578. (b) Goldfinger, M. B.; Swager, T. M. J. Am. Chem. Soc. 1994, 116, 7895. (c) Forster, M.; Annan, K. O.; Scherf, U. Macromolecules 1999, 32, 3159. (d) Scherf, U. J. Mater. Chem. 1999, 9, 1853. (e) Chmil, K.; Scherf, U. Acta Polym. 1997, 48, 208. (f) Jacob, J.; Sax, S.; Piok, T.; List, E. J. W.; Grimsdale, A. C.; Mullen, K. J. Am. Chem. Soc. 2004, 126, 6987. (g) Zheng, Q.; Jung, B. J.; Sun, J.; Katz, H. E. J. Am. Chem. Soc. 2010, 132, 5394. (h) Facchetti, A. Chem. Mater. 2011, 23, 733. (i) Guo, X.; Ortiz, R. P.; Zheng, Y.; Hu, Y.; Noh, Y.-Y.; Baeg, K.-J.; Facchetti, A.; Marks, T. J. J. Am. Chem. Soc. 2011, 133, 1405. (j) Cheng, Y.-J.; Ho, Y.-J.; Chen, C.-H.; Kao, W.-S.; Wu, C.-E.; Hsu, S.-L.; Hsu, S. Macromolecules 2012, 45, 2690. (k) Chang, C.-Y.; Cheng, Y.-J.; Hung, S.-H.; Wu, J.-S.; Kao, W.-S.; Lee, C.-H.; Hsu, C.-S. Adv. Mater. 2012, 24, 549.
(11) Cheng, Y.-J.; Cheng, S.-W.; Chang, C.-Y.; Kao, W.-S.; Liao, M.-H.; Hsu, C.-S. Chem. Commun. 2012, 48, 3203.
(12) (a) Boudreault, P.-L. T.; Najari, A.; Leclerc, M. Chem. Mater. 2011, 23, 456. (b) Peng, Q.; Liu, X.; Su, D.; Fu, G.; Xu, J.; Dai, L. Adv. Mater. 2011, 23, 4554. (c) Hou, J. H.; Chen, H. Y.; Zhang, S. Q.; Yang, Y. J. Phys. Chem. C 2009, 113, 21202. (d) Wang, X. C.; Sun, Y. P.; Chen, S.; Guo, X.; Zhang, M. J.; Li, X. Y.; Li, Y.; Wang, H. Q. Macromolecules 2012, 45, 1208.
(13) (a) Nielsen, C. B.; Bjørnholm, T. Org. Lett. 2004, 6, 3381. (b) Chen, C. M.; Amb., S.; Graham, K. R.; Subbiah, J.; Small, C. E.; So, F.; Reynolds, J. R. J. Am. Chem. Soc. 2011, 133, 10062. (c) Chu, T. Y.; Lu, J.; Beaupré, S.; Zhang, Y.; Pouliot, J. R.; Wakim, S.; Zhou, J.; Leclerc, M.; Li, Z.; Ding, J.; Tao, Y. J. Am. Chem. Soc. 2011, 133, 4250. (d) Berrouard, P.; Najari, A.; Pron, A.; Gendron, D.; Morin, P.-O.; Pouliot, J.-R.; Veilleux, J.; Leclerc, M. Angew. Chem., Int. Ed. 2012, 51,
2068. (e) Zou, Y.; Najari, A.; Berrouard, P.; Beaupré, S.; Aïch, B. R.; Tao, Y.; Leclerc, M. J. Am. Chem. Soc. 2010, 132, 5330. (f) Zhang, Y.; Hau, S. K.; Yip, H. L.; Sun, Y.; Acton, O.; Jen, A. K. Y. Chem. Mater. 2010, 22, 2696. (g) Piliego, C.; Holcombe, T. W.; Douglas, J. D.; Woo, C. H.; Beaujuge, P. M.; Fréchet, J. M. J. J. Am. Chem. Soc. 2010, 132, 7595. (h) Najari, A.; Beaupré, S.; Berrouard, P.; Zou, Y.; Pouliot, J. R.; Pérusse, C. L.; Leclerc, M. Adv. Funct. Mater. 2011, 21, 718.
(14) (a) Zhang, Y.; Chien, S.-C.; Chen, K.-S.; Yip, H.-L.; Sun, Y.; Davies, J. A.; Chen, F.-C.; Jen, A. K. Y. Chem. Commun. 2011, 47, 11026. (b) Zhou, H.; Yang, L.; Stuart, A. C.; Price, S. C.; Liu, S.; You, W. Angew. Chem., Int. Ed. 2011, 50, 2995. (c) Schroeder, B. C.; Huang, Z.; Ashraf, R. S.; Smith, J.; D’Angelo, P.; Watkins, S. E.; Anthopoulos, T. D.; Durrant, J. R.; McCulloch, I. Adv. Funct. Mater. 2012, 22, 1663. (d) Li, Z.; Lu, J.; Tse, S.-C.; Zhou, J.; Du, X.; Tao, Y.; Ding, J. J. Mater. Chem. 2011, 21, 3226.
(15) O’Boyle, N. M.; Tenderholt, A. L.; Langner., K. M. J. Comput. Chem. 2008, 29, 839.