Synthesis and Characterization of Dendritic Poly(ether imide)s
Chyi-Ming Leu, Yao-Te Chang, and Ching-Fong Shu*Department of Applied Chemistry, National Chiao Tung University, Hsin-Chu, Taiwan, 30035, R.O.C. Chia-Fang Teng and Jentaie Shiea
Department of Chemistry, National Sun Yat-Sen University, Kaohsiung, Taiwan, 80424, R.O.C. Received October 8, 1999; Revised Manuscript Received February 3, 2000
ABSTRACT: New dendritic poly(ether imide)s were synthesized by the convergent growth approach, using 1-(4-aminophenyl)-1,1-bis(4-hydroxyphenyl)ethane, as the building block. The aromatic nucleophilic substitution of the building block with 3-nitro-N-phenylphthalimide led to the first-generation dendron with aminophenyl group at the focal point, which was subsequently reacted with 3-nitrophthalic anhydride to yield the dendritic wedge containing an activated nitro group. The resulting nitro functionality was allowed to react with the building block to give the second-generation dendron, which was then condensed with 3-nitrophthalic anhydride, followed by ring closure to re-form the phthalimide ring and restore a reactive nitro group. Through an aromatic nucleophilic substitution, the dendritic wedges with an activated nitro group were coupled to the polyfunctional core to form the dendritic macromolecules. Structures of the ether-imide dendrimers were fully characterized by use of a combination of techniques including1H NMR,13C NMR, IR, and mass spectrometry. The glass transition temperature of the dendrimers increased with molecular weight, and the variation was consistent with theoretical calculations. These dendritic poly(ether imide)s are soluble in various organic solvents and had a thermal stability comparable to linear poly(aryl ether imide)s.
Introduction
Recently, dendritic macromolecules have received considerable attention due to the expectation that their unique highly branched polymeric structure and well-defined three-dimensional architecture will impact un-usual properties.1These dendritic macromolecules are characterized by a central polyfunctional core, from which arise successive layers of monomer units with branching points in each monomer unit. This results in a nearly entanglement-free highly branched structure that may adopt a spherical shape and where the periphery consists of a large number of chain ends.
Two main synthetic strategies have been used to synthesize dendritic macromolecules: the divergent2 and convergent approaches.3Since the synthesis of the dendritic macromolecules is based on the interactive strategy, control over architectural properties is main-tained at each synthesis step. The synthesis of well-defined, highly branched macromolecules with con-trolled molecular size has sparked much interest. Dendrimers such as polyether,3a,4 polyester,5 poly-amide,6poly(amido-amine),2a,7poly(carbosilane),8 pol-ysiloxane,9 poly(carbosilazane),10 poly(phenylene),3c,11 poly(arylacetylene),3b,12poly(triarylamine),13and poly-(ether ketone)14 have been synthesized, giving new materials with desirable properties.
In this paper, we report the synthesis and character-ization of dendritic poly(ether imide)s, which were prepared by the stepwise convergent approach and possess ether-imide units located at the peripheries and within their interior layers. The convergent meth-odology for the dendritic poly(ether imide)s involved the synthesis of the respective dendritic wedges, possessing ether-imide units as the structural components, fol-lowed by their attachment to the core component. Structural components that constitute the branching
regions of the dendritic wedges were derived from 1-(4-aminophenyl)-1,1-bis(4-hydroxyphenyl)ethane (2) and 3-nitrophthalic anhydride, while the core was derived from 1,1,1-tris(4-hydroxyphenyl)ethane. The synthetic sequence for the ether-imide dendrons was based on (i) the aromatic nucleophilic displacement of activated nitro group with phenoxide nucleophile to form the ether linkage15and (ii) the condensation of aminophenyl group with nitro-substituted phthalic anhydride, followed by ring-closure to yield the imide ring containing the reactive nitro functionality.16
Experimental Section
General Directions. The CH3ONa/CH3OH solution was prepared by dissolving sodium metal in absolute MeOH. Other starting materials and reagents were used as obtained from the suppliers. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Unity 300 MHz or a Bruker-DRX 300 MHz spectrometer. Differential scanning calorimetry (DSC) was performed on a SEIKO SSC 5200 DSC using a heating/ cooling rate of 10 °C min-1. Samples were scanned from 30 to 300 °C and then cooled to 30 °C and scanned a second time from 30 to 300 °C. The glass transition temperature was determined from the second heating scan. Thermogravimetric analysis (TGA) was made on a SEIKO TG/DTA 200 instru-ment. The thermal stability of samples was determined in nitrogen by measuring weight loss while heating at a rate of 10 °C min-1. Size-exclusion chromatography (SEC) was carried out on a Waters chromatography connected to a Waters 410 Differential refractometer. Three 5 µm Waters styragel col-umns (300× 7.8 mm) connected in series in order of decreasing pore size (105, 104, and 103Å) were used with DMF/0.05 M LiBr, as the eluant. Infrared spectra were taken on a Nicolet 520 FTIR spectrometer. Mass spectra were obtained on a JEOL JMS-SX/SX 102A mass spectrometer. Analytical TLC was performed on commercial Merck plated coated with silica gel GF254. Silica gel for column chromatography was Merck Kieselgel 60 (70-230 mesh). Matrix-assisted laser desorption/ ionization (MALDI) mass spectra were recorded on a linear 10.1021/ma991695v CCC: $19.00 © 2000 American Chemical Society
time-of-flight mass spectrometer (TOF MS) (HP 2025G) equipped with a pulsed nitrogen laser (337 nm). Minimum laser power was used to desorb the analyte from the sample solution. The solution of analyte (1 mM) and matrix (0.1 M) in DMF was prepared, and 1 µL of this matrix/analyte solution was transferred to the sample probe and dried under reduced pressure. The measurement was performed in positive ion mode using 2,5-dihydroxybenzoic acid, as the matrix.
1,1-Bis(4-hydroxyphenyl)-1-(4-nitrophenyl)ethane 1. A mixture of p-nitroacetophenone (21.0 g, 127 mmol), phenol (150 mL), and trifluoromethanesulfonic acid (0.4 mL) was heated at 185 °C for 24 h. An additional portion of trifluoromethane-sulfonic acid (0.2 mL) was added, and then the resulting mixture was heated at 185 °C for another 24 h. Then, excess phenol was removed in vacuo at 80 °C. The residue was poured into water (300 mL) and extracted with EtOAc (3× 200 mL). The combined extract was dried (MgSO4), and the solvent was removed in vacuo. The crude product was purified by column chromatography (hexane/EtOAc 2:1) and purified by recrys-tallization from EtOAc/hexane to give 1 (17.1 g, 40.2%).1H NMR (DMSO-d6): δ 2.04 (s, 3 H), 6.67 (d, 4 H, J ) 8.7 Hz), 6.81 (d, 4 H, J ) 8.7 Hz), 7.28 (d, 2 H, J ) 8.7 Hz), 8.11 (d, 2 H, J ) 8.7 Hz), 9.37 (s, 2 H).13C NMR (DMSO-d
6): δ 30.1, 51.2, 114.8, 123.0, 129.2, 129.6, 138.3, 145.5, 155.6, 158.0. MS (m/e): found, 335.1169; calculated, 335.1158 for C20H17NO4. 1-(4-Aminophenyl)-1,1-bis(4-hydroxyphenyl)ethane, 2. To a solution of 1 (11.5 g, 34.2 mmol) in EtOH (50 mL) was added 10% Pd/C (575 mg). The resulting mixture was flushed three times with hydrogen to remove oxygen and stirred vigorously at 25 °C under hydrogen for 24 h. The reaction mixture was then filtered, and the filtrate was evaporated to give 2 (10.2 g, 97.5%).1H NMR (DMSO-d 6): δ 1.92 (s, 3 H), 4.86 (s, 2 H), 6.44 (d, 2 H, J ) 7.8 Hz), 6.64 (m, 6 H), 6.79 (d, 4 H, J ) 8.4 Hz), 9.20 (s, 2 H).13C NMR (DMSO-d 6): δ 30.5, 49.8, 113.3, 114.3, 128.7, 129.2, 137.1, 140.5, 146.1, 155.0; MS (m/e): found, 305.1421; calculated, 305.1416 for C20H19NO2. General Procedure for the Synthesis of [G-X]NH2(X
) 1 and 2). A mixture of 2 and CH3ONa/CH3OH was stirred under nitrogen for 1 h at 25 °C. The MeOH was removed in vacuo. The solid was then further dried at 60 °C in vacuo for 2 h. To the resulting solid was added anhydrous DMF and the appropriate nitrophthalimide derivative. The mixture was stirred at 60 °C for 12 h, and then poured into water. The precipitated solid was filtered and purified as outlined in the following text.
General Procedure for the Synthesis of [G-X]NO2(X
) 0, 1, and 2). A solution of the appropriate phenylamine derivative and 3-nitrophthalic anhydride in AcOH was re-fluxed under nitrogen for 3 h. The mixture was cooled to 25 °C and poured into water. The precipitate was filtered and purified, as outlined in the following text.
[G-0]NO2. This was prepared from the reaction of aniline
(30.0 g, 322 mmol) and 3-nitrophthalic anhydride (62.2 g, 322 mmol) and was purified by recrystallization from EtOAc/ hexane to give [G-0]NO2(65.0 g, 75.3%).1H NMR
(DMSO-d6): δ 7.44-7.57 (m, 5 H), 8.11 (dd, 1 H, J ) 7.2, 7.8 Hz) 8.24 (d, 1 H, J ) 7.2 Hz), 8.33 (d, 1 H, J ) 7.8 Hz).13C NMR (DMSO-d6): δ 122.9, 127.0, 127.5, 128.4, 128.5, 128.9, 131.5, 133.5, 136.36, 144.5, 162.6, 165.2.
[G-1]NH2. This was prepared from 2 (3.0 g, 9.8 mmol) and
CH3ONa (41.0 mL, 20.9 mmol, 0.51 M in CH3OH). The resulting phenoxide derivative was then reacted with [G-0]-NO2(5.52 g, 20.6 mmol). The product was purified by column
chromatography (CHCl3) to give [G-1]NH2(4.5 g, 61%).1H NMR (DMSO-d6): δ 2.08 (s, 3 H), 4.98 (s, 2 H), 6.49 (d, 2 H, J ) 8.4 Hz), 6.74 (d, 2 H, J ) 8.4 Hz), 7.09 (d, 4 H, J ) 8.7 Hz), 7.13 (d, 4 H, J ) 8.7 Hz), 7.23 (d, 2 H, J ) 8.1 Hz), 7.42-7.54 (m, 10 H), 7.67 (d, 2 H, J ) 7.5 Hz), 7.82 (dd, 2 H, J ) 7.5, 8.1 Hz).13C NMR (DMSO-d 6): δ 30.4, 50.7, 113.6, 118.0, 119.0, 123.6, 127.6, 128.2, 128.8, 129.0, 130.3, 131.9, 134.0, 135.5, 137.0, 146.3, 146.8, 152.8, 154.0, 164.9, 166.5. MS (m/e): found, 747.2368; calculated, 747.2369 for C48H33N3O6.
[G-1]NO2. This was prepared from the reaction of
[G-1]-NH2(2.60 g, 3.48 mmol) and 3-nitrophthalic anhydride (701
mg, 3.63 mmol) and was purified by column chromatography
(EtOAc/CHCl31:2) to give [G-1]NO2(2.2 g, 69%).1H NMR
(DMSO-d6): δ 2.23 (s, 3 H), 7.12 (d, 4 H, J ) 7.9 Hz), 7.23 (d, 4 H, J ) 9.0 Hz), 7.28 (d, 4 H, J ) 8.3 Hz), 7.39-7.55 (m, 12 H), 7.68 (d, 2 H, J ) 7.5 Hz), 7.83 (dd, 2 H, J ) 7.5, 7.5 Hz), 8.10 (dd, 1H, J ) 7.8, 7.8 Hz), 8.25 (dd, 1 H, J ) 0.8, 7.5 Hz), 8.33 (dd, 1 H, J ) 0.9, 7.6 Hz).13C NMR (DMSO-d 6): δ 30.2, 51.5, 79.2, 118.1, 119.1, 122.9, 123.8, 126.9, 127.0, 127.5, 128.1, 128.4, 128.8, 129.5, 130.2, 131.8, 133.5, 133.9, 136.4, 137.0, 144.5, 144.8, 148.8, 153.2, 153.7, 162.6, 164.7, 165.2, 166.4. MS (m/e): found, 923.2339; calculated, 923.2353 for C56H35N4O10. [G-2]NH2. This was prepared starting from the reaction
of 2 (561 mg, 1.84 mmol) and CH3ONa (7.2 mL, 3.7 mmol, 0.51 M in CH3OH). The resulting phenoxide derivative was then reacted with [G-1]NO2(3.40 g, 3.68 mmol). The product was
purified by column chromatography (EtOAc/CHCl31:50) to give [G-2]NH2(1.6 g, 42%).1H NMR (DMSO-d6): δ 2.07 (s, 3 H), 2.21 (s, 6 H), 4.97 (s, 2 H), 6.49 (d, 2 H, J ) 8.4 Hz), 6.74 (d,2 H, J ) 8.4 Hz), 7.06-7.30 (m, 34 H), 7.41-7.30 (m, 24 H), 7.65 (d, 2 H, J ) 7.2 Hz), 7.66 (d, 4 H, J ) 7.2 Hz), 7.80 (dd, 2 H, J ) 7.2, 7.8 Hz), 7.82 (dd, 4 H, J ) 7.8, 8.1 Hz).13C NMR (DMSO-d6): δ 30.6, 50.7, 51.5, 113.7, 118.2, 119.0, 119.2, 124.0, 127.0, 127.6, 128.3, 128.8, 129.0, 129.9, 130.3, 131.9, 134.0, 137.1, 145.0, 146.3, 146.7, 148.5, 152.9, 153.4, 153.8, 154.0, 164.9, 166.6. MS (m/e): found, 2055.5918; calculated, 2055.5951 for C132H85N7O18.
[G-2]NO2. This was prepared from the reaction of
[G-2]-NH2(1.20 g, 0.584 mmol) and 3-nitrophthalic anhydride (140
mg, 725 µmol) and was purified by column chromatography to give [G-2]NO2(600 mg, 46.1%).1H NMR (DMSO-d6): δ 2.14 (s, 9 H), 7.07 (d, 12 H, J ) 8.2 Hz), 7.13 (d, 12 H, J ) 8.7 Hz), 7.18-7.21(m, 12 H), 7.34-7.46 (m, 26 H), 7.59 (d, 6 H, J ) 7.1 Hz), 7.74 (dd, 6 H, J ) 7.4, 8.0 Hz), 8.02 (dd, 1 H, J ) 7.7, 7.8 Hz), 8.17 (d, 1 H, J ) 7.5 Hz), 8.25 (d, 1 H, J ) 8.0 Hz).13C NMR (DMSO-d 6): δ 30.2, 51.4, 118.0, 119.1, 123.8, 126.8, 127.5, 128.0, 128.8, 129.8, 130.2, 131.5, 131.8, 133.9, 136.4, 136.9, 144.5, 144.8, 148.3, 153.2, 153.7, 162.6, 164.7, 165.2, 166.4. MS (m/e): found, 2230 (M+).
General Procedure for the Synthesis of Dendritic Ether-Imides [G-X]3[C] (X ) 0, 1, and 2). A mixture of
1,1,1-tris(4-hydroxyphenyl)ethane and CH3ONa/CH3OH was stirred under nitrogen for 1 h at 25 °C. The MeOH was removed in vacuo, and then the solid was dried at 60 °C in vacuo for 2 h. To the resulting solid was added anhydrous DMF and the appropriate dendritic nitrophthalimide. The mixture was stirred at 60 °C for 12 h and then poured into water. The precipitated solid was isolated by filtration and purified as outlined in the following text.
[G-0]3[C]. 1,1,1-Tris(4-hydroxyphenyl)ethane (200 mg, 653
µmol) and CH3ONa (4.1 mL, 2.1 mmol, 0.51 M in CH3OH) were reacted according to the general procedure outlined above. The resulting phenoxide derivative was then reacted with [G-0]-NO2(620 mg, 2.31 mmol). The product was purified by column
chromatography (EtOAc/CHCl31:2) to give [G-0]3[C] (402 mg,
63.5%).1H NMR (DMSO-d 6): δ 2.20 (s, 3 H), 7.09 (d, 6 H, J ) 9.0 Hz), 7.21 (d, 6 H, J ) 8.9 Hz), 7.26 (d, 3 H, J ) 8.4 Hz), 7.40-7.54 (m, 15 H), 7.68 (d, 3 H, J ) 7.3 Hz), 7.83 (dd, 3 H, J ) 8.3, 8.3 Hz). 13C NMR (DMSO-d 6): δ 30.2, 51.1, 118.1, 119.0, 123.8, 127.5, 128.1, 128.8, 130.2, 131.8, 133.9, 136.9, 145.0, 153.2, 153.7, 164.7, 166.4. MS (m/e): found, 970.2768; calculated, 970.2764 for C62H40N3O9. [G-1]3[C]. 1,1,1-Tris(4-hydroxyphenyl)ethane (40.0 mg, 131
µmol) and CH3ONa (0.77 mL, 0.39 mmol, 0.51 M in CH3OH) were reacted according to the general procedure outlined above. The resulting phenoxide derivative was then reacted with [G-1]NO2(370 mg, 400 µmol). The product was purified
by column chromatography (EtOAc/CHCl3 ) 1:4) to give [G-1]3[C] (120 mg, 31.3%).1H NMR (DMSO-d6): δ 2.20 (s, 12 H), 7.12-7.27 (m, 51 H), 7.37-7.53 (m, 36 H), 7.66 (d, 9 H, J ) 7.2 Hz), 7.81 (dd, 9 H, J ) 7.4, 8.2 Hz).13C NMR (DMSO-d6): δ 30.2, 51.4, 118.0, 119.0, 123.8, 126.8, 127.4, 128.0, 128.8, 129.8, 130.1, 131.8, 133.8, 136.9, 144.8, 148.3, 153.2, 153.6, 164.7, 166.3. [G-2]3[C]. 1,1,1-Tris(4-hydroxyphenyl)ethane (25 mg, 82
µmol) and CH3ONa (0.50 mL, 0.26 mmol, 0.51 M in CH3OH) were reacted according to the general procedure outlined
above. The resulting phenoxide derivative was then reacted with [G-2]NO2(546 mg, 245 µmol). The product was purified
by column chromatography (EtOAc/CH2Cl2) 1:40) to give [G-2]3[C] (131 mg, 23.2%).1H NMR (DMSO-d6): δ 2.19 (s, 30 H), 7.08 (d, 42 H, J ) 8.8 Hz), 7.21 (d, 42 H, J ) 9.1 Hz), 7.24 (d, 39 H, J ) 8.2 Hz), 7.36-7.51 (m, 78 H), 7.64 (d, 21 H, J ) 7.3 Hz), 7.79 (dd, 21 H, J ) 7.7, 8.1 Hz).13C NMR (DMSO-d 6): δ 30.2, 51.4, 118.0, 119.0, 123.8, 126.8, 127.4, 128.0, 128.8, 130.1, 131.8, 133.8, 136.9, 144.8, 153.2, 153.6, 164.7, 166.3. Results and Discussion
The general synthetic procedure for the poly(ether imide) dendrons is shown in Scheme 1. The synthesis of the building block 2 was performed by the acid-catalyzed condensation reaction of p-nitroacetophenone with excess phenol to give compound 1,17 followed by the reduction of the nitro group of 1 with H2catalyzed by Pd/C. 2 possesses one aminophenyl group and two phenol groups. By reaction with sodium methoxide, the phenol groups of 2 were converted to phenoxides, which subsequently were reacted with 3-nitro-N-phenylphthal-imide, while the amino group remained intact, to yield the ether imide [G-1]NH2. The possibility of nucleo-philic aromatic substitution on 3-nitrophthalimide by the amino group of 2 was further ruled out by a control experiment.18,19The design of the building block 2, in which the phenol groups were isolated by phenyl rings, prevented the formation of complex byproducts due to the transfer redox reaction between the electron-deficient nitrophthalimide and the electron-rich ben-zenediol dianion.20The condensation of the aminophenyl group of [G-1]NH2 with 3-nitrophthalic anhydride, followed by ring-closure, led to the corresponding ph-thalimide [G-1]NO2with the activated nitro function-ality restored at the focal point of the growing dendritic
wedge. Further reaction of [G-1]NO2with the phenox-ides of 2 gave the next-generation ether-imide [G-2]-NH2. Subsequent condensation and cyclization of 3-ni-trophthalic anhydride and [G-2]NH2 resulted in the second-generation phthalimide dendron [G-2]NO2. Structures of the phthalimide dendrons and intermedi-ates synthesized were confirmed by1H NMR,13C NMR, and FT-IR spectroscopy as well as mass spectrometry. The dendritic wedges obtained were attached to a polyfunctional core to afford the dendrimers. Reactions of phenoxide nucleophiles of the core molecule, 1,1,1-tris(4-hydroxyphenyl)ethane, with each generation of nitro-substituted phthalimide dendrons, from [G-0]-NO2, [G-1]NO2, and [G-2]NO2, as shown in Scheme 2, were carried out. Thus, a series of dendritic poly(ether imide)s, dendrimers [G-0]3[C], [G-1]3[C], and [G-2]3 -[C], containing 3, 9, and 21 ether-imide units, were obtained. Characterization of the ether-imide dendrim-ers was accomplished by a combination of techniques including1H NMR, 13C NMR, IR, mass spectrometry, size exclusion chromatography, differential scanning calorimetry, and thermogravimetric analysis.
1H and13C NMR spectroscopy proved valuable in the characterization of the intermediate dendritic fragments and the final dendritic macromolecules.1H NMR spec-troscopy was particularly crucial in confirming the structures produced by our synthetic strategy. Figure 1 shows some distinct features of the1H NMR spectra of [G-1]NH2and [G-1]NO2. For dendritic fragments with aminophenyl group at the focal point, the aromatic protons of the aminophenyl group gave rise to an AB quartet at 6.49 and 6.74 ppm while the resonance for the protons of the amino group appeared at 4.97 ppm, and the resonances for the aromatic protons of the
ether-Scheme 1a
aReagent: (i) phenol, CF
substituted phthalic rings were observed at 7.23, 7.67, and 7.82 ppm. For dendritic fragments with the 3-ni-trophthalimide ring at the focal point, additional reso-nances for the aromatic protons of the nitro-substituted phthalic ring occurred at 8.10, 8.25, and 8.33 ppm, while the resonances associated with protons of the ami-nophenyl group disappeared. The significant differences between these resonances allowed the focal point group to be identified, the dendrons to be characterized, and their purity to be determined. Comparison of the integration values for these groups to the values for other resonances allowed the generation number to be confirmed.
13C NMR provided complementary information. Reso-nances associated with the carbonyl groups of the ether-substituted phthalic rings for dendritic fragments [G-X]-NH2(X ) 1 and 2) appeared at 164.9 and 166.5 ppm, while additional resonances corresponding to the car-bonyl groups of the nitro-substituted phthalic ring
appeared at 162.6 and 165.2 ppm for dendritic wedges [G-X]NO2(X ) 0, 1, and 2).16
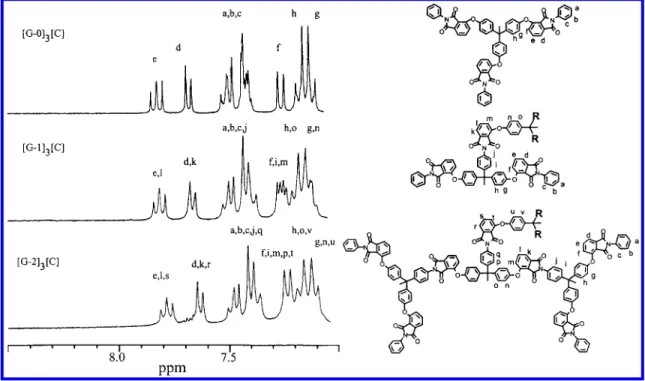
Attachment of the dendritic wedges to the core molecule was readily followed by the disappearance of the resonances for the protons of the nitro-substituted phthalic ring. Figure 2 shows the 1H NMR spectra of dendrimers [G-0]3[C], [G-1]3[C], and [G-2]3[C] shar-ing similar features. The peak assignment was based on the peak position of the dendritic intermediates and the auxiliary of 2D (H,H)-COSY spectra. The absence of resonances at region of 8.0-8.4 ppm for the aromatic protons of the nitro-substituted phthalic ring clearly demonstrated that there was no contamination of residual dendritic wedges. The protons of N-phenyl groups at the peripheries of the dendritic structures gave resonances in the region of 7.34-7.52 ppm. Reso-nances associated with the aromatic protons of the phthalimide rings in different layers were indistinguish-able. This result is consistent with the highly sym-metrical structures of ether-imide dendrimers. In addition, the methyl resonances of the core moiety and of the building blocks in the dendritic wedges were also indistinguishable and appeared as a broad signal at 2.19 ppm. Integration of these areas and comparison with each other confirms not only the generation number but also the number of phthalimide rings on the dendritic macromolecules. Information from 13C NMR spectra added further support for the proposed structure. FTIR spectroscopy also provided complementary evidence for the structures, showing characteristic imide carbonyl absorptions at 1725 and 1778 cm-1.21
Size-exclusion chromatography proved to be useful in the analysis of the purity and polydispersity of den-drimers. Since each generation of the ether-imide dendrimers was synthesized in a stepwise manner, each compound was predicted to be monodisperse. Figure 3 shows a composite of the SEC chromatograms of the various generations of dendritic poly(ether imide)s. All members of the series gave narrow, single-peak sym-metrically shaped chromatograms and showed no evi-dence of an unresolved shoulder. This result revealed that these dendritic macromolecules were pure and had a very narrow distribution of molecular weights. SEC measurements calibrated with narrow-dispersity poly-(methyl methacrylate) standards gave Mnand polydis-persity data for [G-1]3[C] (Mn) 1528 and PDI ) 1.06) and [G-2]3[C] (Mn) 3428 and PDI ) 1.06). Because Scheme 2a
aReagent: (i) 1,1,1-tris(4-hydroxyphenyl)ethane, NaOCH 3.
Figure 1. 300-MHz1H NMR spectra of [G-1]NH
2and
of the overlapping of the trace of [G-0]3[C] with the solvent peak, no SEC data for [G-0]3[C] was obtained. Mass spectrometry was also an effective tool for con-firming the identities and molecular weight distribu-tions of dendritic macromolecules.
Fast atom bombardment (FAB) mass spectrum of [G-0]3[C] established the molecular weight of this ether-imide dendrimer, with molecular ion base peak observed at m/z 970.2768 (calculated, 970.2764). How-ever, mass spectrometry using FAB ionization technique did not provide useful information on dendrimers [G-1]3 -[C] and [G-2]3[C], which have with molecular masses above 2000 Da, presumably because of their involatility. Matrix-assisted laser desorption ionization-time-of-flight mass spectrometry (MALDI-TOF MS) is an attractive alternative to mass analysis of the ether-imide dendrimers. It can provide higher mass determi-nations for intact molecular ions of nonvolatile species and has been explored intensely in recent investigations of dendrimers.8a,b,12b,14b,22 Figure 4 shows the MALDI
spectra of dendrimers [G-1]3[C] and [G-2]3[C]. The dominant signals occurred at m/z values 2973.4 and 6895.8 agreed very closely with calculated values 2973.2 and 6901.3 corresponding, respectively, to the potassium adducts of [G-1]3[C] and [G-2]3[C]. Thus, from the MALDI spectra it could be concluded that these ether-imide dendrimers are pure and monodisperse.
The thermal behavior of these dendritic poly(ether imide)s was evaluated by differential scanning calorim-etry (DSC). The glass transition temperature (Tg) was found to increase with increasing molecular weight. Tg's of dendrimers [G-0]3[C], [G-1]3[C] and [G-2]3[C] were observed at 138, 183, and 208 °C, respectively, where clear melting points were not observed. According to the free-volume theory, free volume around the polymer chain ends is greater than along the polymer chain, due to the poorer packing ability of the chain ends. For linear polymers, the chain-end free volume theory illustrates the dependency of glass transition
Figure 2. Comparison of 300-MHz1H NMR spectra in the region of 7.0-8.5 ppm for ether-imide dendrimers in DMSO-d 6.
Figure 3. SEC traces for ether-imide dendrimers. / signal
due to the solvent of the sample solution. Figure 4. MALDI-TOF mass spectra of [G-1]3[C] and
temperature on molecular weight.23For dendritic mac-romolecules, a new and modified version of the chain-end free volume theory has been derived to account for the large number of chain ends in highly branched structures.24It follows that T
g) Tg∞- K′/(ne/M), where
Tg∞is the value of Tgat infinite molecular weight, neis the number of chain ends, M is the molecular weight, and K′is a constant if the variation of terms such as the density or chain-end free volume with molecular weight is insignificant. It was observed that there was a linear dependence of Tgas a function of ne/M for these ether-imide dendrimers. The experimental variation in
Tg with molecular weight correlated well with the theoretical prediction.
The thermal stability of the dendritic poly(ether imide)s was examined by thermogravimetric analysis (TGA). Because of the aromatic structures, these ether-imide dendrimers were stable up to 430 °C, with a 5% and a 10% weight loss, respectively, at 458 and 487 °C for [G-0]3[C], at 453 and 481 °C for [G-1]3[C], and at 462 and 481 °C for [G-2]3[C]. The results indicate that the thermal stability of these dendrimers is independent of generation and comparable to that of linear poly(aryl ether imide)s (Mn) 10 100, PDI ) 2.2).25
The solubility of the dendrimers was tested in various organic solvents. Because of their highly branched structure, the dendrimers have enhanced solubility in organic solvents and are soluble in typical organic solvents such as dimethyl sulfoxides, amide solvents, tetrahydrofuran, chloroform, and methylene chloride. Summary
A series of dendritic poly(ether imide)s containing 3, 9, and 21 ether-imide units, respectively, was prepared by the stepwise convergent approach. The structural component that constituted the branching regions was derived from 1-(4-aminophenyl)-1,1-bis(4-hydroxyphen-yl)ethane (2), which contains two phenol groups and one aminophenyl group. Key steps of the synthetic sequence of the ether-imide dendrimers were the aromatic nucleophilic displacement of activated nitro groups, which formed the ether linkage and the condensation and cyclization of the aminophenyl group with 3-nitro-phthalic anhydride, which yielded the imide ring con-taining a reactive nitro functionality. Through aromatic nucleophilic substitution, the dendritic wedges with activated nitro group were coupled to the polyfunctional core, 1,1,1-tris(4-hydroxyphenyl)ethane, to form the dendritic macromolecules. Structures of the dendritic poly(ether imide)s were confirmed by1H NMR,13C NMR and FTIR spectroscopy as well as mass spectrometry. The variation of Tg with molecular weight, as deter-mined by DSC, correlated well with the theoretical prediction. The ether-imide dendrimers were soluble in various organic solvents and were thermally stable up to 430 °C. The extension of using 2, as a building block, in the stepwise convergent synthesis of dendritic poly(ether-imide)s with modifiable chain ends as well as in the one-step synthesis of hyperbranched poly(ether imide)s is in progress.
Acknowledgment. We would like to thank the National Science Council (ROC) (NSC 89-2113-M009-001) for financial support.
Supporting Information Available: Figures containing DSC thermograms and the linear plot of Tgas a function of ne/M for these dendrimers. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) (a) Tomalia, D. A.; Naylor, A. M.; Goddard, W. A., III. Angew. Chem., Int. Ed. Engl. 1990, 29, 138. (b) Mekelburger, H.-B.; Jaworek, W.; Vo¨gtle, F. Angew. Chem., Int. Ed. Engl. 1992, 31, 1571. (c) Fre´chet, J. M. J. Science 1994, 263, 1710. (d) Newkome, G. R., Ed. Advances in Dendritic Molecules; JAI Press: Greenwich, CT, 1994; Vol. 1. (e) Newkome, G. R.; Moorefield, C. N.; Vo¨gtle, F. Dendritic Molecules: Concepts, Syntheses, Perspectives; VCH: Weinheim, FRG, 1996. (f) Zeng, F.; Zimmerman, S. C. Chem. Rev. 1997, 97, 1681. (g) Frey, H.; Lach, C.; Lorenz, K. Adv. Mater. 1998, 10, 279. (h) Smith, D. K.; Diederich, F. Chem.sEur. J. 1998, 4, 1353.
(2) (a) Tomalia, D. A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. Polym. J. 1985, 17, 117. (b) Newkome, G. R.; Yao, Z.-Q.; Baker, G. R.; Gupta, V. K. J. Org. Chem. 1985, 50, 2003.
(3) (a) Hawker, C. J.; Fre´chet, J. M. J. J. Am. Chem. Soc. 1990, 112, 7638. (b) Xu, Z. F.; Moore, J. S. Angew. Chem., Int. Ed. Engl. 1993, 32, 1354. (c) Miller, T. M.; Neenan, T. X. Chem. Mater. 1990, 2, 346.
(4) (a) Padias-Buyle, A.; Hall, H. K., Jr.; Tomalia, D. A.; McCo-nnell, J. R. J. Org, Chem. 1987, 52, 5305. (b) Hawker, C. J.; Fre´chet, J. M. J. Macromolecules 1990, 23, 4726. (c) Wooley, K. L.; Hawker, C. J.; Fre´chet, J. M. J. J. Am. Chem. Soc.
1991, 113, 4252. (d) Liu, M.; Kono, K.; Fre´chet, J. M. J. J.
Polym. Sci., Part A: Polym. Chem. 1999, 37, 3492. (5) (a) Miller, J. M.; Kwock, E. W.; Neenan, T. X. Macromolecules
1992, 25, 3143. (b) Hawker, C. J.; Fre´chet, J. M. J. J. Chem.
Soc., Perkin. Trans. 1 1992, 2459. (c) Ihre, H.; Hult, A.; Fre´chet, J. M. J.; Gitsov, I. Macromolecules 1998, 31, 4061.
(6) (a) Newkome, G. R.; Behera, R. K.; Moorefield, C. N.; Baker, G. R. J. Org. Chem. 1991, 56, 7162. (b) Young, J. K.; Baker, G. R.; Newkome, G. R.; Morris, K. F.; Johnson, C. S., Jr. Macromoleules 1994, 27, 3464. (c) Bayliff, P. M.; Feast, W. J.; Parker, D. Polym. Bull. 1992, 29, 265.
(7) (a) Tomalia, D. A.; Berry, V.; Hall, M.; Hedstrand, D. M. Macromoleules 1987, 20, 1164. (b) Moreno-Bundi, M. C.; Orellana, G.; Turro, N. J.; Tomalia, D. A. Macromolecules
1990, 23, 910.
(8) (a) Lorenz, K.; Mu¨ lhaupt, R.; Frey, H.; Rapp, U.; Mayer-Posner, F. J. Macromolecules 1995, 28, 6657. (b) Krska, S. K.; Seyferth, D. J. Am. Chem. Soc. 1998, 120, 3604. (c) Kriesel, J. W.; Tilley, T. D. Chem. Mater. 1999, 11, 1190. (9) Uchida, H.; Kabe, Y.; Yoshino, K.; Kawmata, A.; Tsumuyama,
T.; Masamune, S. J. Am. Chem. Soc. 1990, 112, 7077. (10) Hu, J.; Son, D. Y. Macromolecules 1998, 31, 8644.
(11) Miller, T. M.; Neenan, T. X.; Zayas, R.; Bair, H. E. J. Am. Chem. Soc. 1992, 114, 1018.
(12) (a) Moore, J. S.; Xu, Z. Macromolecules 1991, 24, 5893. (b) Kawaguchi, T.; Walker, K. L.; Wilkins, C. L.; Moore, J. S. J. Am. Chem. Soc. 1995, 117, 2159.
(13) Louie, J.; Hartwig, J. F.; Fry, A. J. J. Am. Chem. Soc. 1997, 119, 11695.
(14) (a) Morikawa, A.; Kakimoto, M.; Imai, Y. Macromolecules
1993, 26, 6324. (b) Morikawa, A.; Ono, K. Macromolecules 1999, 32, 1062.
(15) Williams, F. J.; Donahue, P. E. J. Org. Chem. 1977, 42, 3414. (16) White, D. M.; Takekoshi, T.; Williams, F. J.; Relles, H. M.; Donahue, P. E.; Klopfer, H. J.; Louks, G. R.; Manello, J. S.; Matthews, R. O.; Schluenz, R. W. J. Polym. Sci., Polym. Chem. Ed. 1981, 19, 1635.
(17) Morgan, P. W. Macromolecules 1970, 3, 536.
(18) Hall, H. K., Jr.; Polis. D. W. Polym. Bull. 1987, 17, 409. (19) A mixture of 3-nitro-N-phenylphthalimide and
1-(4-ami-nophenyl)-1,1-bis(4-benzyloxyphenyl)ethane, which is a de-rivative of 2 with the phenol groups being protected in DMF was heated at 60 °C for 24 h, and no reaction product was observed. This result indicated that as the nucleophilic aromatic substitution was carried out in DMF at 60 °C, the nitro-substituted phthalimide was attacked exclusively by the phenoxide groups of 2, while the amino group of 2 remained intact.
(20) Takekoshi, T. Other Synthetic Routes to Polymides. In Polymides; Wilson, D.; Stenzenberger, H. D.; Hergenrother, P. M., Eds.; Blackie, Chapman and Hall: New York, 1990; Chapter 2.
(21) Hsiao, S.-H.; Li, C.-T. J. Polym. Sci., Part A: Polym. Chem.
1999, 37, 1403.
(22) (a) Walker, K. L.; Kahr, M. S.; Wilkins, C. L.; Xu, Z.; Moore, J. S. J. Am. Soc. Mass Spectrom. 1994, 5, 731. (b) Newkome, G. K.; Guther, R.; Moorefield, C. N.; Gardullo, F.; Echegoyen, L.; Pe´rez-Cordero, E.; Luftmann, H.; Angew. Chem., Int. Ed. Engl. 1995, 34, 2023. (c) Leon, J. W.; Kawa, M.; Fre´chet, J. M. J. J. Am. Chem. Soc. 1996, 118, 8847. (d) Wu, Z. C.;
Biemann, K. Int. J. Mass Spectrom. 1997, 165, 349. (e) Yu, D.; Valdimirov, N.; Fre´chet, J. M. J. Macromolecules 1999, 32, 2, 5186.
(23) (a) Turner, D. T. Polymer 1978, 19, 789. (b) Aklonis, J. J.; MacKnight, W. J. Introduction to Polmer Viscoelascity, 2nd ed.; John Wiley & Sons: New York, 1983; pp 78-79. (24) Wooley, K. L.; Hawker, C. J.; Pochan, J. M.; Fre´chet, J. M.
J. Macromolecules 1993, 26, 1514.
(25) Facinelli, J. V.; Gardner, S. L.; Dong, L.; Sensenich, C. L.; Davis, R. M.; Riffle, J. S. Macromolecules 1996, 29, 7342.
![Figure 1. 300-MHz 1 H NMR spectra of [G-1]NH](https://thumb-ap.123doks.com/thumbv2/9libinfo/7935493.157359/4.918.495.816.68.335/figure-mhz-h-nmr-spectra-of-g-nh.webp)