Editorial Manager(tm) for Medical & Biological Engineering & Computing Manuscript Draft
Manuscript Number: MBEC1712R3
Title: Characterization and Prediction of mRNA Polyadenylation Sites in Human Genes Article Type: Original article
Keywords: bioinformatics data mining
polyadenylation poly(A)
support vector machines (SVMs)
Corresponding Author: Jorng-Tzong Horng, PhD Corresponding Author's Institution:
First Author: Tzu-Hao Chang
Order of Authors: Tzu-Hao Chang;Li-Ching Wu;Yu-Ting Chen;Hsien-Da Huang;Baw-Jhiune Liu;Kuang-Fu Cheng;Jorng-Tzong Horng, PhD
Abstract: The accurate identification of potential poly(A) sites has contributed to all many studies with regard to alternative polyadenylation. The aim of this study was the development of a
machine-learning methodology that will help to discriminate real polyadenylation signals from randomly occurring signals in genomic sequence. Since previous studies have revealed that RNA secondary structure in certain genes has significant impact, we tried to computationally pinpoint common structural patterns around the poly(A) sites and to investigate how RNA secondary structure may influence polyadenylation. This involved an initial study on the impact of RNA structure and it was found using motif search tools that hairpin structures might be important. Thus, we propose that, in addition to the sequence pattern around poly(A) sites, there exists a widespread structural pattern that is also employed during human mRNA polyadenylation. In this study, we present a computational model that uses support vector machines (SVMs) to predict human poly(A) sites. The results show that this predictive model has a comparable performance to the current prediction tool. In addition, we identified common structural patterns associated with polyadenylation using several motif finding programs and this provides new insight into the role of RNA secondary structure plays in
polyadenylation.
Response to Reviewers: <COMMENTS FOR THE AUTHOR> editor in chief:
The manuscript improved in structure. However, as clearly stated in the instructions to authors we allow only a total of 8 figures and tabels combined.
You have 8 tables and 11 figures. This will take to much space in the journal which has a limited page allowance with the publisher.
As exception i will allow you a total of 10 (figures plus tables).
Please also read the instructions to authors concernig page charges for overpages papers. You must be willing and able to pay those charges in case your paper will be over 8 printed pages. Only in rare cases I may be able to compromise on that.
---
We revise this manuscript based on the reviewers' comments. 1. The abstract is reduced to 200 words.
2. The introduction is reduced to 1 page. We use the introduction to formulate shortly the background and the objectives.
3. The heading material and methods is changed into : Methods.
4. We reorganize the discussion. The discussion starts out with the major finding. 5. The manuscript is reduced to 8 pages, 5 tables and 4 figures.
1
Characterization and Prediction of mRNA Polyadenylation
Sites in Human Genes
Tzu-Hao Chang
1,Li-Ching Wu
2,
Yu-Ting Chen
1, Hsien-Da Huang
3, Baw-Jhiune Liu
4,
Kuang-Fu Cheng
5, and Jorng-Tzong Horng
1,2,6,*1Department of Computer Science and Information Engineering, National Central University, Taiwan; 2
Institute of Systems Biology and Bioinformatics, National Central University, Taiwan; 3Department of Biological Science and Technology, Institute of Bioinformatics, National Chiao-Tung University, Taiwan;
4Department of Computer Science and Information Engineering, Yuan Ze University, Taiwan;
5Biostatistics Center and Department of Public Health, and Graduate Institute of Statistics, China
Medical University; 6Department of Bioinformatics, Asia University, Taiwan
*To whom correspondence should be addressed: JT Horng: Email: [email protected]
ABSTRACT
The accurate identification of potential poly(A) sites has contributed to all many studies with regard to alternative polyadenylation. The aim of this study was the development of a machine-learning methodology that will help to discriminate real polyadenylation signals from randomly occurring signals in genomic sequence. Since previous studies have revealed that RNA secondary structure in certain genes has significant impact, we tried to computationally pinpoint common structural patterns around the poly(A) sites and to investigate how RNA secondary structure may influence polyadenylation. This involved an initial study on the impact of RNA structure and it was found using motif search tools that hairpin structures might be important. Thus, we propose that, in addition to the sequence pattern around poly(A) sites, there exists a widespread structural pattern that is also employed during human mRNA polyadenylation. In this study, we present a computational model that uses support vector machines (SVMs) to predict human poly(A) sites. The results show that this predictive model has a comparable performance to the current prediction tool. In addition, we identified common structural patterns associated with polyadenylation using several motif finding programs and this provides new insight into the role of RNA secondary structure plays in polyadenylation.
1 INTRODUCTION
The polyadenylation of mRNA is an essential cellular process by which most eukaryotic pre-mRNAs form their 3’ ends (histone mRNAs are the major exceptions). Previous studies have indicated that several cis elements of great importance participate in signaling most events of human polyadenylation. In general, there are two core elements essential for polyadenylation. One is the highly conserved AAUAAA hexamer (or a close variant), which is usually referred to as the polyadenylation signal (PAS) and is located 10-40 nucleotides (nt) upstream of the poly(A) site [1-4]. The other element is often referred to as a poorly conserved GU- or U-rich downstream element and is located 20-40 nt downstream of the poly(A) sites [4-5].
Traditional bioinformatics collects a large amount of cDNA sequences and Expressed Sequenced Tags (ESTs) with the aim of aligning the cDNA/ESTs and the genome sequence [4, 6-7]. This has provided a systematic approach to the identification of poly(A) sites in genomes. A substantial amount of data generated computationally via cDNA/ESTs alignment is considered valid and, consequently, the dataset serves as an excellent resource for the studies related to polyadenylation machinery. The prediction of poly(A) sites takes advantage of cDNA/ESTs and because of their availability on a large scales, this approach has became practical. In early studies, the problem of poly(A) site prediction was transformed to the identification of a putative polyadenylation signal, which was thought to be primarily defined by the location of the poly(A) sites [5, 8]. Since PASes are highly conserved elements in the region upstream of a poly(A) sites, a correctly identified PAS indicates that a real poly(A) sites is not far away. In view of this, recognition of PAS is considered to be an alternative solution to solve the problem of poly(A) site prediction. Reliable prediction of poly(A) sites plays a enhancing role in the exploration of the complex mechanism of alternative polyadenylation, since it involves the identification of cis elements and characterization of the flanking regions. The information revealed via prediction can be of great value when studying the mechanisms involved in polyadenylation as well as how gene regulation occurs due to alternative polyadenylation. The objective of this study was the development of a machine-learning
Manuscript
methodology that will help to discriminate real polyadenylation signals from randomly occurring signals in genomic sequence.
2
METHODS
2.1 Datasets
In this study, we used a large number of positive sequences and negative sequences to train and test our model. A positive sequence is the human genomic sequence surrounding a poly(A) site. All the positive sequences were obtained from the PolyA_DB 2 database [9], which contains poly(A) sites identified for genes from several vertebrate species using alignments between cDNA/ESTs and the genome sequences. We retrieved 33745 positive sequences from PolyA_DB 2 in total, which correspond to 14078 human genes. Each positive sequence is 250nt in length, spanning -125 to +125 nt relative to the poly(A) site. A sequence was defined as a single-site type if its associated poly(A) site was unique, otherwise it was be defined as a multiple-site type. Among all the positive sequences, 5275 sequences are denoted as single-site type and 28470 sequences were denoted as multiple-site type. In addition, we obtained 2327 sequences from the Erpin training data [5] to perform an independent test. Each of these sequences is 200nt in length with a candidate PAS in the middle. To test our model, we prepared several types of negative sequences, that is sequences without real poly(A) sites. Each of negative sequences is also 250nt in length. The negative sets we used 6000 sequence included randomized poly(A) regions (produced by randomizing the sequence surrounding a poly(A) site), 313454 human mRNA coding sequences (CDS), 25700 human 5’-untranslated regions (5’-UTRs) and randomized genome sequences. The human
RefSeq mRNA coding sequences were obtained from NCBI Build 36 [10]
(ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/). The 5’-untranslated regions were downloaded from UTRdb release 22 [11] (http://www.ba.itb.cnr.it/UTR/). The chromosome 1 sequence of the human genome (hg17 version) was downloaded from the UCSC genome bioinformatics site (http://genome.ucsc.edu). We also generated randomized sequences of same first order Markov model as human CDS, 5’-UTRs and chromosome 1 sequence of human genome.
2.2 Test procedure
In this study, we tested our prediction model using all the positive sequences and all the categories of negative sequences, and compared its performance with polya_svm [12], the most current tool for poly(A) site prediction. It must be noted that our prediction model uses PAS location for prediction while polya_svm predicts the location of a potential poly(A) site directly. Therefore, the procedure used to evaluate our model and polya_svm should be clearly defined. Since our model could easily reject a sequence without a PAS, this would cause a large number of true negative or false negative predictions depending on the testing data. To avoid possible bias relative to the testing data, only sequences with PASes were taken into account. For positive and negative sequences, we filtered out those without PASes through the simple approach of putative PAS detection, which will be illustrated later. The testing data we used is shown in Table 3. A total of 27573 sequences (4908 single type, 22665 multiple-type) were detected to have putative PASes. For each negative set, 500 sequences were randomly selected (from 14958 Poly(A) region sequences randomized by 1st order Markov chain, 45203 CDS, 16368 CDS randomized by 1st order Markov chain, 3156 5’-UTR sequences, 4645 5’-UTR sequences randomized by 1st order Markov chain, 10113 Genomic sequences randomized by 1st order Markov chainand) predicted by our model and polya_svm, which was repeated 10 times to calculate mean values. Predictive accuracy was then measured as follows: Sensitivity: SN=(TP/(TP+FN)), Specificity:
SP=(TP/(TP+FP)) Correlation Coefficient:
(TP
FP)(TP
FN)(TN
FP)(TN
FN)
FN
FP
TN
TP
CC
where TP is true positive, TN is true negative, FN is false negative and FP is false positive.
Due to the differences between the predictive models, for polya_svm, a prediction was considered to be TP if the reported site is within 24 nt of a real poly(A) site, and is otherwise FN. For our model, a sequence was predicted as positive if a PAS is detected within 40 nt upstream of a real poly(A) site according to a previous study [4]. To yield Specificity, the number of TP derived from all of the positive testing set was scaled so that the size of the positive testing set was equal to the size of negative testing set.
2.3 Detection of candidate PASes
To prepare for training and testing, only the sequences with candidate PASes were retained. We referred to previous studies and selected frequently occurring PASes [2, 4]. The candidate PASes consist of the
3 canonical AAUAAA hexamer, other 11 single-base variants of AAUAAA and one two-base variant of AAUAAA. A sequence having any of the 13 hexamers within 40 nt upstream of a poly(A) site was retained, otherwise the sequence was discarded. To this end, we implemented a filter to sequentially detect these 13 types of candidate PAS; they are shown in Table 1 in terms of their frequencies. For single-type poly(A) sites, ~70% of them have AAUAAA, 14% have AUUAAA, ~12% have one of the other 11 types of PAS hexamers and ~7% of them do not have any known PAS. The pattern of PAS usage is consistent with observation reported in previous studies [4-5] and the ranking is approximately the same. Thus, in this study, we focused on the detection of these 13 hexamers, which are found in 81.7% of all poly(A) sites.
2.4 Extraction of k-mer features
For the upstream and downstream regions of a candidate PAS, the features we used are occurrences of k-length contiguous subsequences, that is k-mers [13]. In this study, we used k-mer nucleotide patterns (k = 1,2,3) as our features, each of which has a frequency value. The same patterns, but appearing on different sides of a candidate PAS, were treated as two distinct features. For example, the frequency of GC, a 2-mer pattern, should be counted separately when upstream and when downstream. Thus, a total
number of 168 (= (
2 3
4 4
4
2
)) possible words, that is features, were used in the first training stage. For a sequence with a candidate PAS, we retrieved 78 bases upstream as well as 78 bases downstream of the PAS for generation of the k-mer features.
2.5 Characterization of the sub-regions around the PAS
The nucleotide composition of positive sequences as well as negative sequences was examined in order to characterize the sub-regions upstream and downstream of the polyadenylation signals. The nucleotide frequencies were visualized at each position in order to highlight regions that can significantly discriminate real PASes from look-alikes.
2.6 Detection of the core elements involved in the RNA secondary structure
To discover the structural patterns around poly(A) sites, we used several well-known motif finding programs, including Sfold [14], RNAfold [15] and RNAMotif [16], to identify possible RNA secondary structures that may be involved in polyadenylation. Based on previous observations in the literature, we assumed that there exists simple structures that flank the poly(A) sites and are to a certain extent currently unknown. Thus, we focused on a simple hairpin structure that contains a PAS in its loop or has a U-rich stem; this was because stem-loop structures commonly define protein-RNA binding sites. 2.7 Machine learning
In this study, we used support vector machines (SVMs) as the machine learning method. As state-of-the-art classifiers, SVMs have been shown to have excellent empirical performance in prediction tasks. In addition, machine learning via SVMs is known to achieve good performance when identifying biological signals, such as translation initiation sites [17] and splice sites [18-20]. Thus, we used the SVM library LIBSVM for binary classification (http://www.csie.ntu.edu.tw/~cjlin/libsvm) in which the C-support vector classification (C-SVC) method and the radial basis kernel function (RBF) were applied at the default settings, i.e. cost = 1 and gamma = 1/15.
2.8 Integration of the different types of features
We designed a predictive model that was constructed using two SVMs. In this model, the first SVM employs k-mer features (k = 1,2,3) and outputs a probability value, which serves as an input value for the second SVM. The second SVM employs the contents of the characteristic sub-regions as features, which will be mentioned in Results. To train our model, we randomly selected 4000 positive sequences in addition to 6000 negative sequences from the six types of negative set (1000*6).
3
RESULTS
3.1 Characterization of the polyadenylation signals
First, we examined the nucleotide composition of the genomic sequences of the single-type and multiple-type poly(A) sites. For each poly(A) site, we selected terminal sequences spanning -125 to +125 nt surrounding the poly(A) site (Fig. 1(a) and Fig. 1(b)). Both types of poly(A) sites have similar patterns in the -35 to +35 region, in which the curve for each nucleotide acid reveals quick rises and falls. Upstream of the poly(A) site, an A-rich region is located from -25 to -15 and causes a drop in U-content (%U); this is closely followed by a U-rich region (-15 to -5). Downstream of the poly(A) site, there is a visible rise in %U with a peak at around +20; this spans a wider region and is closely mirrored by a
decline in %A. This U-rich region is generally regarded as the area containing CstF-binding sites. Meanwhile, a sudden rise in %A at -1 indicates that cleavage preferentially occurs next to an Adenine. When multiple-type poly(A) sites are compared with single-type poly(A) sites, the difference between the AU- and GC-ratio is larger at almost each position in the vicinity of the former type of site with the exception of the cleavage site and the region containing PAS hexamers, which are both shown to be highly conserved (Fig. 1(c)).
As the next step, in order to discriminate positive sequences from the various categories of negative sequences, we analyzed nucleotide composition in each type. As Fig. 3(a) shows, the Adenine peak at around position 1 corresponds to the upstream A-rich region in Fig. 1(a) and Fig. 1(b). Similar peaks are found in negative sequences, especially those from CDS and hs_MC; however, these seem to reflect how often a randomly occurring PAS hexamer is found within an A-rich region. There is a decline in %A in the downstream region, which corresponds to the U-rich region (Fig. 3(b)), and this can help significantly to discriminate real sequences from negative sequences. In addition, we noticed a minor U-rich region was located between the PAS and the major U-rich region and that this results in a lower %C and %G relative to the whole sequence, as shown in Fig. 3(c) and Fig. 3(d).
To summarize, we identified the characteristics of polyadenylation signals as made up of the following sub-regions, which are shown in Fig. 2:
(1) A non-G-rich region, spanning -20 to +20 across the PAS. (2) A major U-rich region, spanning +20 to +45.
(3) A minor U-rich region, spanning +3 to +12. (4) A non-C-rich region, spanning +6 to +15. (5) A non-A-rich region, spanning +17 to +55. Note that the positions described are relative to the PAS.
The content of these five sub-regions, for example, the G-richness in the non-G-rich regions, was used by our SVM model for prediction.
3.2 Prediction of poly(A) sites by the SVM
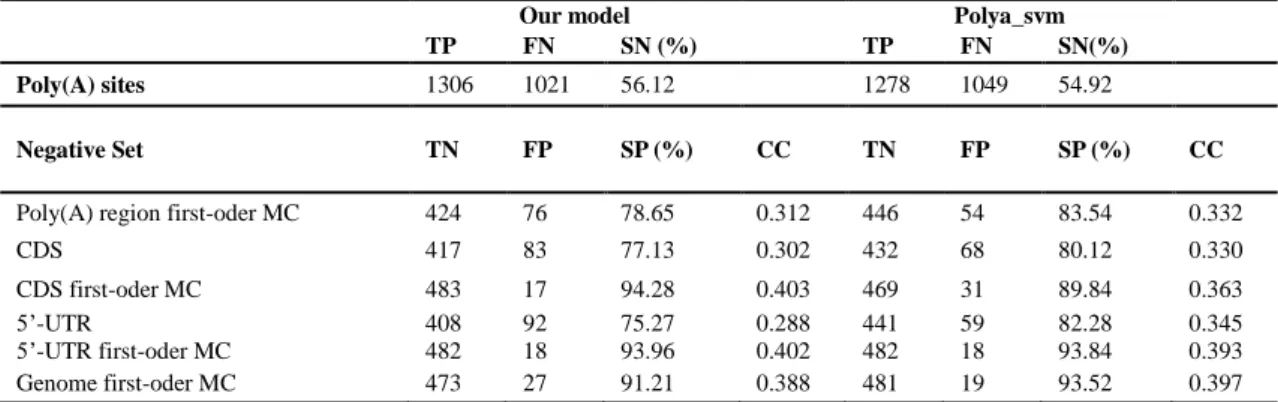
We conducted an independent test using 2327 positive sequences and six types of negative sequences as previously described in Materials and Methods. For each negative set, 500 sequences were generated and predicted by our model and polya_svm version 1.1 using the default settings. The process was repeated 10 times and mean values are presented. As shown in Table 2, our model is more sensitive than polya_svm, but only by a small amount. Comparable false positive (FP) levels were predicted by our model and by polya_svm for the randomized sequences. Using most types of randomized sequences, our model showed a high Specificity and Correlation Coefficient, the exception being randomized poly(A) region sequences. Interestingly, our model outperforms polya_svm when randomized CDS and randomized 5’-UTRs are used but shows an unexpected difference with real CDS and 5’-UTRs, which requires further discussion.
3.3 RNA secondary structure
Here, we firstly tested the hypothesis that it is RNA secondary structures that make a real polyadenylation signal what it is, one key factor being recognition by the CPSF. To this end, several computer programs were used to pinpoint possible secondary structures. We used RNAfold [15] with default parameters for the structure folding. Fig. 4 (a)shows the probability distribution at each site along the -40/+40 region of PAS. In contrast with CDS, the result suggests that the PAS hexamers in the poly(A) sites and 5’-UTRs have a high probability of being involved in a single-stranded structure, for instance, lying in a loop. This result was then verified using Sfold [14] with default setting to assess statistical folding. As shown in Fig. 4 (b), we found that the AAUAAA hexamers (the middle of the sequence) tend to be unpaired when sequences were compared across all datasets, including single-type poly(A) sites, CDS and 5’-UTRs. The same pattern was revealed when window sizes of 2 nt and 4 nt were used (data not shown).
Based on the above, it seems likely that polyadenylation signals may stay unpaired during processing if no other factors interfere. However, the results for CDS and 5’-UTRs showed that this property did not distinguish positive sequences from negative ones. Consequently, we turned the spotlight on the downstream U-rich region and used RNAMotif [16], which is a common RNA secondary structure search program. In this test, we focused on simple hairpin structures in which the loop and the stem both have a flexible length of 6 to 10. G:U pairing was permitted in the stem in addition to the default Watson/Crick paring rule. Mispairs are allowed in the stem, with the base-paired limit set at 80% for the stem. Based on the positions in the literature, we counted the occurrence of PAS hexamers being entirely
5 in a loop or just being part of it. As Table 3 shows, for example, the value 50.16 represents 50.16% of AAUAAA hexamers in poly(A) sites being present in the loop and therefore the results suggests that there is no obvious preference for AAUAAA and AUUAAA to lie in the loop. Again, we found a high percentage was found in negative sequences, especially randomized poly(A) region sequences.
For the downstream U-rich regions in the stem, we tried several Fig.s and eventually choose to set the threshold of U-richness at 60%. As shown in Table 4, about 60% of real polyadenylation signals have downstream U-rich regions that form the stems of hairpins and this is a relatively high correlation compared to other negative sequences. The value of Diff varies form ~10% to ~43%. We supposed such differences are mainly due to the U-content downstream of the different types of sequences.
Finally, we explored the interaction between the two cases, that is, we identified those poly(A) sites with a PAS hexamer involved in the loop of one hairpin structure and a downstream U-rich region forming the stem of another loop. Such an arrangement is exemplified by the SV40 L polyadenylation signal [21]. As a result, out of 27573 sequences there were 8977 matches, which is approximately one-third of the sequences that have a PAS. When examined in detail, most of the matches are found to be multiple-type poly(A) sites (
Table 5). Given that the number of multiple-type sites is five times that of single-type sites, the association with this structure can not be inferred rigorously. For genes related to those matches, we observed a 46% coverage of 13756 human genes, which suggests that such a structural pattern might commonly exist around poly(A) sites. In addition, preference in usage could be found in multiple-type genes and this could be associated with the role RNA structure plays in the selection of multiple poly(A) sites.
4
DISCUSSION
In order to discriminate real polyadenylation signals from false ones, the characteristic of both the whole 3’-UTR region and in its sub-regions motifs are curial. The traditional conserved AAUAAA PAS hexamer located 10-40 nucleotides (nt) upstream of the poly(A) site [1-4] is not the only factor. Other features such as structure and small sequence variants are also important. In our predictive model, we took into account not only the general AU-rich environment around the poly(A) site but also the characteristic sub-regions, which reveal significant positional dependency. In a manner consistent with previous findings, those sub-regions are supposed to harbor simple but important cis elements. A notable example is the specific AU-rich elements known as AREs, which represent the most common determinant of RNA stability in mammalian cells [22-23]. Our predictive model was found to be comparable to the most current prediction tool, polya_svm [12] and may have in many cases a higher sensitivity and specificity depending on the context of sequences in evalutaion. It is noteworthy that when testing with CDS and 5’UTRs, both our model and polya_svm predicted a surprising number of false positives, but this was not the case with randomized CDS and 5’-UTRs, where both showed excellent specificity. To explain this result, we presume that a large number of “real sites” might in fact exist that are capable of satisfying the feature definitions of our predictive model and of polya_svm. For CDS, this would be consistent with previous findings that there are poly(A) sites in internal exons [4, 7]. On the other hand, the false positives in the 5’-UTRs may actually act as some form of regulatory element. However, this hypothesis will require considerable experimental evaluation to assess its validity.
The prediction result indicates that other unidentified features, such as RNA structures, may account for polyadenylation activity among the false negative sequences. In our analysis of RNA secondary structure, it seems that PAS hexamers tend to be in single-stranded form and unlikely to be affected by surrounding sequences. Notably, the U-rich region downstream of polyadenylation signals probable form the stem of a simple hairpin structure, which may be regarded as a feature that will be able to improve the prediction of poly(A) sites in the future. In the last test, it was found that 32% of poly(A) sites with PAS hexamers have their PAS and downstream U-rich region involved in two separate hairpins. Overall, we found that 46% of 13756 human genes would seem to have this structural pattern around their poly(A) sites; clearly this might be related to functionality. Based on these observations, we suggest that this simple hairpin structure is common across human genes and such this structural pattern could be one of a number of functional RNA structures associated with polyadenylation. Since this structural pattern has not been pinpointed as important in the past, an extensive study is needed to delineate the significance of hairpin structures during mRNA polyadenylation. We hope our present study has shed some light on the role that common RNA structures play in the complex mechanism of polyadenylation.
In most cases, the PAS serves as the binding site for the CPSF as soon as it is transcribed, while the GU- or U–rich element is bound by the CstF. There may be multiple GU/U–rich downstream elements
associated with a single poly(A) site, suggesting configuration may control the efficiency of polyadenylation [21]. When bound, cooperation between the CstF and the CPSF produces a greatly enhanced binding to the pre-mRNA substrate, because a weak interaction of the PAS with a CPSF can be compensated for by a strong interaction of the GU/U –rich element with a CstF, and vice versa [24-25]. Several human disease have been reported to be caused by a malfunction of polyadenylation. The system involved include simian virus 40 (SV40), human immunodeficiency virus type 1 (HIV-1), human C2 complement, collagen and cyclooxygenase-2 [26-32]. One examples is the FOXP3 gene, where a point mutation with a polyadenylation signal (AAUAAA to AAUGAA) can lead to IPEX syndrome [33]. Furthermore, some diseases may be ascribed to an abnormal level of mRNA 3’end formation during the process of polyadenylation, such as hereditary thrombophilia [34]. We developed a comprehensive methodology for human poly(A) site prediction in this study and we hope our study assist the current understanding of features related to the polyadenylation.
REFERENCES
1. Graber, J.H., et al., In silico detection of control signals: mRNA 3'-end-processing sequences in diverse species. Proc Natl Acad Sci U S A, 1999. 96(24): p. 14055-60.
2. Beaudoing, E., et al., Patterns of variant polyadenylation signal usage in human genes. Genome Res, 2000. 10(7): p. 1001-10.
3. MacDonald, C.C. and J.L. Redondo, Reexamining the polyadenylation signal: were we wrong about AAUAAA? Mol Cell Endocrinol, 2002. 190(1-2): p. 1-8.
4. Tian, B., et al., A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res, 2005. 33(1): p. 201-12.
5. Legendre, M. and D. Gautheret, Sequence determinants in human polyadenylation site selection. BMC Genomics, 2003. 4(1): p. 7.
6. Brockman, J.M., et al., PACdb: PolyA Cleavage Site and 3'-UTR Database. Bioinformatics, 2005. 21(18): p. 3691-3. 7. Yan, J. and T.G. Marr, Computational analysis of 3'-ends of ESTs shows four classes of alternative polyadenylation in human,
mouse, and rat. Genome Res, 2005. 15(3): p. 369-75.
8. Tabaska, J.E. and M.Q. Zhang, Detection of polyadenylation signals in human DNA sequences. Gene, 1999. 231(1-2): p. 77-86.
9. Lee, J.Y., et al., PolyA_DB 2: mRNA polyadenylation sites in vertebrate genes. Nucleic Acids Res, 2007. 35(Database issue): p. D165-8.
10. Pruitt, K.D. and D.R. Maglott, RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res, 2001. 29(1): p. 137-40.
11. Mignone, F., et al., UTRdb and UTRsite: a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res, 2005. 33(Database issue): p. D141-6.
12. Cheng, Y., R.M. Miura, and B. Tian, Prediction of mRNA polyadenylation sites by support vector machine. Bioinformatics, 2006. 22(19): p. 2320-5.
13. Liu, H., et al., An in-silico method for prediction of polyadenylation signals in human sequences. Genome Inform, 2003. 14: p. 84-93.
14. Ding, Y., C.Y. Chan, and C.E. Lawrence, Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res, 2004. 32(Web Server issue): p. W135-41.
15. Hofacker, I.L., Vienna RNA secondary structure server. Nucleic Acids Res, 2003. 31(13): p. 3429-31.
16. Macke, T.J., et al., RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res, 2001. 29(22): p. 4724-35.
17. Zien, A., et al., Engineering support vector machine kernels that recognize translation initiation sites. Bioinformatics, 2000.
16(9): p. 799-807.
18. Zhang, M.Q., Discriminant analysis and its application in DNA sequence motif recognition. Brief Bioinform, 2000. 1(4): p. 331-42.
19. Zhang, X.H., et al., Sequence information for the splicing of human pre-mRNA identified by support vector machine classification. Genome Res, 2003. 13(12): p. 2637-50.
20. Yeo, G., et al., Variation in alternative splicing across human tissues. Genome Biol, 2004. 5(10): p. R74.
21. Zarudnaya, M.I., et al., Downstream elements of mammalian pre-mRNA polyadenylation signals: primary, secondary and higher-order structures. Nucleic Acids Res, 2003. 31(5): p. 1375-86.
22. Shaw, G. and R. Kamen, A conserved AU sequence from the 3' untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell, 1986. 46(5): p. 659-67.
23. Chen, C.Y. and A.B. Shyu, AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci, 1995. 20(11): p. 465-70.
24. Wahle, E., 3'-end cleavage and polyadenylation of mRNA precursors. Biochim Biophys Acta, 1995. 1261(2): p. 183-94. 25. Colgan, D.F. and J.L. Manley, Mechanism and regulation of mRNA polyadenylation. Genes Dev, 1997. 11(21): p. 2755-66. 26. Carswell, S. and J.C. Alwine, Efficiency of utilization of the simian virus 40 late polyadenylation site: effects of upstream
sequences. Mol Cell Biol, 1989. 9(10): p. 4248-58.
27. Brown, P.H., L.S. Tiley, and B.R. Cullen, Efficient polyadenylation within the human immunodeficiency virus type 1 long terminal repeat requires flanking U3-specific sequences. J Virol, 1991. 65(6): p. 3340-3.
28. Valsamakis, A., et al., The human immunodeficiency virus type 1 polyadenylylation signal: a 3' long terminal repeat element upstream of the AAUAAA necessary for efficient polyadenylylation. Proc Natl Acad Sci U S A, 1991. 88(6): p. 2108-12. 29. Moreira, A., et al., Upstream sequence elements enhance poly(A) site efficiency of the C2 complement gene and are
phylogenetically conserved. EMBO J, 1995. 14(15): p. 3809-19.
30. Arhin, G.K., et al., Downstream sequence elements with different affinities for the hnRNP H/H' protein influence the processing efficiency of mammalian polyadenylation signals. Nucleic Acids Res, 2002. 30(8): p. 1842-50.
31. Natalizio, B.J., et al., Upstream elements present in the 3'-untranslated region of collagen genes influence the processing efficiency of overlapping polyadenylation signals. J Biol Chem, 2002. 277(45): p. 42733-40.
7 32. Hall-Pogar, T., et al., Alternative polyadenylation of cyclooxygenase-2. Nucleic Acids Res, 2005. 33(8): p. 2565-79. 33. Bennett, C.L., et al., A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA-->AAUGAA) leads to the IPEX
syndrome. Immunogenetics, 2001. 53(6): p. 435-9.
34. Gehring, N.H., et al., Increased efficiency of mRNA 3' end formation: a new genetic mechanism contributing to hereditary thrombophilia. Nat Genet, 2001. 28(4): p. 389-92.
Table 1. Top detected PAS hexamers
Frequency (%) Rank Single Multiple Hs Hs Hs.B Hs.B* AAUAAA 67.00 41.52 45.51 1 1 1 AUUAAA 14.14 14.57 14.51 2 2 2 UAUAAA 2.26 4.21 3.91 3 3 3 AGUAAA 2.60 3.49 3.35 4 4 4 AAGAAA 0.74 3.01 2.65 5 10 5 AAUAUA 1.00 2.13 1.96 7 5 6 AAUACA 1.19 2.02 1.89 8 8 7 CAUAAA 1.23 1.67 1.60 9 6 8 GAUAAA 0.89 1.50 1.40 10 7 9 AAUGAA 0.64 1.54 1.40 11 11 10 UUUAAA 0.74 2.39 2.13 6 9 11 ACUAAA 0.28 0.91 0.81 12 13 12 AAUAGA 0.32 0.64 0.60 13 12 13 coverage 93.04 79.61 81.71
Human sequences located -40 to -1 nt upstream of poly(A) sites were used to detect hexamers that may function as polyadenylation signals. Single, single-type poly(A) sites; Multiple, multiple-type poly(A) site; Hs, all human poly(A) sites in our material; Hs.B, human result reported by Beaudoing et al. [2]; Hs.B*, human result reported by Tian et al. [4].
Table 2. Comparison of our predictive model with the polya_svm approach
Our model Polya_svm
TP FN SN (%) TP FN SN(%)
Poly(A) sites 1306 1021 56.12 1278 1049 54.92
Negative Set TN FP SP (%) CC TN FP SP (%) CC
Poly(A) region first-oder MC 424 76 78.65 0.312 446 54 83.54 0.332
CDS 417 83 77.13 0.302 432 68 80.12 0.330
CDS first-oder MC 483 17 94.28 0.403 469 31 89.84 0.363
5’-UTR 408 92 75.27 0.288 441 59 82.28 0.345
5’-UTR first-oder MC 482 18 93.96 0.402 482 18 93.84 0.393 Genome first-oder MC 473 27 91.21 0.388 481 19 93.52 0.397 MC, Markov chain. Poly(A) region first-oder MC, randomized sequences surrounding poly(A) sites; CDS, coding region sequences; CDS first-oder MC, randomized CDS; 5’-UTR, 5’-UTR sequences; 5’-UTR first-oder MC, randomized 5’-UTRs. TP, true positives; FP, false positives; TN, true negatives; FP, false positives. SN, sensitivity; SP, specificity; CC, correlation coefficient.
Table 3. Statistics of PAS involvement in hairpin loops for the different types of sequences
Percentage (%) AAUAAA AUUAAA Other 11 types All types
all_hs 50.16 53.99 50.08 50.82 hsCDS 37.07 44.01 40.23 40.23 hsCDS_MC 33.35 39.55 39.03 38.42 hs_MC 45.28 49.57 48.82 48.52 5UTR_nr 36.60 43.88 38.96 39.13 5UTR_nr_MC 27.09 37.11 36.56 35.89 chr1_MC 30.84 40.82 35.87 35.84
all_hs, human poly(A) site; hsCDS, human CDS; hsCDS_MC, randomized CDS; hs_MC, randomized sequence of poly(A) region; 5UTR_nr, 5’-UTRs; 5UTR_nr_MC, randomized 5’-UTRs; chr1_MC, randomized genomic sequence of chromosome 1.
Table 4. Statistics of downstream U-rich region involvement in hairpin stems for the different types of sequences
#Reported #All Percentage (%) Diff (%)
all_hs 16532 27573 59.96 hsCDS 16027 45203 35.46 24.50 hsCDS_MC 5477 16368 33.46 26.50 hs_MC 7498 14958 50.13 9.83 5UTR_nr 1096 3156 34.73 25.23 5UTR_nr_MC 792 4645 17.05 42.91 chr1_MC 1717 10113 16.98 42.98
#Reported, the number of sequences reported by RNAMotif [16]; #All, the size of dataset; Diff, difference between all_hs and each negative sets.
Table 5. Statistics of poly(A) sites with PAS involvement in hairpin loops and the presence of downstream U-rich regions in hairpin
stems
#Reported sites Percentage of all reported sites (%) #Related genes #Genes Percentage of all genes (%) All 8977 6390 13756 46.45 Single-type 1378 15.35 1378 5272 26.14 Multiple-type 7599 84.65 5012 8484 59.08
Note that the percentage of all reported sites (column 3) derives from values in column 2, e.g., 15.35 = 1378 / 8977 * 100. Percentage of all genes (column 6) is derived from the same row, e.g., 45.39 = 6390 / 14078 * 100.
Fig. 1. Nucleotide composition across the -125/+125 region
of (a) single-type poly(A) sites (b) multiple-type poly(A) sites.
(c)The difference between AU-ratio and GC-ratio. The
difference at each position is calculated from (AU-ratio – GC-ratio). single_hs, single-type poly(A) sites; multiple_hs, multiple-type poly(A) sites.
Fig. 2. Characteristic sub-regions. PAS, polyadenylation
signal.
Fig. 3. (a) Adenine (b) Uracil (c) Cytosine (d) Guanine
frequencies at each position. In the vicinity of the poly(A) site (all_hs), CDS (hsCDS), randomized human CDS (hsCDS_MC), randomized poly(A) region (hs_MC), 5’-UTRs (5UTR_nr), randomized 5’-UTRs (5UTR_nr_MC) and randomized genomic sequences (chr1_MC).
Fig. 4. Probability profiling of the loops by (a)RNAfold [15]
and (b) Sfold [14]. single_hs, single-type poly(A) site (4908 sequences); hsCDS, human CDS (5000 sequences); 5UTR_nr, 5’-UTRs (3156 sequences). Note that in this test each sequence has the AAUAAA hexamer in the middle.