A synthetic approach to tacamonine. Access to 3-epitacamonine
and 14-epitacamonine
Tse-Lok Ho* and Chun-Yuan Su
Department of Applied Chemistry, National Chiao Tung University, Hsinchu, Taiwan, ROC Received 21 August 2000; revised 12 October 2000; accepted 26 October 2000
AbstractÐ3-Epitacamonine and 14-epitacamonine have been synthesized starting from the dinitrile 3. The synthetic route was designed on the basis of symmetric considerations which emphasized the establishment of the relative con®guration in two of the three stereocenters. q 2001 Elsevier Science Ltd. All rights reserved.
Tacamonine (1) is an indole alkaloid which initially made its appearance as a synthetic racemate named pseudovinca-mone I.1Two years later, in a paper describing an
exami-nation of the constituents of Tabernaemontana eglandulosa Stapf, which is widely distributed in Central Africa, taca-monine was identi®ed2along with a number of known and
new compounds. At that time the tacaman class represented a novel skeleton and tacamonine is an isomer of eburnamo-nine 2. The attachment of the ethyl group away from the D/E ring junction of the pentacyclic network engenders an additional stereocenter. That all three methine hydrogen atoms are cis related is a point signi®cant to our interest in pursuing a synthetic study of tacamonine.
After the isolation of tacaman alkaloids was reported, several syntheses towards them have appeared. Thus, an asymmetric synthesis of 1 featured free radical cyclization to form the D ring,3whereas another approach relied on the
development of the D ring component from a substituted pyridine.4,5 On the other hand, our interest in these
substances was based on an analysis of the stereochemical characteristics of tacamonine, that is the possibility of exploiting the symmetrical nature of synthetic inter-mediates. We believed that reward may be garnered by choosing the dinitrile 3 as starting material.
cis-1,3-Bis(tosyloxymethyl)-4-cyclopentene is readily avail-able from norbornadiene in two steps, i.e. ozonolysis with reductive work up and tosylation.6 Displacement of the
ditosylate with KCN in DMF led to dinitrile 3 which was submitted to another ozonolysis to furnish the unstable dialdehyde dinitrile 4. Direct condensation of 4 with trypta-mine in acetic acid and subsequent treatment with formic acid permitted the isolation of the tetracyclic b-carboline derivatives. This reaction protocol was designed to facilitate product isolation and minimize the disturbance of the two existing stereocenters while establishing a new one. Formic acid was employed to reduce the tetracyclic iminium species as they were formed. We found that two stereo-isomers in an 8:1 ratio were generated, therefore we have to conclude that either equilibration of a small amount of the iminium ion (5a5b) occurred via the enamine 6 (Eq. (1)),
Tetrahedron 57 (2001) 507±510
Pergamon
TETRAHEDRON
0040±4020/01/$ - see front matter q 2001 Elsevier Science Ltd. All rights reserved. PII: S0040-4020(00)01025-5
1
Keywords: tacamonine; epitacamonine; synthetic.
* Corresponding author. Present address: Department Chemie, LMU-Munchen, Batenandstr. 5-13 (Haus F), 81377 LMU-Munchen, Germany. Tel.: 1886-35712121; fax: 1886-35723764; e-mail: tlho@cc.nctu.edu.tw
T.-L. Ho, C.-Y. Su / Tetrahedron 57 (2001) 507±510 508
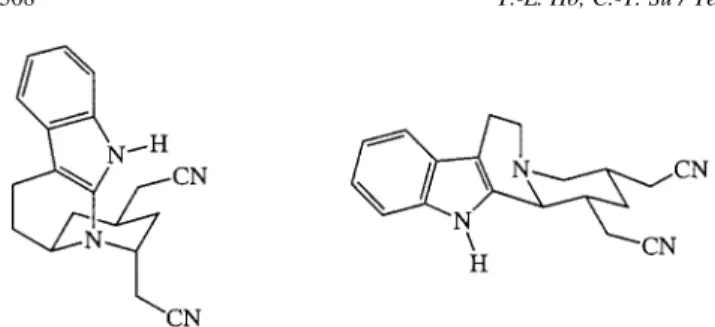
or the carbinolamine intermediate derived from the Pictet± Spengler cyclization underwent ring-chain tautomerization and epimerization of the aldehydo form. At this point the relative con®guration of the ring junction in either dinitrile 7a or the major and desired isomer 7b could not be deter-mined, but no Bohlmann±Wenkert bands were evident in their infrared spectra. Accordingly, we followed our plan to carry on the cyclization reaction. On re¯uxing with NaOMe in methanol and then aqueous hydrochloric acid the tetra-cyclic lactams 8a and 8b were obtained. Both products displayed distinct and complex absorption bands in the region of 2700±2800 cm21, indicating the C/D ring moiety
in each compound is a trans-quinolizidine.7The absence of
such bands in the precursors 7a and 7b would suggest the cis ring junction is more favorable, and such is possible because it only involved inversion of the lone-pair electrons on the nitrogen atom (Fig. 1).
We decided to proceed further with our synthetic work as we entertained a notion that inversion of 3-epitacamonine may still be possible at the end. Moreover, the new stereo-isomers of tacaminone (and tacamine) may possess some useful physiological properties. To convert the nitrile func-tion to a methyl group the ®rst step involved reducfunc-tion with diisobutylaluminum hydride. As part of the product from either 8a or 8b also suffered reduction of the lactam
car-bonyl which could not be avoided by limiting the quantities of the reducing agent or varying reaction temperature, the crude product was reoxidized with pyridinium dichromate/ Celite. Deoxygenation of the aldehyde in 9a and 9b was separately accomplished via the ethylene dithioacetal by reaction with Raney nickel in re¯uxing ethanol. 3-Epitaca-minone and 14-epitaca3-Epitaca-minone were obtained (Scheme 1). The NMR data of our synthetic 14-epitacaminone are in complete agreement with those reported.4
The attempt at isomerizing 3-epitacaminone to tacaminone by heating with pivalic acid was unsuccessful. An indirect method consisting of N-oxidation, Potier±Polonovski rearrangement using tri¯uoroacetic anhydride8 and
reduc-tion (with NaBH4or catalytic hydrogenation) also failed to
deliver any tacaminone. The behavior is contrasting to a tetracyclic ester with the trans±trans arrangement.5
In summary, we have demonstrated the advantage of a synthesis based on symmetry considerations.9 This effort
serves to stimulate other modi®cations for alternative stereochemical manipulation of C-3.
1. Experimental
NMR spectra were recorded with CDCl3as solvent, at 300
and 75 MHz, respectively for 1H and 13C absorptions.
Chemical shifts are reported in ppm relative to 0 for TMS. Electron impact mass spectra were measured at 70 eV. Drying of organic solutions used anhydrous Na2SO4.
Merck Silica gel (70±230 mesh) was used for chroma-tography. Melting points, determined with a Laboratory Devices apparatus, are uncorrected.
1.1. cis-1,3-Bis(cyanommethyl)-4-cyclopentene (3) A solution of ditosylate (10.0 g, 22.9 mmol) and KCN
Figure 1. Apparent preferred conformations of 7a and 7b.
Scheme 1. Reagents and conditions: (i) O3; Me2S; (ii) tryptamine; HOAc, D; HCOOH, 808C. For 2 steps: 7a: 5%, 7b: 40%; (iii) NaOMe, MeOH, D; aq. HCl,
T.-L. Ho, C.-Y. Su / Tetrahedron 57 (2001) 507±510 509
(4.5 g, 91.8 mmol) in DMF (50 mL) was heated at 608C for 16 h. The solvent was evaporated in vacuo and the residue was taken up in EtOAc and washed successively with water, 0.5N HCl, and dried. After concentration and chroma-tography over silica gel (eluent: hexane±EtOAc 1:1) the dinitrile 3 (3.1 g, 93%) was obtained as a colorless oil.
nmax (®lm)/cm21 2247; dH 1.20 (2H, m), 2.47 (4H, d, J6.6 Hz), 3.10 (2H, m), 5.74 (2H, s); dC 23.2 (t), 34.8 (t), 42.0 (d), 118.3 (s), 133.9 (d); M1 (EI) 146.0841 (146.0845 calcd for C9H10N2). 1.2. 1,3-Bis(cyanommethyl)-1,2,3,4,6,7,12,12b-octa-hydropyrido[2,1-a]-b-carbolines (7a/7b)
Slightly more than 1 equiv. of ozone was bubbled into a solution of dinitrile 3 (2.79 g, 19.11 mmol) in methanol (90 mL), which contained a small amount of Sudan Red as indicator, at 2788C. The excess ozone was removed with a stream of nitrogen and the resulting ozonide was treated with dimethyl sul®de (2.16 mL, 29.54 mmol). On warming to room temperature, tryptamine (3.06 g, 19.11 mmol) was added, the mixture was stirred for 1 h, re¯uxed with acetic acid (5.73 g, 95.45 mmol) for 24 h and evaporated. On heating the residue with 88% formic acid (10.0 g, 191.3 mmol) at 808C for 8 h the reaction was completed. After evaporation in vacuo and basi®ed with saturated NaHCO3 solution the products were extracted
into dichloromethane. The combined extracts were dried, concentrated, and chromatographed (silica gel, eluent: CH2Cl2±MeOH 1±2:100) to give the trans±trans isomer
7a (0.29 g, 5%) and the trans±cis isomer 7b (2.32 g, 40%). 1.2.1. Isomer 7a: mp 174±1758C;nmax (®lm)/cm212247;
dH1.13±1.32 (1H, m), 1.87±1.94 (1H, m), 1.96±2.10 (3H, m), 2.10±2.45 (2H, m), 2.45±2.84 (4H, m), 2.84±3.16 (2H, m), 3.16±3.23 (1H, m), 4.08 (1H, s), 7.13 (2H, m), 7.35 (1H, d, J7.8 Hz), 7.47 (1H, d, J7.8 Hz), 8.36 (1H, s);dC17.3 (t), 20.6 (t), 21.5 (t), 28.5 (d), 30.1 (t), 32.8 (d), 51.1 (t), 51.4 (t), 57.2 (d), 108.6 (s), 111.1 (d), 117.9 (s), 118.1 (d), 119.1 (s), 119.6 (d), 121.8 (d), 127.4 (s), 130.7 (s), 135.9 (s); M1
(EI) 304.1676 (304.1690 calcd for C19H20N4).
1.2.2. Isomer 7b: mp 1748C; nmax (®lm)/cm21 2246; dH 1.25±1.57 (1H, m), 2.11±2.40 (5H, m), 2.42±2.77 (2H, m), 2.77±2.95 (4H, m), 3.05±3.13 (2H, m), 3.63 (1H, d, J9.6 Hz), 7.14 (2H, m), 7.35 (1H, d, J7.8 Hz), 7.48 (1H, d, J7.8 Hz), 7.84 (1H, s);dC21.3 (t), 21.4 (t), 21.9 (t), 29.2 (d), 33.9 (d), 36.7 (t), 47.7 (t), 59.1 (t), 59.7 (d), 109.7 (s), 111.2 (d), 117.7 (s), 118.07 (d), 118.13 (s), 119.4 (d), 121.8 (d), 126.8 (s), 132.6 (s), 136.5 (s); M1 (EI) 304.1683 (304.1690 calcd for C19H20N4). 1.3. (2RS, 13aRS, 13bRS)-2-Cyanommethyl-12-oxo-2,3,5, 6,2,13,13a,13b-octahydro-1H-[1,7]naphthyridino[7,8,1-lma]-b-carboline (8a)
A mixture of dinitrile 7a (260 mg, 0.86 mmol) and NaOMe (from Na, 40 mg, 1.74 mmol) in methanol (5 mL) was re¯uxed for 2 h, evaporated and treated with aqueous hydro-chloric acid (from conc HCl, 1.4 mL, H2O, 5 mL). After
warming to 808C for 0.5 h the reaction mixture was quenched with saturated NaHCO3 solution and extracted
with dichloromethane. The combined organic extracts
were dried, concentrated and subjected to ¯ash chromato-graphy over silica gel (eluent: CH2Cl2±MeOH 1:100) to
afford 8a as colorless crystals (mp 1568C; 236 mg, 90%);
nmax(®lm)/cm212245, 1707;dH1.28±1.52 (1H, m), 1.72± 2.07 (2H, m), 2.18±2.39 (2H, m), 2.40±2.71 (7H, m), 2.71± 3.08 (3H, m), 7.25 (2H, m), 7.35 (1H, d, J8.6 Hz), 8.29 (1H, d, J8.6 Hz);dC20.1 (t), 21.0 (t), 31.6 (d), 32.5 (d), 32.7 (t), 38.7 (t), 51.3 (t), 56.6 (t), 61.3 (d), 111.5 (s), 115.9 (d), 118.1 (d), 119.1 (s), 123.7 (d), 124.0 (d), 129.5 (s), 133.3 (s), 134.8 (s), 166.9 (s); M1 (EI) 305.1524
(305.1530 calcd for C19H19N3O).
1.4. (2SR, 13aRS, 13bRS)-2-Cyanommethyl-12-oxo-2,3, 5,6,12,13,13a,13b-octahydro-1H-[1,7]naphthyridino[7,8, 1-lma]-b-carboline (8b)
A mixture of dinitrile 7b (2.22 g, 7.30 mmol) and NaOMe (from Na, 0.336 g, 14.6 mmol) in methanol (40 mL) was re¯uxed for 4 h, evaporated and treated with aqueous hydro-chloric acid (from conc HCl, 14 mL, H2O, 50 mL). After
warming at 808 for 1 h the reaction mixture was quenched with saturated NaHCO3 solution and extracted with
dichloromethane. The combined organic extracts were dried, concentrated and subjected to ¯ash chromatography over silica gel (eluent: CH2Cl2±MeOH 2:100) to afford 8b
as colorless crystals (mp 1588C; 1.96 g, 88%);nmax(®lm)/
cm21 2357, 1705; d H 1.09±1.23 (1H, m), 1.94±2.17 (2H, m), 2.18±2.26 (2H, m), 2.36±2.41 (2H, m), 2.43±2.54 (1H, m), 2.46±2.81 (5H, m), 2.81±3.00 (1H, m), 3.16 (1H, d, J8.4 Hz), 7.32 (2H, m), 7.43 (1H, d, J7.5 Hz), 8.35 (1H, d, J7.5 Hz); dC21.3 (t), 21.9 (t), 33.0 (d), 35.4 (t), 37.1 (d), 39.0 (t), 51.8 (t), 58.9 (t), 61.0 (d), 112.0 (s), 116.2 (d), 117.6 (s), 118.4 (d), 124.1 (d), 124.5 (d), 129.6 (s), 133.1 (s), 135.1 (s), 167.1 (s); M1 (EI) 305.1527
(305.1530 calcd for C19H19N3O).
1.5. 2-[(2RS, 13aRS, 13bRS)-12-oxo-2,3,5,6,12,13,13a, 13b-octahydro-1H-[1,7]naphthyridino[7,8,1-lma]-b-carbolin-yl]acetaldehyde (9a)
To a stirred solution of 8a (174 mg, 0.57 mmol) in dry dichloromethane (10 mL) at 2788C was added diisobutyl-aluminum hydride (1 M in hexane, 1.71 mL, 1.71 mmol) dropwise. After 0.5 h the reaction mixture was quenched with saturated NaHCO3 solution and allowed to warmed
to room temperature. The crude product was extracted into chloroform, the extracts were evaporated and heated with pyridinium dichromate (858 mg, 2.28 mmol) and Celite (850 mg) in chloroform (30 mL) for 48 h. The cooled mixture was ®ltered through a bed of Celite, concentrated in vacuo, and chromatographed over silica gel (eluent: CH2Cl2±MeOH 2:100) to furnish the product 9a as colorless
crystals (mp 1358C; 67 mg, 38%);nmax(®lm)/cm21 1706; dH 1.41±1.50 (1H, m), 1.64 (1H, m), 2.01±2.19 (1H, m), 2.30±2.40 (1H, m), 2.48±2.79 (7H, m), 2.79±2.99 (2H, m), 2.99±3.15 (2H, m), 7.23 (2H, m), 7.38 (1H, d, J9 Hz), 8.29 (1H, d, J9 Hz), 9.79 (1H, br s); dC 21.0 (t), 27.9 (d), 33.2 (d), 34.1 (t), 39.1 (t), 46.7 (t), 51.9 (t), 57.7 (t), 61.7 (d), 111.7 (s), 116.1 (d), 118.3 (d), 123.9 (d), 124.3 (d), 129.6 (s), 133.1 (s), 135.0 (s), 167.2 (s), 201.6 (s); M1(EI) 308.1498 (308.1525 calcd for C19H20N2O2).
T.-L. Ho, C.-Y. Su / Tetrahedron 57 (2001) 507±510 510
1.6. 2-[(2SR, 13aRS, 13bRS)-12-oxo-2,3,5,6,12,13,13a, 13b-octahydro-1H-[1,7]naphthyridino[7,8,1-lma]-b-carbolin-yl]acetaldehyde (9b)
To a stirred solution of 8b (0.40 g, 1.31 mmol) in dry dichloromethane (20 mL) at 2788C was added diisobutyl-aluminum hydride (1 M in hexane, 3.90 mL, 3.90 mmol) dropwise. After 0.5 h the reaction mixture was quenched with saturated NaHCO3 solution and allowed to warmed
to room temperature. The crude product was extracted into chloroform, the extracts were evaporated and heated with pyridinium dichromate (1.97 g, 5.24 mmol) and Celite (1.97 g) in chloroform (60 mL) for 72 h. The cooled mixture was ®ltered through a bed of Celite, concentrated in vacuo, and chromatographed over silica gel (eluent: CH2Cl2±
MeOH 2:100) to furnish the product 9b as colorless crystals (mp 1768C; 0.129 g, 32%);nmax(®lm)/cm211717;dH0.82± 0.95 (1H, m), 1.82±2.08 (3H, m), 2.31±2.42 (3H, m), 2.42± 2.72 (4H, m), 2.82±2.93 (1H, m), 2.93±3.15 (3H, m), 7.24 (2H, m), 7.36 (1H, d, J9.2 Hz), 8.29 (1H, d, J9.2 Hz), 9.70 (1H, t, J1.8 Hz);dC21.2 (t), 30.9 (d), 36.1 (t), 37.2 (d), 39.1 (t), 48.0 (t), 51.8 (t), 59.7 (t), 61.1 (d), 111.8 (s), 116.1 (d), 118.3 (d), 123.9 (d), 124.2 (d), 129.7 (s), 133.6 (s), 135.0 (s), 167.4 (s), 200.9 (s); M1 (EI) 308.1522 (308.1525 calcd for C19H20N2O2). 1.7. 14-Epitacamonine (10a)
A mixture of aldehyde 9a (60 mg, 0.19 mmol), ethane-dithiol (0.03 mL, 0.38 mmol) and boron tri¯uoride etherate (0.03 mL, 0.23 mmol) in dry dichloromethane (5 mL) was stirred at 08C for 1 h, poured into saturated NaHCO3
solu-tion and extracted with dichloromethane. The solusolu-tion was evaporated, re¯uxed with Raney nickel (ca. 0.5 mL, 50% slurry) in ethanol (10 mL) for 8 h. The cooled reaction mixture was ®ltered through Celite, and the ®ltrate was evaporated. The residue was chromatographed over silica gel (eluent: CH2Cl2) to give racemic 14-epitacamonine (mp
1328C; 54 mg, 96%);nmax(®lm)/cm211706;dH0.90 (3H, t, J7.2 Hz), 1.20±1.53 (3H, m), 1.58±1.81 (3H, m), 1.94± 2.20 (1H, m), 2.37±2.59 (3H, m), 2.60±2.76 (2H, m), 2.81± 2.94 (2H, m), 2.98±3.06 (1H, m), 7.26 (2H, m), 7.38 (1H, d, J7.8 Hz), 8.32 (1H, d, J7.8 Hz); dC12.5 (q), 21.4 (t), 25.0 (t), 33.4 (d), 34.0 (t), 36.2 (d), 39.7 (t), 52.3 (t), 58.0 (t), 62.6 (d), 111.7 (s), 116.1 (d), 118.2 (d), 123.8 (d), 124.0 (d), 130.0 (s), 134.4 (s), 135.1 (s), 168.0 (s); M1(EI) 294.1732
(294.1732 calcd for C19H20N2O).
1.8. 3-Epitacamonine (10b)
A mixture of aldehyde 9b (90 mg, 0.29 mmol), ethane-dithiol (0.05 mL, 0.59 mmol) and boron tri¯uoride etherate (0.04 mL, 0.32 mmol) in dry dichloromethane (8 mL) was stirred at 08C for 1 h, poured into saturated NaHCO3
solu-tion and extracted with dichloromethane. The solusolu-tion was evaporated, re¯uxed with Raney nickel (ca. 1 mL, 50% slurry) in ethanol (15 mL) for 8 h. The cooled reaction mixture was ®ltered through Celite, and the ®ltrate was evaporated. The residue was chromatographed over silica gel (eluent: CH2Cl2) to give racemic 3-epitacamonine (mp
1748C; 78 mg, 91%);nmax (®lm)/cm21 1708;dH0.81 (2H, m), 0.87 (3H, t, J 7.2 Hz), 1.16±1.32 (2H, m), 1.64±2.05 (4H, m), 2.30±2.40 (1H, m), 2.45±2.70 (3H, m), 2.83±3.00 (1H, m), 3.00±3.13 (2H, m), 7.20 (2H, m), 7.33 (1H, d, J9 Hz), 8.26 (1H, d, J9 Hz);dC11.3 (q), 21.1 (t), 27.0 (t), 36.1 (t), 37.3 (d), 37.6 (d), 39.3 (t), 52.0 (t), 60.2 (t), 61.5 (d), 111.7 (s), 116.1 (d), 118.3 (d), 123.9 (d), 124.2 (d), 129.7 (s), 133.5 (s), 135.1 (s), 167.6 (s); M1 (EI)
294.1731 (294.1732 calcd for C19H20N2O).
Acknowledgements
This work was generously supported by a grant from the National Science Council, ROC.
References
1. Massiot, G.; Oliveira, F. S.; Levy, J. Bull. Soc. Chim. Fr. 1982, (II), 185.
2. van Beek, T. A.; Verpoorte, R.; Svendsen, A. B. Tetrahedron 1984, 40, 737.
3. Ihara, M.; Setsu, F.; Shohda, M.; Taniguchi, N.; Tokunaga, Y.; Fukumoto, K. J. Org. Chem. 1994, 59, 5317.
4. Din Belle, D.; Tolvanen, A.; Lounasmaa, M. Tetrahedron 1996, 52, 11361.
5. Lounasmaa, M.; Karinen, K.; Din Belle, D.; Tolvanen, A. Tetrahedron 1998, 54, 157.
6. Grob, C. A.; Pfaendler, H. R. Helv. Chim. Acta 1970, 53, 2156. 7. Wenkert, E.; Roychaudhari, D. K. J. Am. Chem. Soc. 1956, 78,
6418; Bohlmann, F. Chem. Ber. 1958, 91, 2157.
8. Langlois, N.; Gueritte, F.; Langlois, Y.; Potier, P. J. Am. Chem. Soc. 1976, 98, 7017.
9. Ho, T.-L. Symmetry. A Basis for Synthesis Design; Wiley: New York, 1995.