Theoretical Study of the Structure, Energetics, and the n
-
π* Electronic Transition of the
Acetone
+
nH
2O (n)
1
-
3) Complexes
Dai-Wei Liao,† Alexander M. Mebel,*,‡Yit-Tsong Chen,‡,§and Sheng-Hsien Lin‡,§,|
Institute of Physical Chemistry, Department of Chemistry and State Key Lab of Physical Chemistry on Solid Surfaces, Xiamen UniVersity, Xiamen, Fujian 361005, China, Institute of Atomic and Molecular Sciences, Academia Sinica, P.O. Box 23-166, Taipei 10764, Taiwan, ROC, Department of Chemistry,
National Taiwan UniVersity, Taipei 106, Taiwan, ROC, and Department of Chemistry and Biochemistry, Arizona State UniVersity, Tempe, Arizona 85287-1604
ReceiVed: June 30, 1997; In Final Form: October 14, 1997X
The structure, energetics, and vibrational spectra of the (CH3)2CO‚(H2O)n(n)1-3) complexes have been studied using density functional and ab initio B3LYP, MP2, and CCSD(T) methods. The excitation energies and the oscillator strength for the n-π* electronic transition in acetone and acetone-water complexes have been calculated using the CIS, CASSCF, and CASPT2 approaches. The results show that the first water molecule is coordinated to the carbonyl group of acetone, while the oxygen atom of H2O forms a weak hydrogen bond with a methyl hydrogen. The second H2O occupies a position between the first water and a methyl group, and the third H2O occupies a position between the second H2O and the methyl hydrogen of acetone. The energies of the coordination of the first, second, and third water molecules to the complexes are 3.7, 5.7, and 6.7 kcal/mol, respectively. The formation of the (CH3)2CO‚(H2O)ncomplexes results in the shift of vibrational frequencies for acetone and water, particularly, red shifts for the OH stretching vibrations (up to 358 cm-1
) and CO stretching vibrations, as well as a blue shift for the HOH bending vibrations. A small but noticeable red shift (∼30 cm-1) of the C
-H stretch can be observed in the (CH3)2CO‚(H2O)2complex 2a. The excitation energy of the n-π* electronic transition is blue shifted by 0.25-0.30 eV, which is in agreement with the experimental blue shift observed in acetone/H2O. The oscillator strength for the n-π* transition increases from zero to∼10-4
in (CH3)2CO‚(H2O)3. The effect of the coordination of water molecules on the spectral intensity is expected to be weaker than the effect due to vibronic coupling.
1. Introduction
The theoretical investigation of excited states of molecules, especially the location, assignments, and analysis of the spectra for excited states, is increasingly important and interesting in applied and computational quantum chemistry. Acetone (C3H6O), as the simplest aliphatic ketone, has been used as a probe in photochemistry, photophysics, and biochemistry, in both ex-perimental and theoretical research, and scientists have obtained a deeper insight into the excited states of acetone in the past decades.1-17 Experimentally, for example, the stepwise
dis-sociation dynamics of acetone has been investigated by molec-ular beam photofragment translational spectroscopy (MBPTS)9 and femtosecond pump-probe experiments.
3 The electronic spectra, especially the Rydberg excited states, have been studied by optical UV,16electron-impact spectroscopy,4resonant mul-tiphoton enhanced ionization (REMPI) spectroscopy,1,2 and absorption spectroscopy.6,7,10 Theoretically, the multiphoton absorption properties5and the electronic spectra1,5,14,15of acetone have been studied by ab initio calculations at various levels such as configuration interaction (CI),14random phase approximation (RPA),5equation-of-motion coupled cluster (EOM-CC),15and the CASPT2 method.1 The dynamics of liquid acetone has been studied by molecular dynamics simulation (MDS)11,12and the Monte Carlo method.13
However, there still remain a lot of important problems to be solved for acetone, especially for the complexes of acetone
with water, (CH3)2CO + nH2O (n > 1). They in particular include the structure and energetics of the acetone-water complexes and the spectroscopic properties of these complexes. Traditionally, the interaction of solvent molecules with a solute molecule has been studied spectroscopically, by monitoring the spectral shifts in vibrational bands and the shifts in electronic spectra. Using ab initio calculations, we have investigated how water molecules (up to three) surround the acetone molecule, i.e., the structure and energetics of different arrangements of (CH3)2CO‚(H2O)n (n) 1-3). The calculations also provide information on the change of vibrational spectra upon the coordination of water molecules.
Experimentally, the FT-infrared absorption spectrum of the hydrogen-bonded (CH3)2CO‚H2O complex in solid argon ma-trixes has been reported.18 Red shifts were observed for the CdO stretching mode of acetone and the O-H stretching modes of water indicating that water is hydrogen-bonded to the carbonyl oxygen of acetone. Ab initio HF/6-31G** calcula-tions18of (CH
3)2CO‚H2O revealed a cyclic hydrogen-bonded structure involving the interaction between water and acetone at both the carbonyl oxygen and methyl hydrogen. Good agreement was found between the observed and calculated vibrational frequency shifts. Williams and Lowrey19considered structures and vibrational frequencies for some conformations of (CH3)2CO‚(H2O)n (n) 1-3) at the HF/4-31G level with the goal to determine the scale factors for ab initio force constants of acetone in aqueous solution.
The electronic transition to the first excited n-π* ( 1A
2) state of acetone is symmetry-forbidden. In the gas phase, a weak band corresponding to this transition has been observed,1,4,14,15,17 which is due to the vibronic coupling and intensity-borrowing †Xiamen University.
‡Institute of Atomic and Molecular Sciences. §National Taiwan University.
|Arizona State University.
XAbstract published in AdVance ACS Abstracts, December 1, 1997.
S1089-5639(97)02102-6 CCC: $14.00 © 1997 American Chemical Society
Downloaded by NATIONAL TAIWAN UNIV on September 3, 2009 | http://pubs.acs.org
mechanism. In water solution, the n-π* transition can also become allowed; if the acetone-water complexes are formed, the C2V symmetry of the molecule is broken. In the present
study we quantitatively investigate how the intensity and the excitation energy of this transition change from acetone to (CH3)2CO‚(H2O)n(n)1-3).
2. Calculation Methods
The geometries of the acetone molecule and the acetone -water complexes have been optimized using the hybrid density functional B3LYP method20with the 6-311G** basis set.21The B3LYP method is known to give accurate (within 0.01 Å and 1°for bond lengths and bond angles, respectively) geometries, in particular, for the first-row molecules.22,23 Vibrational frequencies, calculated at the B3LYP/6-311G** level and scaled by 0.963 as recommended by Rauhut and Pulay,22have been used for characterization of stationary points, zero-point energy (ZPE) corrections, and prediction of vibrational spectra. All the stationary points have been positively identified as minima (number of imaginary frequencies NIMAG)0) or transition states (NIMAG)1).
To obtain more reliable energies of the complex formation, we used the G2M(rcc,MP2*) method.23 G2M(rcc,MP2*) is a modification of the Gaussian-2 (G2) approach,24similar to G2-(MP2,SVP),25and uses B3LYP/6-311G** optimized geometries and ZPE corrections. The total energy in G2M(rcc,MP2*) is calculated as follows:
∆E(HLC) is an empirical “higher level correction” depending on the numbers of the paired and unpaired electrons in the system. Since the acetone-water complex formation does not change these numbers,∆E(HLC) cancels out in the calculations of relative energies.
Various methods were used to calculate the excitation ener-gies and oscillator strengths of the n-π* electronic transition in acetone and acetone-water complexes. Preliminary calcu-lations were carried out using the CIS method26 with the 6-31G**, 6-311G**, and 6-311+G** basis sets.
21 More accurate calculations were performed at the state-averaged CASSCF level27with two electrons distributed in six active orbitals, s/a-CASSCF(2,6), and in the CASPT2 approximation,28 both with the 6-31G** basis set. The GAUSSIAN 9429 and MOLPRO 9630programs were employed in the calculations. The natural charges of the atoms and the Wiberg bond indices31 were obtained as a result of natural population analysis32using GAUSSIAN 94.
3. Results and Discussion
Structure and Energetics of the (CH3)2CO‚(H2O)n (n) 1-3) Complexes. The optimized geometries of acetone and acetone-water complexes are presented in Figure 1. For the acetone molecule we considered three configurations, two eclipsed C2V-symmetric, cis-HCCO (0a) and trans-HCOO (0b),
as well as a staggered 0c of Cssymmetry. 0a is the most stable
structure at the B3LYP/6-311G** level with ZPE correction and has no imaginary frequencies. This structure could be stabilized by two intramolecular hydrogen bonds between the carbonyl oxygen (natural charge-0.55e) and methyl hydrogens (natural charge+0.22e) with the O‚‚‚H distances of 2.52 Å and the Wiberg bond index (WBI) of 0.005. Also, in structure 0a the methyl groups are rotated such that the distance between
two hydrogens located in the OCCC plane is maximized. 0c has one imaginary frequency and corresponds to a transition state for the internal rotation of methyl groups, while 0b has two imaginary frequencies and corresponds to a second-order hilltop. As shown in Table 1, the calculated relative energies of 0b and 0c are low, 1.9 and 0.6 kcal/mol, respectively. The present result agrees with earlier studies of acetone; the doubly eclipsed conformation 0a has been recognized in the literature as the most stable on the basis of experimental33and theoreti-cal1,5,14evidence.
Two isomers of the (CH3)2CO‚H2O complex have been found. The more stable structure, 1a, is similar to that previously reported by Zhang et al.18 The water molecule interacts with acetone at the carbonyl oxygen and one methyl hydrogen in a cyclic form. The hydrogen-bond distance between H of H2O and the carbonyl oxygen is 1.97 Å and WBI is 0.03, as shown in Figure 2. The bond between the O of H2O and methyl hydrogen is weaker, with the distance of 2.42 Å and WBI of 0.02. The OH bond in water and the CO bond in acetone are stretched by∼0.01 Å, and the corresponding WBIs decrease by 0.05-0.06. The CH bond of the methyl group is not stretched due to the hydrogen atom interacting with the oxygen of H2O, but WBI decreases by 0.02. The CC bonds in acetone are slightly shortened. The natural population analysis gives results similar to the Mulliken population analysis;18 the net charges on atoms become greater on the formation of the complex; that is, the negative charges on oxygens and the positive charges on interacting hydrogens increase. The energy of the complex formation is not sensitive to the theoretical methods but changes significantly with the basis set improve-ment, from 6.2 at MP2/6-31G** to 4.0 kcal/mol at MP2/ 6-311+G(3df,2p). The most reliable G2M(rcc,MP2*) energy is 3.7 kcal/mol,∼3 kcal/mol lower than the complexation energy calculated by B3LYP/6-311G** with ZPE correction. The complexation energies calculated with small basis sets have significant basis set superposition errors (BSSE). We estimated BSSE for the energy of formation of 1a from acetone and water using the full counterpoise (CP) method34 at the MP2 level. With the 6-31G** basis set the CP correction is as high as 4.1 kcal/mol. With 6-311+G(3df,2p), it decreases to 0.8 kcal/mol. Despite some controversy whether the CP procedure yields an overestimation of the actual BSSE,35this correction is usually considered as an upper limit of the error. Since the G2M energy approximates the CCSD(T) energy with the 6-311+G(3df,2p) basis set, we expect that the calculated G2M energies of complex formation, discussed in this section, have BSSE lower than 1 kcal/mol.
The second isomer of (CH3)2CO‚H2O, 1b, is weakly bound. The O atom of the water molecule is coordinated with two hydrogens of the two methyl groups of acetone. The O‚‚‚H distances corresponding to the two hydrogen bonds are long, 2.52 and 2.53 Å, and the WBIs are as low as 0.01. The O atom of H2O slightly accumulates negative charge,-0.90e vs -0.88e in a free water molecule. The positive charges of the hydrogen-bonded hydrogens increase by 0.02-0.03e. The geometry of the acetone molecule in 1b is less perturbed than that in 1a. The complex formation energy of 1b is only 1.0 kcal/mol at the G2M level. The B3LYP/6-311G** + ZPE approximation overestimates this energy by ∼2 kcal/mol. Hydrogen bonds to C-H hydrogen, like those in 1b, are un-usual but have been reported in the literature. For example, Fraser et al. observed the weakly bound complex NH3 -acetylene, in which the acetylene hydrogen bonds to the ammonia,36Erickson and McLoughlin found an intramolecular CF2H‚‚‚OdC hydrogen bond in pyrazole carboxamide fungi-E[G2M(rcc,MP2*)])E[RCCSD(T)/6-31G(d,p)]+
E[MP2/6-311+G(3df,2p)]-E[MP2/6-31G(d,p)]+ ZPE+∆E(HLC)
Downloaded by NATIONAL TAIWAN UNIV on September 3, 2009 | http://pubs.acs.org
cide,37and Masunov and Dannenberg predicted the C-H‚‚‚O H-bonding interactions between water and various para-substituted phenylacetylenes.38
Four isomers of (CH3)2CO‚(H2O)2have been calculated. In the most favorable structure, 2a, acetone and two water molecules form an eight-member cycle. Three hydrogen bonds have been formed: between the carbonyl oxygen and a hydrogen of the first water molecule (the O‚‚‚H distance)1.85 Å and WBI)0.05), between the oxygen of the first water molecule and a hydrogen of the second H2O (O‚‚‚H)1.83 Å, WBI) 0.05), and between the O atom of the second H2O and a methyl hydrogen (O‚‚‚H)2.25 Å, WBI)0.02). The hydrogen bonds
in 2a are stronger than those in 1a. The negative and positive charge accumulation on the interacting oxygen and hydrogen atoms, respectively, is also more pronounced. Geometries of the acetone and water molecules are perturbed to a higher extent in 2a than those in 1a by stretching the CO and OH bonds and shrinking the CC bonds. The CH distance for the methyl hydrogen bonded with the O of the second H2O is elongated to 1.10 Å, and its WBI decreases by 0.07. The energy of the formation of 2a, from acetone and two water molecules, is calculated to be 9.4 kcal/mol at the G2M level. The B3LYP+ ZPE method overestimates this value by 6.4 kcal/mol. The use of the larger basis set, 6-311+G(3df,2p), is critical. For Figure 1. Optimized geometries (B3LYP/6-311G**, bond lengths are in angstroms, bond angles are in degrees) of acetone and acetone-water
complexes. 2
Downloaded by NATIONAL TAIWAN UNIV on September 3, 2009 | http://pubs.acs.org
instance, at the MP2 level the energy of the complex formation changes from 14.5 kcal/mol with the 6-31G** basis set to 10.2 kcal/mol with 6-311+G(3df,2p). The complex formation energy of 2a from 1a and H2O is 5.7 kcal/mol. Thus, the addition of the second water molecule (2a) brings a higher energy gain than the addition of the first one (1a).
The complex 2b can be formed from 1a by adding the second H2O between the two methyl groups of acetone, or from 1b by adding H2O between the carbonyl oxygen and a methyl hydrogen. At various levels of theory, 2b is about 5 kcal/mol less stable than 2a. At the B3LYP and MP2 levels the energy of formation of 2b from the free acetone and two water molecules approximately equals the sum of the energies of formation of 1a and 1b. Thus, the complexation energies of the second H2O to 1a and 1b are the same as the complexation
energies of the first H2O to the methyl and carbonyl sites of acetone, respectively. In this sense, the complexation of two water molecules to the different sites of acetone is additive energetically. We estimate the energy of formation of 2b to be 4.8 kcal/mol at the G2M level.
For the third isomer of (CH3)2CO‚(H2O)2, 2c, the second water molecule is added to the H2O of the (CH3)2CO‚H2O complex 1b. 2c can also be described as a complex of the water dimer and acetone. The second H2O does not directly interact with acetone but forms a O‚‚‚H hydrogen bond with a hydrogen of the first H2O. The energy of 2c with respect to acetone+ 2H2O is-4.1 kcal/mol at our best G2M level. The energy of the complex formation from 1b and H2O is 3.1 kcal/mol. For comparison, we carried out calculations of the water dimer at the same levels of theory. In the B3LYP + ZPE and G2M approximations, the energy of (H2O)2formation from two H2O [∆H°(0)] is found to be 5.5 and 3.0 kcal/mol, respectively. The experimental value of∆H°for water dimerization at 373 K is 3.6(0.5 kcal/mol.
39 Using the calculated B3LYP geometries and vibrational frequencies, one can compute the thermal correction for the enthalpy. Adding the thermal correction to the G2M calculated∆H°(0) we obtain∆H°(373))3.2 kcal/ mol, in close agreement with experiment. The geometry and the binding energy of the water dimer, calculated at the G2M/ /B3LYP level, also agree with previous theoretical results.40 Therefore, we believe that the G2M method provides high accuracy for the complexation energies of (CH3)2CO‚(H2O)n. The energy of formation for 2c from acetone and water dimer is only 1.1 kcal/mol, which is similar to the complexation energy of 1b, where a single water molecule is attached to the methyl groups of acetone.
The fourth isomer of (CH3)2CO‚(H2O)2, 2d, suggested by Williams and Lowrey,19 has a symmetric C
2structure. Both water molecules are attached to the carbonyl oxygen. The geometry of 2d is similar to that of 1a, but with an additional H2O molecule. Two H2O’s are situated symmetrically with respect to the CO axis of acetone. The CO‚‚‚HOH hydro-gen bonds in 2d are slightly weaker than that in 1a, as can be judged from the comparison of the geometries and energies. The O‚‚‚H distances in 2d (1.99 Å) are longer than that in 1a (1.97 Å), and the stretch of the OH bonds participating in the CO‚‚‚HOH hydrogen bonding is smaller. On the other hand, the CO bond stretch in 2d is larger than in 1a because the carbonyl oxygen takes part in two hydrogen bonds. The coordination of the second water molecule to 1a to form 2d
TABLE 1: Relative Energies (kcal/mol) of the Acetone-Water Complexes Erel
species ZPEa B3LYP/6-311G** CCSD(T)/6-31G** MP2/6-31G** MP2/6-311
+G (3df,2p) G2M 0a, C2V b 50.24 (0) -193.21302 -192.62100 -192.56399 -192.76813 0b, C2V 50.08 (2) 1.9 c 0c, Cs 50.23 (1) 0.6c H2O, C2V b 12.88 (0) -76.44745 -76.22912 -76.21728 -76.31826 (H2O)2, Csd 27.87 (0) -5.5 -4.6 -4.8 -3.2 -3.0 0a+3H2O 88.88 (0) 0.0 0.0 0.0 0.0 0.0 1a+2H2O e 91.02 (0) -6.5 -5.9 -6.2 -4.0 -3.7 1b+2H2O e 90.15 (0) -3.2 -2.5 -2.6 -1.1 -1.0 2a+H2O e 93.50 (0) -15.8 -13.7 -14.5 -10.2 -9.4 2b+H2O e 92.26 (0) -10.1 -9.1 -5.3 (-4.8) f 2c+H2O e 92.34 (0) -9.8 -8.1 -8.6 -4.6 -4.1 2d+H2O e 93.08 (0) -12.5 -11.4 -11.8 -7.6 -7.2 3ae 96.13 (0) -25.4 -23.3 -22.9 -15.7 -16.1 3be 94.68 (0) -18.9 -17.6 -11.1 (-10.4) f 3ce 94.44 (0) -17.0 -15.4 -9.1 (-7.8) f aZero-point energies, calculated at the B3LYP/6-311G** level and scaled by 0.963. In parentheses: number of imaginary frequencies.bTotal
energies in hartrees.cWith respect to 0a.dThe relative energies are given with respect to 2H
2O.eThe relative energies are given with respect to 0a+3H2O.
fThe energy is estimated on the basis of additivity.
Figure 2. Calculated natural atomic charges and Wiberg bond indices
for acetone and acetone-water complexes.
Downloaded by NATIONAL TAIWAN UNIV on September 3, 2009 | http://pubs.acs.org
brings the energy gain of 3.5 kcal/mol at the G2M level. The complexation energy per each H2O is 3.6 kcal/mol in 2d, slightly lower than the energy of the formation of 1a from acetone and water. Isomer 2d is more stable than 2b and 2c, but less favorable than 2a by 2.2 kcal/mol. The coordination of additional H2O to the carbonyl oxygen weakens the hydrogen bonding, while the formation of the acetone-H2O-H2O cyclic structure results in increasing hydrogen bond strengths.
We also considered three isomers of (CH3)2CO‚(H2O)3. In structure 3a, three water molecules and acetone form a 10-member ring. The structure has four hydrogen bonds: between the carbonyl oxygen and an H of the first H2O (the bond length is 1.76 Å and the Wiberg bond index is 0.06); between the O of the first H2O and a hydrogen of the second water (O‚‚‚H) 1.85 Å, WBI)0.05); between the O of the second H2O and a hydrogen of the third H2O (O‚‚‚H )2.03 Å, WBI )0.03); and between the O of the third water molecule and a hydrogen of the methyl group (O‚‚‚H )2.31 Å, WBI) 0.02). There also exists a weak interaction between the O atom of the second H2O and a second methyl hydrogen (O‚‚‚H)2.59 Å, WBI) 0.004). The OH bonds participating in the hydrogen bonds are stretched to 0.970-0.984 Å; the shorter and the stronger the hydrogen bond, the larger the stretch of the corresponding OH bonds. The CO bond in acetone is elongated to 1.222 Å, which is the longest value in the row from free acetone, 1a, 2a to 3a. The CC bonds are contracted to 1.506 and 1.509 Å, the shortest values among all (CH3)2CO‚(H2O)n(n)1-3) complexes. The stretch of the CH bond in the hydrogen bonding of 3a is smaller than that in 2a, and the O‚‚‚HCH2 hydrogen bond in 3a is weaker than that in 2a. The charge distribution is characterized by a negative charge accumulation on the oxygens and a positive charge accumulation on the hydrogen-bonded hydrogens. The charge accumulation in 3a is enhanced in comparison with 2a and 1a.
The complexation energy of 3a with respect to acetone and three water molecules is 16.1 kcal/mol at the G2M level. Coordination of the third H2O to 2a brings an energy gain of 6.7 kcal/mol, which is 1.0 kcal/mol higher than the energy of the coordination of the second H2O to 1a. At the B3LYP + ZPE level, the complexation energy of 3a is overestimated by about 9 kcal/mol. This is due to the fact that the energy of complex formation is additive with respect to the number of coordinating H2O ligands. The higher the number of ligands, the larger the error accumulated at the B3LYP level.
Structure 3b can be described as a combination of 2a and 1b, while 3c is a combination of 1a and 2c. At the MP2 and B3LYP levels the relative energy of 3b is close to the sum of the relative energies of 2a and 1b, and the energy of 3c is close to the sum of the energies of 1a and 2c. On the basis of additivity, our best estimates for the complexation energies at the G2M level of theory are 10.4 and 7.8 kcal/mol for 3b and 3c, respectively. 3b and 3c are weakly bound, in considering that only∼1 kcal/mol is required for the elimination of a water molecule (3b f 2a) or a water dimer (3c f 1a). More isomers of (CH3)2CO‚(H2O)3can be speculated on, and their energies can be estimated on the basis of additivity. For instance, a structure that is a combination of 2a and 1a and can be formed from 2d + H2O or 2a + H2O is expected to have the complexation energy of ∼13 kcal/mol. Another structure, a combination of 2d and 1b, can be formed from 2d+H2O or 2b+H2O, and its complexation energy is estimated to be 8-9 kcal/mol.
The most likely scenario for the aqueous solution of acetone involves the complexes 1a, 2a, and 3a. The first water molecule is coordinated to the carbonyl group of (CH3)2CO. The second
H2O occupies a position between the first water and a methyl group of acetone, etc. How many water molecules can be accommodated in the (CH3)2CO-nH2O rings is yet to be answered. The complexes 1b, 2b, 2c, 3b, and 3c are weakly bound and might exist only at very low temperatures. Upon temperature increase, they should eliminate H2O or (H2O)2to go back to acetone, 1a, or 2a.
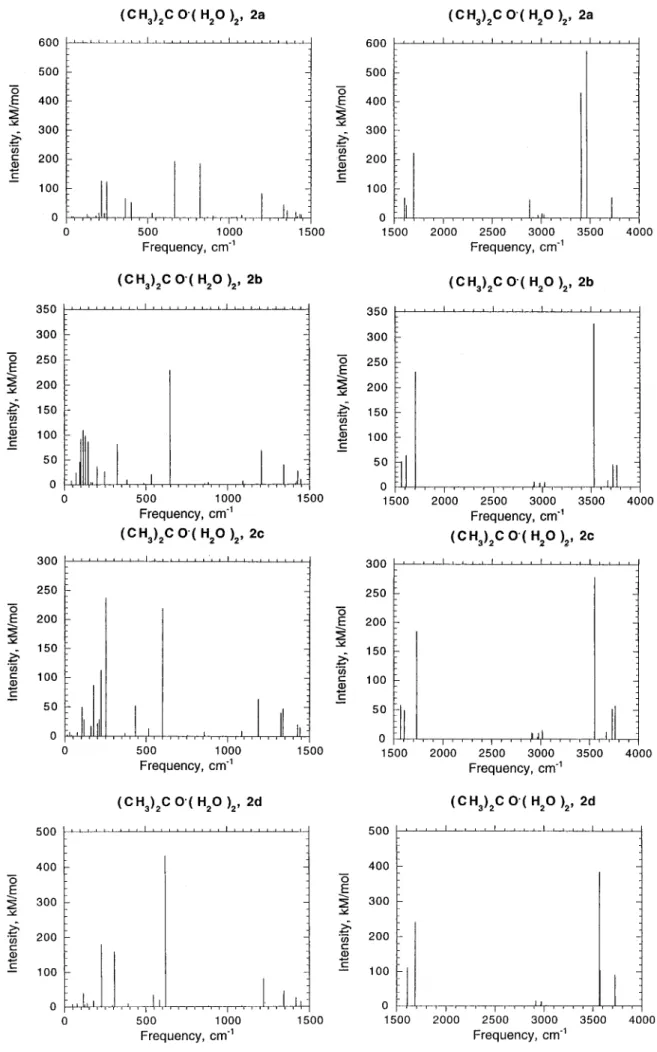
Vibrational Spectra. Calculated IR spectra of acetone, water, and the acetone-water complexes are shown in Figures 3-5, and the frequencies of the most intense vibrations are presented in Table 2. The deviations of the calculated frequen-cies for (CH3)2CO, H2O, and (CH3)2CO‚H2O from the frequen-cies measured experimentally in solid argon18are in the range 20-30 cm
-1
. Frequencies in the complexes are shifted as compared to those in isolated acetone and water. The largest shift is found for the OH stretching modes of H2O. In 1a, two OH stretching frequencies exhibit red shifts of 39 and 121 cm-1.
The lower frequency of 3549 cm-1
corresponds to the vibration of the stretched (0.971 Å) OH bond participating in the hydrogen bonding. In the weakly bound complex 1b, the OH stretch frequencies are not shifted. In 2a, the red shifts are 40, 204, and 261 cm-1
. The longer the OH bond, the larger the red shift of the corresponding stretching frequency. The red-shifted vibrations in 1a and 2a have high intensities. In 3a, one of the OH bonds is stretched to 0.984 Å and the corresponding OH frequency is shifted by 358 cm-1
to 3312 cm-1
with an intensity as high as 961 km/mol. The OH stretch frequencies of 2b are similar to those of water and 1a, with a slightly higher red shift of the lowest frequency (3531 vs 3549 cm-1 in 1a). The
frequencies of 2c are close to those of the water dimer, but the lowest OH stretch is slightly (13 cm-1) red shifted. While the
spectrum of 3b is a combination of the spectra of 2a and water, the spectrum of 3c is a combination of 1a and 2c.
The frequencies of the HOH bending vibrations are blue shifted in the complexes. The shift reaches 30, 43, and 72 cm-1
in 1a, 2a, and 3a, respectively. The frequency of the CO stretching vibration decreases in parallel with the elongation of this bond in the complex. The red shift constitutes 27 cm-1
in 1a, 41 cm-1 in 2a, and 51 cm-1 in 2d and 3a. The C
-H stretching vibrations are less affected. A noticeable shift can be observed for 2a, where one of the C-H frequencies decreases by 31 cm-1
. This is attributed to the stretch to 1.100 Å of the C-H bond taking part in hydrogen bonding. The spectrum of 3b is similar to that of 2a, and one of the CH stretchings is red shifted by 25 cm-1. In 3a, where the hydrogen bond between
a methyl hydrogen and the third water oxygen is weak, the CH bond is stretched to a lesser extent than that in 2a and the frequency shift is only 12 cm-1
.
Table 3 displays the most IR intense frequencies, character-istic for the complexes, which are not observed for the isolated acetone and water molecules. The frequencies lie between 87 and 824 cm-1
. For example, the three intense vibrational features for 1a and 2d are calculated at 621-624, 306-313, and 225-227 cm
-1
. For 2a, the most prominent vibrations are found at 822, 667, 249, and 215 cm-1, and for 3a at 783, 674,
461, and 406 cm-1
. The weak complexes, 1b, 2b, 2c, 3b, and 3c, are characterized by the group of intense vibrations in the 100-200 cm
-1
region.
The n-π* Electronic Transition. The n-π* excitation energies and oscillator strengths for isolated acetone and the acetone-water complexes are collected in Table 3. In acetone, the1A
1 -1A
2transition is symmetry-forbidden, and the oscillator strength is zero. The calculated vertical excitation energy at our best CASPT2/6-31G** level, 4.38 eV, reproduces the experimental value.4 The CASSCF approach slightly under-2
Downloaded by NATIONAL TAIWAN UNIV on September 3, 2009 | http://pubs.acs.org
estimates this energy, but the agreement with experiment is still good. The CIS approach does not provide a satisfactory accuracy; the excitation energy is overestimated by∼0.8 eV. The CIS calculation with the 6-31G**, 6-311G**, and 6-311+G** basis sets demonstrated that the excitation energy does not change with increasing basis set. For the (CH3)2CO‚ H2O complexes we have also carried out calculations at the CASSCF and CASPT2 levels. The CASSCF and CASPT2 energies agree with each other to within 0.1 eV and the cal-culated oscillator strengths are also close. Hence, we analyze the whole set of data based on the CASSCF/6-31G** results. The trends observed with the complex formation are the increase of the intensity and the blue shift of the excitation energy upon increasing the number of water molecules coor-dinated to acetone. The coordination of the first H2O breaks the C2Vsymmetry. The n
-π* transition becomes
symmetry-allowed and the oscillator strength increases to 5.3× 10-6in 1a and 6.8× 10-7
in 1b. The coordination of the second and the third H2O brings the oscillator strengths to 5.4× 10-5
in 2a and 7.1× 10-5
in 3a. Therefore, we expect that the n-π* transition can be observed in the water solution of acetone, and its intensity should increase with increasing the number of H2O molecules surrounding (CH3)2CO. The energy of the transition is shifted to higher values by 0.25 eV in 1a (0.16 eV at the CASPT2 level) and 0.26 and 0.31 eV in 2a and 3a, respectively. This result is in good agreement with experiment; the electronic spectra of liquid acetone/H2O show large blue shifts of∼0.25 eV.41 Structure 2d exhibits the largest blue shift of 0.40 eV. Coordination of H2O between two methyl groups, when very weak complexes are formed, affects the excitation energy and the oscillator strength to a minor extent. For instance, from 0a to 1b, the energy increases by 0.066 eV (0.117 eV at the Figure 3. Calculated IR spectra of acetone-water complexes 1a and 1b in comparison with the combined spectrum of acetone and water [(CH3)2
-CO and H2O].
Downloaded by NATIONAL TAIWAN UNIV on September 3, 2009 | http://pubs.acs.org
Figure 4. Calculated IR spectra of acetone-water complexes 2a, 2b, 2c, and 2d. 2
Downloaded by NATIONAL TAIWAN UNIV on September 3, 2009 | http://pubs.acs.org
CASPT2 level); from 1a to 2b, the energy and the oscillator strength decrease by 0.028 eV and 1.6× 10-6
, respectively. From 2a to 3b, the energy increases by 0.019 eV, but the oscillator strength slightly drops. The effect of the water-dimer coordination between two methyl groups (from 0a to 2c and from 1a to 3c) is similarly small.
The oscillator strength for the n-π* transition of acetone due to the formation of the acetone-water complexes does not exceed 10-4
. The n-π* transition in the gas-phase acetone is observable due to the vibronic coupling of the1A
2state with other excited states, such as n-σ* (1B1) andπ-π* (1A1), and the subsequent intensity borrowing from these symmetry-allowed transitions. Our calculations for the intensities of the n-π* vibronic transitions for acetone are now under way. For H2CO, the oscillator strength of ∼2 × 10-4
for the n-π* transition has been reported.42,43 Thus, the effect of the vibronic
coupling on the spectral intensity is stronger than that of the coordination of water molecules (up to three).
The blue shifts of the n-π* electronic excitation energy in the (CH3)2CO‚(H2O)n(n)1-3) complexes can be explained on the basis of a simple perturbation treatment for the spectral shifts on the HOMO-LUMO level, as those by Gerhards et al. for p-cresole‚H2O and H2CO‚H2O complexes.41 The n-π* transition in acetone is a transition of one electron from HOMO to LUMO. In a crude approximation, the larger the gap between HOMO and LUMO, the higher the excitation energy. In the acetone-water complexes, H2O is a proton-donating substituent which stabilizes theπ system of acetone. The calculations show
that both n (HOMO) and π* (LUMO) levels are indeed
stabilized by the hydrogen bond with H2O. The spectral shift depends on which molecular orbital is more stabilized. Gerhards et al.41 described the change in the π-system energy of the Figure 5. Calculated IR spectra of acetone-water complexes 3a, 3b, and 3c.
Downloaded by NATIONAL TAIWAN UNIV on September 3, 2009 | http://pubs.acs.org
ground and excited states, due to an inductive effect on the O atom of the carbonyl group, in terms of the first-order perturba-tion theory:
where q0and q*0are the O-atom charge orders defined by∑bj
CjO2. bjis the population number and CjOis the O atom orbital
coefficient in the jth MO; dR0is the change in the Coulomb integral at the oxygen atom, which is negative for the proton-donating solvents. The infinitesimal shift after addition of the solvent is
where CLOand CHOare the LUMO and HOMO MO coefficients at the O atom. All the calculations show that CHO>CLOfor (CH3)2CO‚H2O. Therefore, the n orbital (HOMO) is more
stabilized and the increasing gap between HOMO and LUMO results in a blue shift.
4. Conclusions
The calculations of the structure and energetics of the (CH3)2 -CO‚(H2O)n (n ) 1-3) complexes show that the first water molecule is coordinated to the carbonyl group of acetone and the oxygen atom of H2O forms a weak hydrogen bond with a methyl hydrogen. The complexation energy for (CH3)2CO‚H2O is predicted to be 3.7 kcal/mol. The second H2O occupies a position between the first water and a methyl group; the third H2O occupies a position between the second H2O and the methyl hydrogen of acetone. The energies of the coordination of the second and third water molecules to the complexes are 5.7 and 6.7 kcal/mol, respectively. Coordination of the second H2O to the carbonyl oxygen leads to an energy gain of 3.5 kcal/mol. The water molecule or water dimer can also be attached between two methyl groups, but the corresponding coordination energy is very low, about 1 kcal/mol.
The formation of the (CH3)2CO‚(H2O)ncomplexes results in a shift of vibrational frequencies for acetone and water. The
TABLE 2: Characteristic Vibrational Frequencies (cm-1
) and Intensities (in brackets, km/mol) of Acetone, Water, and Acetone-Water Complexes
(CH3)2CO+H2O 1a 1b 2a 2b 2c 2d 3a 3b 3c (H2O)2 C-H Stretch in Acetone a1 3024 [9.8] 3026 [9.6] 3021 [8.5] 3028 [12] 3023 [9.7] 3020 [11] 3022 [1.5] 3027 [13] 3027 [10] 3025 [10] b2 3023 [13] 3021 [1.3] 3020 [16] 3008 [14] 3022 [2.3] 3015 [15] 3022 [2.6] 3009 [14] 3019 [4.7] 3021 [3.6] b1 2972 [25] 2973 [17] 2975 [12] 2969 [8.2] 2973 [7.9] 2978 [10] 2973 [13] 2968 [6.8] 2971 [6.2] 2976 [6.5] a2 2965 [0] 2966 [2.9] 2969 [1.1] 2967 [11] 2968 [1.4] 2968 [3.8] 2966 [3.6] 2960 [6.1] 2969 [18] 2969 [4.5] a1 2919 [7.1] 2919 [8.5] 2917 [9.1] 2915 [2.9] 2917 [10] 2917 [9.9] 2917 [15] 2916 [4.0] 2911 [2.7] 2915 [14] b2 2912 [2.0] 2911 [4.4] 2910 [5.7] 2881 [63] 2910 [3.1] 2909 [11] 2910 [2.1] 2900 [46] 2887 [36] 2908 [4.5]
CdO Stretch in Acetone
a1 1737 [174] 1710 [216] 1732 [184] 1696 [222] 1735 [231] 1732 [185] 1686 [242] 1686 [236] 1692 [234] 1734 [235] O-H Stretch in Water b2 3763 [25] 3724 [52] 3763 [42] 3723 [31] 3764 [44] 3761 [57] 3726 [30] 3738 [67] 3763 [42] 3760 [58] 3758 [53] a1 3670 [3.6] 3549 [281] 3668 [12] 3722 [70] 3723 [45] 3729 [52] 3725 [91] 3733 [50] 3727 [61] 3729 [51] 3730 [43] 3466 [574] 3669 [13] 3669 [12] 3575 [104] 3651 [136] 3720 [33] 3721 [44] 3664 [11] 3409 [431] 3531 [327] 3552 [279] 3566 [385] 3577 [164] 3669 [13] 3669 [12] 3565 [273] 3462 [273] 3455 [534] 3539 [320] 3312 [961] 3397 [507] 3526 [313] HOH Bend in Water
a1 1578 [58] 1608 [59] 1564 [54] 1621 [44] 1613 [64] 1606 [49] 1606 [111] 1650 [116] 1625 [56] 1615 [65] 1606 [48] 1606 [69] 1564 [52] 1569 [58] 1602 [0.1] 1613 [82] 1608 [60] 1607 [52] 1571 [74] 1595 [51] 1567 [52] 1571 [56] Complex Frequencies 624 [240] 227 [23] 822 [185] 648 [230] 600 [220] 621 [433] 783 [150] 824 [195] 654 [106] 644 [141] 313 [84] 181 [13] 667 [193] 323 [82] 433 [52] 306 [159] 674 [358] 670 [183] 637 [342] 375 [77] 225 [92] 143 [23] 398 [53] 145 [87] 252 [238] 227 [180] 667 [128] 404 [57] 420 [61] 197 [24] 136 [188] 363 [67] 128 [99] 222 [112] 531 [48] 366 [67] 323 [87] 170 [154] 99 [88] 249 [124] 115 [110] 176 [87] 461 [87] 256 [64] 256 [213] 149 [286] 95 [56] 215 [125] 99 [92] 105 [50] 406 [123] 214 [108] 245 [59] 64 [52] 94 [46] 177 [287] 212 [48] 164 [79] 202 [102] 110 [59] 152 [132] 87 [41] TABLE 3: n-π* Excitation Energies (eV) and Oscillator Strengths for Acetone and Acetone-Water Complexes
CIS/ 6-31G** 6-311G** 6-311+G** CASSCF/6-31G** CASPT2/6-31G** 0a 5.2012 (0) 5.1974 (0) 5.2131 (0) 4.3481 (0) 4.3781 (0) 1a 5.3692 (6.2× 10-6) 5.3637 (5.8× 10-6) 5.3727 (9.5× 10-6) 4.6004 (5.3× 10-6) 4.5413 (4.8× 10-6) 1b 5.2401 (3.2× 10-6) 5.2334 (3.0× 10-6) 5.2487 (3.0× 10-6) 4.4140 (6.8× 10-7) 4.4950 (3.3× 10-7) 2a 5.4911 (1.1× 10-4) 5.4845 (2.9× 10-5) 5.4857 (7.0× 10-5) 4.6036 (5.4× 10-5) 2b 5.4187 (5.3× 10-6) 5.4111 (4.0× 10-6) 5.4173 (7.0× 10-5) 4.5729 (3.7× 10-6) 2c 5.2563 (8.9× 10-6) 5.2502 (1.0× 10-5) 5.2668 (8.6× 10-6) 4.4272 (1.8× 10-6) 2d 5.5287 (7.9× 10-7) 5.5246 (2.4× 10-8) 5.5287 (7.2× 10-7) 4.7479 (1.0× 10-5) 3a 5.5468 (3.1× 10-5 ) 5.5445 (4.0× 10-5 ) 5.5436 (5.3× 10-5 ) 4.6568 (7.1× 10-5 ) 3b 5.5076 (1.4× 10-4 ) 5.4996(1.6× 10-4 ) 5.4995 (1.3× 10-4 ) 4.6225 (4.7× 10-5 ) 3c 5.4334 (8.0× 10-6) 5.4253 (5.8× 10-6) 5.4309 (7.2× 10-6) 4.5858 (6.8× 10-6) dEπ)q 0dR 0 dE*π)q*0dR0 ∆dEπ)(CLO 2 -CHO 2 )dR0 2
Downloaded by NATIONAL TAIWAN UNIV on September 3, 2009 | http://pubs.acs.org
calculated vibrational shifts are in good agreement with available experimental data. Red shifts up to 358 cm-1
are predicted for the OH stretching vibrations. The HOH bending vibrations are blue shifted and the CO stretching frequencies are red shifted in complexes. A small but noticeable red shift of the C-H stretch can be observed in the (CH3)2CO‚(H2O)2complex 2a. In the complexes, the excitation energy of the n-π* electronic transition is blue shifted by 0.25-0.40 eV, which is in agreement with the experimental blue shift observed in liquid acetone/ H2O. The oscillator strength for the n-π* transition increases from zero (symmetry-forbidden in free acetone) to ∼10-4
in (CH3)2CO‚(H2O)3. However, the effect of the coordination of water molecules on the spectral intensity is expected to be weaker than the effect due to vibronic coupling.
Acknowledgment. This work is supported in part by the National Science Council of ROC (NSC-86-2113-M-001-043CT). A.M.M. is grateful to Academia Sinica for the fellowship at IAMS. D.W.L. gratefully acknowledges an invita-tion from IAMS. We are thankful to Dr. H. C. Chang for a valuable discussion.
References and Notes
(1) Merchan, M.; Roos, B. O.; McDiarmid, R.; Xing, X. J. Chem. Phys.
1996, 104, 1791.
(2) Xing, X.; McDiarmid, R.; Philis, J. G.; Goodman, L. J. Chem. Phys.
1993, 99, 7565.
(3) Buzza, S. A.; Snyder, E. M.; Castleman, A. W. J. Chem. Phys.
1996, 104, 5040.
(4) Walzl, K. N.; Koerting, C. F.; Kuppermann, A. J. Chem. Phys.
1987, 87, 3796.
(5) Galasso, V. J. Chem. Phys. 1990, 92, 2495.
(6) Gaines, G. A.; Donaldson, D. J.; Strickler, S. J.; Vaida, V. J. Phys.
Chem. 1988, 92, 2762.
(7) Donaldson, D. J.; Gaines, G. A.; Vaida, V. J. Phys. Chem. 1988,
92, 2766.
(8) Buzza, S. A.; Snyder, E. M.; Card, D. A.; Folmer, D. E.; Castleman, A. W. Jr. J. Chem. Phys. 1996, 105, 7425.
(9) North, S. W.; Blank, D. A.; Gezelter, J. D.; Longfellow, C. A.; Lee Y. T. J. Chem. Phys. 1995, 102, 4447.
(10) Hall, G. E.; Bout, D. V.; Sears, T. J. Chem. Phys. 1991, 94, 4182. (11) Brodka, A.; Zerda, T. W. J. Chem. Phys. 1996, 104, 6313. (12) Brodka, A.; Zerda, T. W. J. Chem. Phys. 1996, 104, 6319. (13) Jorgensen, W. L.; Briggs, J. M.; Contreras, M. L. J. Phys. Chem.
1990, 94, 1683.
(14) Hess, B.; Bruna, P. J.; Buenker, R. J.; Peyerimhoff, S. D. Chem.
Phys. 1976, 18, 267.
(15) Gwaltney, S. R.; Bartlett, R. J. Chem. Phys. Lett. 1995, 241, 26. (16) Suto, M.; Wang, X.; Lee, L. C. J. Chem. Phys. 1986, 85, 4228. (17) Herzberg, G. Molecular Spectra and Molecular Structure. III.
Electronic Spectra and Electronic Structure of Polyatomic Molecules; Van
Nostrand Reinhold: New York, 1966; p 658.
(18) Zhang, X. K.; Lewars, E. G.; March, R. E.; Parnis, J. M. J. Phys.
Chem. 1993, 97, 4320.
(19) Williams, R. W.; Lowrey, A. H. J. Comput. Chem. 1991, 12, 761. (20) (a) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. (b) Lee, C.; Yang, W.; Parr, R. G. Phys. ReV. 1988, B 37, 785.
(21) Hehre, W. J.; Radom, L.; Schleyer, P. v. R.; Pople, J. Ab Initio
Molecular Orbital Theory; Wiley: New York, 1986.
(22) Rauhut, G.; Pulay, P. J. Phys. Chem. 1995, 99, 3093.
(23) (a) Mebel, A. M.; Morokuma, K.; Lin, M. C. J. Chem. Phys. 1995,
103, 7414. (b) Liu, R.; Morokuma, K.; Mebel, A. M.; Lin, M. C. J. Phys. Chem. 1996, 100, 9314.
(24) Curtiss, L. A.; Jones, C.; Trucks, G. W.; Raghavachari, K.; Pople, J. A. J. Chem. Phys. 1990, 93, 2537.
(25) Curtiss, L. A.; Redfern, P. C.; Smith, B. J.; Radom, L. J. Chem.
Phys. 1996, 104, 5418.
(26) Foresman, J. B.; Head-Gordon, M.; Pople, J. A.; Frisch, M. J. J.
Phys. Chem. 1992, 96, 135.
(27) (a) Werner, H.-J.; Knowles, P. J. J. Chem. Phys. 1985, 82, 5053. (b) Knowles, P. J.; Werner, H.-J. Chem. Phys. Lett. 1985, 115, 259.
(28) Werner, H.-J. Mol. Phys. 1996, 89, 645.
(29) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.; Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T.; Petersson, G. A.; Montgomery, J. A.; Raghavachari, K.; Al-Laham, M. A.; Zakrzewski, V. G.; Ortiz, J. V.; Foresman, J. B.; Cioslowski, J.; Stefanov, B. B.; Nanayakkara, A.; Challacombe, M.; Peng, C. Y.; Ayala, P. Y.; Chen, W.; Wong, M. W.; Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Binkley, J. S.; Defrees, D. J.; Baker, J.; Stewart, J. P.; Head-Gordon, M.; Gonzalez, C.; Pople, J. A. GAUSSIAN 94, Revision D.4; Gaussian, Inc.: Pittsburgh, PA, 1995.
(30) MOLPRO is a package of ab initio programs written by H.-J. Werner and P. J. Knowles, with contributions from J. Almlo¨f, R. D. Amos, M. J. O. Deegan, S. T. Elbert, C. Hampel, W. Meyer, K. Peterson, R. Pitzer, A. J. Stone, P. R. Taylor and R. Lindh. Information how to obtain MOLPRO is available on the WWW site http://www.tc.bham.ac.uk/molpro/mol-pro.html.
(31) Wiberg, K. B. Tetrahedron 1968, 24, 1083.
(32) Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. ReV. (Washington,
D.C.) 1988, 88, 899.
(33) Nelson, R.; Pierce, L. J. Mol. Spectrosc. 1965, 18, 344. (34) Boys, S. F.; Bernardi, F. Mol. Phys. 1970, 19, 533.
(35) (a) Gutowski, M.; Chalasinski, G. J. Chem. Phys. 1993, 98, 533. (b) Schwenke, D. W.; Truhlar, D. G. J. Chem. Phys. 1985, 82, 2418.
(36) Fraser, G. T.; Leopold, K. R.; Klemperer, W. J. Chem. Phys. 1984,
80, 1423.
(37) Erickson, J. A.; McLoughlin, J. I. J. Org. Chem. 1995, 60, 1626. (38) Masunov, A.; Dannenberg, J. J. J. Mol. Struct. (THEOCHEM) 1996,
371, 17.
(39) Curtiss, L. A.; Blander, M. Chem. ReV. (Washington, D.C.) 1988,
88, 827.
(40) (a) Frisch, M. J.; Del Bene, J. E.; Binkley, J. S.; Schaefer, H. F., III. J. Chem. Phys. 1986, 84, 2279. (b) van Duijneveldt-van de Rijdt, J. G. C. M.; van Duijneveldt, F. B. J. Chem. Phys. 1992, 97, 5019. (c) Mo´, O.; Ya´nez, M.; Elguero, J. J. Chem. Phys. 1992, 97, 6628. (d) Xantheas, S. S.; Dunning, T. H., Jr. J. Chem. Phys. 1993, 99, 8774. (e) Suhai, S. J. Phys.
Chem. 1995, 99, 1172. (f) Kim, J.; Lee, J. Y.; Mhin, B. J.; Kim, K. S. J. Chem. Phys. 1995, 102, 310. (g) Gonza´lez, L.; Mo´, O.; Ya´nez, M.; Elguero,
J. J. Mol. Struct. (THEOCHEM) 1996, 371, 1.
(41) Gerhards, M.; Kimpfel, B.; Pohl, M.; Schmitt, M.; Kleinermanns, K. J. Mol. Struct. 1992, 270, 301.
(42) Pople, J. A.; Sidman, J. W. J. Chem. Phys. 1957, 27, 1270. (43) Van Dijk, J. M. F.; Kemper, M. J. H.; Kerp, J. H. M.; Buck, H. M.
J. Chem. Phys. 1978, 69, 2453.
Downloaded by NATIONAL TAIWAN UNIV on September 3, 2009 | http://pubs.acs.org